
Introduction
Metabolic syndrome is a multi-pathological manifestation of syndrome, comprising obesity, dyslipidemia, insulin resistance, and type 2 diabetes mellitus (T2MD), etc. These diseases are in a close connection with increased risk of hepatocellular carcinoma (HCC) occurrence and development (Figure 1) [1]. Based on the existing study, insulin resistance, along with its associated adipocyte cytokines, hyperglycemia, and hyperinsulinemia may lead to hypertension and abnormal lipid profile, both of which will accelerate the progress of HCC [2]. With the wildly performing of genome-wide association studies (GWASs) and gene sequencing, substantial genetic component compositions were proposed [3]. For fat and lean mass in different body regions, including whole body and trunk fat mass, the heritability have been estimated to about 65% [4]. It is supposed that distinction in fat distribution and illustration of genetic factors predisposing to adiposity could contribute to a further exploration of the phenotypic diversity and eventually make a more accurate disease sub-classification possible. In the past few decades, huge efforts have been made to explore the common genetic factors in metabolic syndrome. However, it is a challenging task at last for the extensive heterogeneity. Nevertheless, the pattern which have found common genetic variations associated with various symptoms and diseases, had been broken by the emergence of GWASs [5]. Furthermore, pinpointing genes implicated in metabolic syndrome could be helpful to discover the latent biological pathways, which could be exploited in the development of medical treatment. Here, we briefly summarize the relationship between metabolic syndrome and HCC, focusing on the genetic factors contributed to metabolic syndrome and HCC.
Association between metabolic syndrome and HCC
Obesity and hepatocellular carcinoma (HCC)
Over the past few decades, the prevalence of obesity has sharp increased [6]. Overweight and obesity have been identified as independent risk factors for various cancers including liver cancer, breast cancer, endometrial cancer, colorectal cancer and esophagus cancer. There is also a possible association between obesity and gallbladder, pancreas, thyroid and hematologic malignancies [7]. For HCC, research showed that the body mass index (BMI) more than 30 kg/m2 could increase HCC risk [8]. Besides, HCC mortality rates in male with high BMI are 5 times higher than men of normal weight [7]. Comprehensive analysis including 11 studies from Asia, Europe and United States evidenced that both obesity (RR = 1.89) and overweight (RR = 1.17) were closely associated with the development of HCC [9]. SEER-Medicare data analysis showed that adjusted OR of obesity for HCC was 1.93 [10]. The Metabolic Syndrome and Cancer Project (Me-Can) examined 578700 subjects from Austria, Norway and Sweden, showed RR of obesity for the development of HCC was 1.39 [11]. Danish and Korean boffins obtained similar conclusions after analyzing large cohorts of overweight patients [12]. In addition, in a large retrospective cohort including 342 patients who transplanted liver for HCC, BMI was identified as an independent predictor of capillaries invasion [13].
Type 2 diabetes mellitus (T2DM) and HCC
For developing chronic liver disease and HCC, T2DM act as an independent risk factor [14]. In different cohort studies, the morbidity of HCC among those with diabetes rose from 1.5 to 4 times [10, 15]. A recent meta-analysis of 21 cohort studies demonstrated that the summary RR of HCC with T2DM was 1.86 (95% CI: 1.49-2.31) for patients with chronic liver disease [16]. T2DM combined with its relative hyperinsulinemia and hyper-insulin-like growth factor 1 (IGF-1) production may also accelerate the development of HCC [17]. Diabetes seems to increase the risk of liver cancer by cooperating with other variables, such as viral hepatitis and alcohol [18]. Furthermore, coexistent with diabetes appears to make the HCC relapse rate rise after curative therapy [19].
Insulin resistance and HCC
Insulin resistance is characterized by reduced sensitivity to insulin and, as a result, impaired ability of insulin to reduce peripheral blood glucose concentration [20]. It is well established that insulin resistance act as a primary dominator that connects all the sections of metabolic syndrome [21]. To overcome insulin resistance and maintain normal metabolic functions, insulin secretion is increased, leading to a state of compensatory hyperinsulinemia [22]. Insulin resistance may directly accelerate hepatocarcinogenesis via promoting cellular proliferation, inhibiting apoptosis and stimulating hepatic neovascularization [23]. However, a 10-year prospective study from Korea of 1,298,385 patients showed that the risk of tumor was influenced by fasting blood glucose concentrations rather than insulin resistance, which challenges the theory that insulin directly effects on promotion of cancers [24].
Non-alcoholic fatty liver disease (NAFLD) and HCC
NAFLD is the clinical manifestation in liver of metabolic syndrome [6]. In Western countries and Asia, NAFLD is considered as the most common cause of chronic liver disease [25]. The clinic pathological classification of NAFLD include from extensive isolated steatosis (a milder form of hepatic adipose infiltration) to nonalcoholic fatty liver disease (a more severe form of hepatic adipose infiltration). The clinical progress of simple steatosis is relatively gentle, but there are about one third of non-alcoholic steatohepatitis (NASH) affected subjects can develop into liver cirrhosis [26], and about 27% of NASH-associated hepatic cirrhosis eventually progresses to HCC [27, 28]. In recent years, NASH has been considered as an important pathogenic factor of HCC. Thus, since the prevalence of obesity, diabetes and metabolic syndrome continues to rise, a large proportion of the population will have a risk of developing NASH and cirrhosis, and ultimately liver cancer [29, 30].
Susceptibility loci for metabolic syndrome and HCC
The advances in genome technology, human genome deep sequencing, and the establishment of human variation cataloging, which helped the human genome analysis to create a new era, have further promoted the complex disease genetics research. It was not until 2007 that the genetic map of complex diseases, such as obesity and HCC, had been preliminarily studied by genetic linkage analysis and candidate gene association studies. But for the potential limitations of the design, their application was limited. However, since 2007, through genome-wide association studies, there is a rocket increase in our understandings of specific genetic risk factors for adiposity, insulin resistance, T2MD, NAFLD and HCC. These advances contributed a lot of convincing genetic loci related to the complex metabolic symptom and HCC [31].
Obesity
The inheritance of obesity has been paid large attention and the genetic research of obesity has been the focus of research. The heritability of obesity is now universally thought to be range from 40% to 70% [32]. In general, as of 2016, GWASs have successfully identified about 400 different loci associated with adiposity phenotypes [http://www.ebi.ac.uk/gwas/]. The discoveries are mainly derived from individual studies and meta-analysis based on BMI as an indicator of obesity. Through GWAS, Frayling et al. reported the first BMI gene locus with significant statistical significance, which is a byproduct of the study for FTO gene associated with T2MD by GWAS [33]. In 2008, a meta-analysis of GWASs from 16,876 European individuals replicated the FTO finding and also found another strong signal 188 kb downstream of the MC4R locus [34]. In 2009, a paper reported a meta-analysis of GWASs from GIANT consortium, which included more than 32,000 individuals with independent replication from >59,000 individuals. They replicated the MC4R and FTO loci and reported six novel loci: TMEM18, SH2B1, GNPDA2, KCTD15, NEGR1, and MTCH2 [35]. In 2012, a meta-analysis of GWASs for BMI in East Asians was performed. They replicated seven previously identified loci (FTO, SEC16B, MC4R, GIPR-QPCTL, ADCY3-DNAJC27, BDNF and MAP2K5) and identified three additional loci in or near the CDKAL1, GP2, and PCSK1 genes [36]. In 2013, Monda et al. conducted a meta-analysis in African individuals and identified two new loci in GALNT10 and MIR148A-NFE2L3 [37]. Then, Pei et al. carried out a meta-analysis of seven GWASs for BMI-related traits from diverse ethnic populations. They confirmed three previously reported obesity susceptibility loci in FTO, MC4R and TMEM18; and identified two novel loci in CTSS and NLK associated with fat body mass [38]. Recently, another meta-analysis among Asians was conducted by Wen et al. and four loci near the KCNQ1, ITIH4, NT5C2 and ALDH2/MYL2 genes were identified [39]. FTO is a common susceptibility gene for BMI in Asian, African and European populations. And it remains the most influential gene, imposing an allelic 0.39 kg/m2 increase in BMI [34].
Besides, in 2011, Heid et al. published a meta-analysis of 32 GWASs of Waist-hip ratio (WHR) adjusted for BMI with follow-up of 16 loci in an additional 29 studies. They identified 13 novel loci in or near RSPO3, NFE2L3, VEGFA, TBX15-WARS2, ITPR2-SSPN, GRB14, DNM3-PIGC, HOXC13, ADAMTS9, LY86, ZNRF3-KREMEN1, CPEB4, and NISCH-STAB1 and the known signal at LYPLAL1 unique to fat distribution independently of BMI. In 2013, a GWAS of WHR in up to 27,350 African individuals was performed and rs6931262 at RREB1 was identified for WHR adjusted for BMI [40].
T2DM
Having a sibling with T2DM will increases the risk of T2DM by two to three times [41], suggesting genetic factors play an important role in T2DM. As of 2016, more than 200 genetic loci have been identified as T2DM risk loci [http://www.ebi.ac.uk/gwas/]. In 2006, the risk variant in TCF7L2 was discovered by a positional linkage strategy in Iceland population [42], which remains the most influential common T2DM variant (allelic OR = ~1.46) [43]. Then it was successfully validated by several other GWASs [44-46]. Soon, multiple T2DM loci were discovered by independent GWASs, such as SLC16A11, CDKAL1, FTO, HHEX, IGF2BP2, KCNJ11, PPARG, SLC30A8 et al.[44, 45, 47]. DIAGRAM conducted the first meta-analysis on T2DM comprising 10,128 European individuals at the discovery stage and identified six novel loci including ADAMTS9, CDC123-CAMK1D, JAZF1, NOTCH2, TSPAN8-LGR5, and THADA [48]. The second meta-analysis by DIAGRAM had a sample size of 8130 cases and 38,987 controls of European descent. Notably 12 T2DM loci harboring genes that included BCL11A, CENTD2, CHCHD9, HMGA2, DUSP9, ZBED3, HNF1A, KLF14, PRC1, KCNQ1, TP53INP1, and ZFAND6 were identified [49]. The third effort made by DIAGRAM included 26,488 cases and 83,964 controls of diverse ethnic populations. Eight novel loci were discovered: ANK1, ANKRD55, BCAR1, CILP2, KLHDC5, MC4R, TLE1 and ZMIZ1 [50]. In addition, AGEN-T2DM conducted a meta-analysis including approximately 55,000 individuals in East Asian populations and identified eight novel T2DM loci in European populations: MAEA, FITM2-R3HDML-HNF4A, GCC1-PAX4, GLIS3, KCNK16, PEPD, PSMD6 and ZFAND3 [51].
Although T2DM and obesity are highly interrelated from both epidemiological and pathophysiological viewpoints, they have few genetic risk loci in common. Of 200 loci associated with T2DM and 400 loci associated with standard measures of adiposity, merely 11 loci are shared (CDKAL1, FTO, GIPR, KCNQ1, LINGO2, LYPLAL1, MC4R, TMEM18, GRB14, RREB1 and ZNF608), which may do not include shared associations below the level of genome-wide statistical significance. However, for SNPs primarily associated with BMI, there seems to be a positive correlation between the effect size on BMI and the effect of the same SNP on T2DM [31].
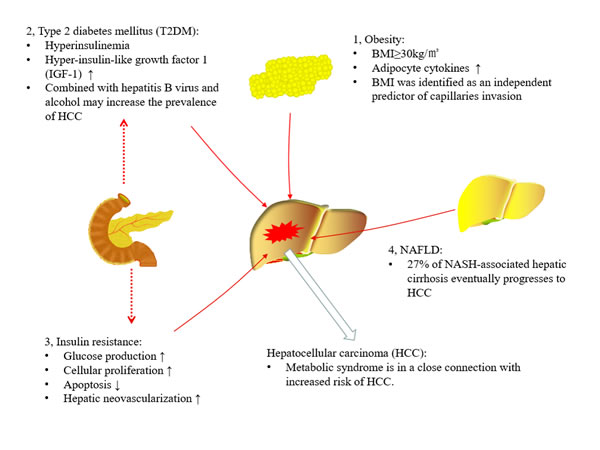
Figure 1: Association between metabolic syndrome and hepatocellular carcinoma (HCC). Metabolic syndrome is in a close connection with an increased risk of HCC. In obese patients, adipocyte cytokines may lead to abnormal lipid profile, and the body mass index (BMI) more than 30 kg/m2 could increase HCC risk. In type 2 diabetes mellitus (T2MD), its relative hyperinsulinemia and hyper-insulin-like growth factor 1 (IGF-1) production, or synergistic actions with other variables, such as viral hepatitis and alcohol, all may contribute to the development of HCC. In addition, insulin resistance impaired the ability of insulin to suppress glucose production, and directly accelerate hepatocarcinogenesis via promoting cellular proliferation, inhibiting apoptosis and stimulating hepatic neovascularization. Moreover, bout 27% of non-alcoholic steatohepatitis (NASH)-associated hepatic cirrhosis eventually progresses to HCC.
Insulin resistance
The T2DM physiologic characteristics are pancreatic β-cell dysfunction and insulin resistance in the peripheral tissues and liver [52]. Interestingly, T2DM susceptibility loci seldom map directly to insulin resistance. Multiple independent studies showed that the insulin resistance susceptibility loci included APOC3, GCKR, IRS1, IGF1, PPARG and so on [53]. By 2016, about 70 genetic loci have been identified as insulin resistance risk loci [http://www.ebi.ac.uk/gwas/]. In 2011, based on the HyperGen study, a GWAS including 1,040 African Americans explored the association between insulin resistance and genetic variation. The results showed SNPs linked with homeostasis model assessment of insulin resistance (HOMA-IR) and fasting insulin near ATP10A (rs6576507 and rs8026527) and CACNA1D (rs1401492) [54].
Obesity is related with insulin resistance, and also a strong risk factor for T2DM. Thus, we can assume reasonably that some loci of obesity would also be insulin resistance loci, or possibly T2DM susceptibility loci. However, there is little overlap among loci for these traits. These results suggest that the etiologically distinct subsets may exist in these extreme phenotypes.
NAFLD
NAFLD appears family genetic predisposition. Compared with overweight children without NAFLD, fatty liver was found to be more common in children whose siblings and parents had NAFLD. [55]. After adjusting age, race, sex, and BMI, the heritability of NAFLD was estimated to be 39%, suggesting close relationship between the development of NAFLD and genetic factors. To date, a number of potential genetic determinants based on GWASs have been proposed. The Dallas Heart Study firstly identified the nonsynonymous rs738409 (I148M) located in PNPLA3, which was the most important genetic variant associated with NAFLD [56]. After adjustment for ethnicity, BMI, diabetes status and alcohol use, PNPLA3 rs738409 was significantly associated with hepatic fat content. The results were subsequently confirmed by many independent studies [57] and other GWASs [58]. A meta-analysis in 2011 including 16 studies also proposed a strong association between PNPLA3 rs738409 and a more aggressive disease. Homozygous GG will lead to 28% increase in serum ALT levels, 3.5-fold greater risk of NASH, and 3.3-fold higher risk of fibrosis. [59]. The second GWAS about NAFLD was performed in 236 women with NAFLD and identified an association between SNP rs2645424 in FDFT1 (an enzyme with a role in cholesterol synthesis) and NAFLD activity score [60]. In 2011, Speliotes et al. conducted the third NAFLD GWAS [58], and identified five SNPs associated with NAFLD in or near PNPLA3 (rs738408), NCAN (rs2228603), PPP1R3B (rs4240624), GCKR (rs780094) and LYPLAL1 (rs12137855). There was a strong linkage disequilibrium between PNPLA3 rs738408 and the previously identified rs738409 [56]. Another recently GWAS in adolescents with NAFLD identified SNPs relevant to two neuron-specific genes (SLC38A8 and LPPR4) and two liver-specific genes (LCP1and GC). This study further confirmed the significant differential expression of GC and LCP1 in a NAFLD cohort [61]. Another GWAS was conducted in the Japanese population involving 392 NAFLD subjects and 934 controls. In addition to those polymorphisms in the PNPLA3 gene, the polymorphisms in SAMM50 and PARVB were also observed to be associated with the occurrence and progression of NAFLD. [62].
HCC
As we known, virus infection, obesity, diabetes mellitus, alcohol and aflatoxin B1 exposure are pivotal risk factors for inducing HCC. Genetic factors of the individual genome may also act a role in liver malignant tumor [63]. In recent years, a plethora of studies have confirmed that the host genetic factors play crucial roles in developing HBV-induced HCC. One of the first GWASs included 355 HBV carriers with HCC and 360 asymptomatic HBV carriers (ASCs) in Chinese population, indicated rs17401966 in KIF1B was significantly associated with HBV-related HCC, and SNPs in UBE4B and PGD genes were also shown to be significant for HCC emergence among patients with HBV-positive [64]. In recent years, a large number of studies have confirmed that the host genetic factors played a key role in the development of HBV-related liver cancer. The other two GWASs from Chinese also found some novel SNPs as risk factors for HBV-related HCC, including rs9272105 in HLA-DQA1/DRB1, rs455804 in GRIK1 [65], rs9275319 in HLA-DQ gene and rs7574865 in STAT4 gene [66]. Two GWASs conducted in large Japanese cohorts concluded that variant rs2596542 in the promoter region of the MICA gene [67], and SNP rs1012068 in DEPDC5 gene [68] was significantly related to HCV-induced HCC.
Ancestry-specific genetic susceptibility loci
Most of the GWASs have been completed in the European, and there are many studies are emerging in other races. Studies of obesity have shown highly comparable effects of common variants across major ancestry groups, strongly supporting shared common BMI and obesity loci across populations, although ancestry-specific loci have also been shown, such as KLHL32 in Africans and KLF9 in Asians [37]. Moreover, of the 14 WHR-associated loci, only 7 were found to have a significant effect in women [69]. For T2DM, studies have reported novel loci such as KCNQ1and C2CD4A in Japanese individuals [70] and a number of loci for T2DM in East Asians [51, 71]. The ancestry-specific genetic susceptibility loci also exist in NAFLD and related cancer, including HCC and pancreatic cancer, suggesting great heterogeneity between the genetic background of different races or populations. It also presented challenges for precise medicine and personalized medicine.
Outlook
We remain in infancy of the research for potential molecular basis in the HCC development. More specifically, in patients with NASH, the genetic variants and mechanisms that drive the development of HCC remains largely mysterious. Based on the current GWASs, we hope to find new genetic components related to metabolic syndrome and liver cancer progression, yet major efforts are still needed to gain biological knowledge from discoveries.
Seeking the missing heritability
The past 8 years of genetic discoveries brought about by the GWASs approach have meant a giant leap for genetic research of complex traits, with more than 288 genetic loci shown to associate with metabolic traits. Yet, the major part of the genetic predisposition to these phenotypes remains unaccounted since the proportion of variance explained by genetic risk variants discovered to date is limited. Implicit in the initial design, GWASs-identified variants in metabolic syndrome and related cancer are common (minor allele frequency >5% in the population). There has been much focus on this missing heritability [72]. The rare alleles with large effects are the primary drivers of common disease, which has received renewed attention. The focus is needed to change to study the individual and combined effect of low-frequency and rare variants in metabolic disease through the analysis of data from microarray-based genotyping and wide-genome sequencing.
In addition, influences other than main effects of SNPs may explain parts of the susceptibility for metabolic disease. Preliminary evidence supports the role of copy number variations and gene-environment interactions in obesity, T2DM and HCC, as solid findings have been reported [73, 74], and future large-scale studies may reveal gene-gene interactions.
Identifying causal variants
GWASs contribute to find the common genetic variants between normal and pathological features, but the major challenge is how to recognize the precise targets of those associations among them. The studies for investigating gene function for the loci from GWASs are needed, including accurate mapping of GWASs signal(s) combined with genetic epidemiology and bioinformatics methods of the integration and optimization of putative functional SNPs, and in vitro and in vivo experimental verification for predicted molecular mechanism to identify the targeted genes [75].
Risk prediction
As discussed above, common genetic variants unanimously impose modest risk increments on metabolic syndrome and related cancer. Furthermore, combining these variants does not enable prediction these disease/traits [76, 77]. Future studies are needed to reveal further important genetic susceptibility elements. In addition, systemic integration of complex date obtained from other “omics” such as transcriptomics, proteomics and metabolomics, and modeling research for composite effect on the integrate common metabolic phenotype, is projected to be a breakthrough in the research of genetic determinants of metabolic traits. Besides gaining biological knowledge and allowing the identification of at-risk individuals, hopes have been high that knowledge of genetic risk factors would lead to personalized treatment based on the genetic profile. With the developing of the polymorphism risk-scoring algorithm and gene sequencing, it is plausible for identification high-risk groups for prevention and healthcare, and early screening for potential biomarkers. And the complex molecular mechanisms for the NASH, related cirrhosis and HCC need more retrospective and prospective studies to underpin. Since there is a strong association between NASH and HCC, the direction of the study about NAFLD/NASH treatment should focus on how to reduce the risk of cancer in these patients, provide long-term benefits and reduce socio-economic pressure. With the continued popularity of obesity, the prevalence of diabetes mellitus and metabolic syndrome is increasing, so it is imperative to screen the high risk groups of HCC in patients with metabolic syndrome, and to provide the appropriate monitoring strategies.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
GRANT SUPPORT
This work was supported in part by the National Key Basic Research Program of China (2013CB530604), the National Natural Science Foundation of China (81600685), the Natural Science Foundation of Jiangsu Province (BK20160141), the Medical Science and technology development Foundation of Nanjing Department of Health (YKK16201), and the Science and Technology Development Fund of the Nanjing Medical University (2015NJMUZD062).
References
1. Scalera A, Tarantino G. Could metabolic syndrome lead to hepatocarcinoma via non-alcoholic fatty liver disease? World J Gastroenterol. 2014;20:9217-9228.
2. Elsayed EY, Mosalam NA, Mohamed NR. Resistin and Insulin Resistance: A Link Between Inflammation and Hepatocarcinogenesis. Asian Pac J Cancer Prev. 2015;16:7139-7142.
3. Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J, Croteau-Chonka DC, Esko T, Fall T, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197-206.
4. Hsu FC, Lenchik L, Nicklas BJ, Lohman K, Register TC, Mychaleckyj J, Langefeld CD, Freedman BI, Bowden DW, Carr JJ. Heritability of body composition measured by DXA in the diabetes heart study. Obes Res. 2005;13:312-319.
5. Sandholt CH, Grarup N, Pedersen O, Hansen T. Genome-wide association studies of human adiposity: Zooming in on synapses. Mol Cell Endocrinol. 2015;418 Pt 2:90-100.
6. Yu J, Shen J, Sun TT, Zhang X, Wong N. Obesity, insulin resistance, NASH and hepatocellular carcinoma. Semin Cancer Biol. 2013;23:483-491.
7. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625-1638.
8. Chen CL, Yang HI, Yang WS, Liu CJ, Chen PJ, You SL, Wang LY, Sun CA, Lu SN, Chen DS, Chen CJ. Metabolic factors and risk of hepatocellular carcinoma by chronic hepatitis B/C infection: a follow-up study in Taiwan. Gastroenterology. 2008;135:111-121.
9. Larsson SC, Wolk A. Overweight, obesity and risk of liver cancer: a meta-analysis of cohort studies. Br J Cancer. 2007;97:1005-1008.
10. Welzel TM, Graubard BI, Zeuzem S, El-Serag HB, Davila JA, McGlynn KA. Metabolic syndrome increases the risk of primary liver cancer in the United States: a study in the SEER-Medicare database. Hepatology. 2011;54:463-471.
11. Borena W, Strohmaier S, Lukanova A, Bjorge T, Lindkvist B, Hallmans G, Edlinger M, Stocks T, Nagel G, Manjer J, Engeland A, Selmer R, Haggstrom C, et al. Metabolic risk factors and primary liver cancer in a prospective study of 578,700 adults. Int J Cancer. 2012;131:193-200.
12. Oh SW, Yoon YS, Shin SA. Effects of excess weight on cancer incidences depending on cancer sites and histologic findings among men: Korea National Health Insurance Corporation Study. J Clin Oncol. 2005;23:4742-4754.
13. Siegel AB, Lim EA, Wang S, Brubaker W, Rodriguez RD, Goyal A, Jacobson JS, Hershman DL, Verna EC, Zaretsky J, Halazun K, Dove L, Brown RS JR, et al. Diabetes, body mass index, and outcomes in hepatocellular carcinoma patients undergoing liver transplantation. Transplantation. 2012;94:539-543.
14. Rosmorduc O, Fartoux L. HCC and NASH: how strong is the clinical demonstration? Clin Res Hepatol Gastroenterol. 2012;36:202-208.
15. Setiawan VW, Hernandez BY, Lu SC, Stram DO, Wilkens LR, Le Marchand L, Henderson BE. Diabetes and racial/ethnic differences in hepatocellular carcinoma risk: the multiethnic cohort. J Natl Cancer Inst. 2014;106.
16. Chen J, Han Y, Xu C, Xiao T, Wang B. Effect of type 2 diabetes mellitus on the risk for hepatocellular carcinoma in chronic liver diseases: a meta-analysis of cohort studies. Eur J Cancer Prev. 2015;24:89-99.
17. Smedile A, Bugianesi E. Steatosis and hepatocellular carcinoma risk. Eur Rev Med Pharmacol Sci. 2005;9:291-293.
18. Donato F, Gelatti U, Limina RM, Fattovich G. Southern Europe as an example of interaction between various environmental factors: a systematic review of the epidemiologic evidence. Oncogene. 2006;25:3756-3770.
19. Teoh NC, Fan JG. Diabetes mellitus and prognosis after curative therapy for hepatocellular carcinoma: alas, still grave for those who are hyperglycemic. J Gastroenterol Hepatol. 2008;23:1633-1634.
20. Siddique A, Kowdley KV. Insulin resistance and other metabolic risk factors in the pathogenesis of hepatocellular carcinoma. Clin Liver Dis. 2011;15:281-296, vii-x.
21. Gaggini M, Morelli M, Buzzigoli E, DeFronzo RA, Bugianesi E, Gastaldelli A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients. 2013;5:1544-1560.
22. Bugianesi E, McCullough AJ, Marchesini G. Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology. 2005;42:987-1000.
23. Kaji K, Yoshiji H, Kitade M, Ikenaka Y, Noguchi R, Yoshii J, Yanase K, Namisaki T, Yamazaki M, Moriya K, Tsujimoto T, Kawaratani H, Akahane T, et al. Impact of insulin resistance on the progression of chronic liver diseases. Int J Mol Med. 2008;22:801-808.
24. Jee SH, Ohrr H, Sull JW, Yun JE, Ji M, Samet JM. Fasting serum glucose level and cancer risk in Korean men and women. JAMA. 2005;293:194-202.
25. Wong VW. Nonalcoholic fatty liver disease in Asia: a story of growth. J Gastroenterol Hepatol. 2013;28:18-23.
26. Serfaty L, Lemoine M. Definition and natural history of metabolic steatosis: clinical aspects of NAFLD, NASH and cirrhosis. Diabetes Metab. 2008;34:634-637.
27. Wong VW, Wong GL, Choi PC, Chan AW, Li MK, Chan HY, Chim AM, Yu J, Sung JJ, Chan HL. Disease progression of non-alcoholic fatty liver disease: a prospective study with paired liver biopsies at 3 years. Gut. 2010;59:969-974.
28. Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820-1832.
29. Fan JG, Farrell GC. Epidemiology of non-alcoholic fatty liver disease in China. J Hepatol. 2009;50:204-210.
30. Toffanin S, Friedman SL, Llovet JM. Obesity, inflammatory signaling, and hepatocellular carcinoma-an enlarging link. Cancer Cell. 2010;17:115-117.
31. Grarup N, Sandholt CH, Hansen T, Pedersen O. Genetic susceptibility to type 2 diabetes and obesity: from genome-wide association studies to rare variants and beyond. Diabetologia. 2014;57:1528-1541.
32. Barsh GS, Farooqi IS, O’Rahilly S. Genetics of body-weight regulation. Nature. 2000;404:644-651.
33. Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JR, Elliott KS, Lango H, Rayner NW, Shields B, Harries LW, Barrett JC, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889-894.
34. Loos RJ, Lindgren CM, Li S, Wheeler E, Zhao JH, Prokopenko I, Inouye M, Freathy RM, Attwood AP, Beckmann JS, Berndt SI, Prostate LC, Ovarian Cancer Screening T, et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet. 2008;40:768-775.
35. Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM, Berndt SI, Elliott AL, Jackson AU, Lamina C, Lettre G, Lim N, Lyon HN, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25-34.
36. Wen W, Cho YS, Zheng W, Dorajoo R, Kato N, Qi L, Chen CH, Delahanty RJ, Okada Y, Tabara Y, Gu D, Zhu D, Haiman CA, et al. Meta-analysis identifies common variants associated with body mass index in east Asians. Nat Genet. 2012;44:307-311.
37. Monda KL, Chen GK, Taylor KC, Palmer C, Edwards TL, Lange LA, Ng MC, Adeyemo AA, Allison MA, Bielak LF, Chen G, Graff M, Irvin MR, et al. A meta-analysis identifies new loci associated with body mass index in individuals of African ancestry. Nat Genet. 2013;45:690-696.
38. Pei YF, Zhang L, Liu Y, Li J, Shen H, Liu YZ, Tian Q, He H, Wu S, Ran S, Han Y, Hai R, Lin Y, et al. Meta-analysis of genome-wide association data identifies novel susceptibility loci for obesity. Hum Mol Genet. 2014;23:820-830.
39. Wen W, Zheng W, Okada Y, Takeuchi F, Tabara Y, Hwang JY, Dorajoo R, Li H, Tsai FJ, Yang X, He J, Wu Y, He M, et al. Meta-analysis of genome-wide association studies in East Asian-ancestry populations identifies four new loci for body mass index. Hum Mol Genet. 2014;23:5492-5504.
40. Liu CT, Monda KL, Taylor KC, Lange L, Demerath EW, Palmas W, Wojczynski MK, Ellis JC, Vitolins MZ, Liu S, Papanicolaou GJ, Irvin MR, Xue L, et al. Genome-wide association of body fat distribution in African ancestry populations suggests new loci. PLoS Genet. 2013;9:e1003681.
41. Kobberling J. [Predictive value of diagnostic procedures]. Dtsch Med Wochenschr. 1982;107:591-595.
42. Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, Styrkarsdottir U, Magnusson KP, Walters GB, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38:320-323.
43. Cauchi S, El Achhab Y, Choquet H, Dina C, Krempler F, Weitgasser R, Nejjari C, Patsch W, Chikri M, Meyre D, Froguel P. TCF7L2 is reproducibly associated with type 2 diabetes in various ethnic groups: a global meta-analysis. J Mol Med (Berl). 2007;85:777-782.
44. Consortium STD, Williams AL, Jacobs SB, Moreno-Macias H, Huerta-Chagoya A, Churchhouse C, Marquez-Luna C, Garcia-Ortiz H, Gomez-Vazquez MJ, Burtt NP, Aguilar-Salinas CA, Gonzalez-Villalpando C, Florez JC, et al. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature. 2014;506:97-101.
45. Saxena R, Saleheen D, Been LF, Garavito ML, Braun T, Bjonnes A, Young R, Ho WK, Rasheed A, Frossard P, Sim X, Hassanali N, Radha V, et al. Genome-wide association study identifies a novel locus contributing to type 2 diabetes susceptibility in Sikhs of Punjabi origin from India. Diabetes. 2013;62:1746-1755.
46. Perry JR, Voight BF, Yengo L, Amin N, Dupuis J, Ganser M, Grallert H, Navarro P, Li M, Qi L, Steinthorsdottir V, Scott RA, Almgren P, et al. Stratifying type 2 diabetes cases by BMI identifies genetic risk variants in LAMA1 and enrichment for risk variants in lean compared to obese cases. PLoS Genet. 2012;8:e1002741.
47. Hara K, Fujita H, Johnson TA, Yamauchi T, Yasuda K, Horikoshi M, Peng C, Hu C, Ma RC, Imamura M, Iwata M, Tsunoda T, Morizono T, et al. Genome-wide association study identifies three novel loci for type 2 diabetes. Hum Mol Genet. 2014;23:239-246.
48. Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, de Bakker PI, Abecasis GR, Almgren P, Andersen G, Ardlie K, Bostrom KB, Bergman RN, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638-645.
49. Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, McCulloch LJ, Ferreira T, Grallert H, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579-589.
50. Replication DIG, Meta-analysis C, Asian Genetic Epidemiology Network Type 2 Diabetes C, South Asian Type 2 Diabetes C, Mexican American Type 2 Diabetes C, Type 2 Diabetes Genetic Exploration by Nex-generation sequencing in muylti-Ethnic Samples C, Mahajan A, Go MJ, Zhang W, Below JE, Gaulton KJ, Ferreira T, Horikoshi M, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46:234-244.
51. Cho YS, Chen CH, Hu C, Long J, Ong RT, Sim X, Takeuchi F, Wu Y, Go MJ, Yamauchi T, Chang YC, Kwak SH, Ma RC, et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet. 2011;44:67-72.
52. Watanabe RM. The genetics of insulin resistance: Where’s Waldo? Curr Diab Rep. 2010;10:476-484.
53. Petersen KF, Dufour S, Hariri A, Nelson-Williams C, Foo JN, Zhang XM, Dziura J, Lifton RP, Shulman GI. Apolipoprotein C3 gene variants in nonalcoholic fatty liver disease. N Engl J Med. 2010;362:1082-1089.
54. Irvin MR, Wineinger NE, Rice TK, Pajewski NM, Kabagambe EK, Gu CC, Pankow J, North KE, Wilk JB, Freedman BI, Franceschini N, Broeckel U, Tiwari HK, et al. Genome-wide detection of allele specific copy number variation associated with insulin resistance in African Americans from the HyperGEN study. PLoS One. 2011;6:e24052.
55. Schwimmer JB, Celedon MA, Lavine JE, Salem R, Campbell N, Schork NJ, Shiehmorteza M, Yokoo T, Chavez A, Middleton MS, Sirlin CB. Heritability of nonalcoholic fatty liver disease. Gastroenterology. 2009;136:1585-1592.
56. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461-1465.
57. Rotman Y, Koh C, Zmuda JM, Kleiner DE, Liang TJ, Nash CRN. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology. 2010;52:894-903.
58. Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, Gudnason V, Eiriksdottir G, Garcia ME, Launer LJ, Nalls MA, Clark JM, Mitchell BD, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7:e1001324.
59. Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. 2011;53:1883-1894.
60. Chalasani N, Guo X, Loomba R, Goodarzi MO, Haritunians T, Kwon S, Cui J, Taylor KD, Wilson L, Cummings OW, Chen YD, Rotter JI, Nonalcoholic Steatohepatitis Clinical Research N. Genome-wide association study identifies variants associated with histologic features of nonalcoholic Fatty liver disease. Gastroenterology. 2010;139:1567-1576, 1576 e1561-1566.
61. Adams LA, White SW, Marsh JA, Lye SJ, Connor KL, Maganga R, Ayonrinde OT, Olynyk JK, Mori TA, Beilin LJ, Palmer LJ, Hamdorf JM, Pennell CE. Association between liver-specific gene polymorphisms and their expression levels with nonalcoholic fatty liver disease. Hepatology. 2013;57:590-600.
62. Kitamoto T, Kitamoto A, Yoneda M, Hyogo H, Ochi H, Nakamura T, Teranishi H, Mizusawa S, Ueno T, Chayama K, Nakajima A, Nakao K, Sekine A, et al. Genome-wide scan revealed that polymorphisms in the PNPLA3, SAMM50, and PARVB genes are associated with development and progression of nonalcoholic fatty liver disease in Japan. Hum Genet. 2013;132:783-792.
63. Hoshida Y, Fuchs BC, Tanabe KK. Genomic risk of hepatitis C-related hepatocellular carcinoma. J Hepatol. 2012;56:729-730.
64. Zhang H, Zhai Y, Hu Z, Wu C, Qian J, Jia W, Ma F, Huang W, Yu L, Yue W, Wang Z, Li P, Zhang Y, et al. Genome-wide association study identifies 1p36.22 as a new susceptibility locus for hepatocellular carcinoma in chronic hepatitis B virus carriers. Nat Genet. 2010;42:755-758.
65. Li S, Qian J, Yang Y, Zhao W, Dai J, Bei JX, Foo JN, McLaren PJ, Li Z, Yang J, Shen F, Liu L, Yang J, et al. GWAS identifies novel susceptibility loci on 6p21.32 and 21q21.3 for hepatocellular carcinoma in chronic hepatitis B virus carriers. PLoS Genet. 2012;8:e1002791.
66. Jiang DK, Sun J, Cao G, Liu Y, Lin D, Gao YZ, Ren WH, Long XD, Zhang H, Ma XP, Wang Z, Jiang W, Chen TY, et al. Genetic variants in STAT4 and HLA-DQ genes confer risk of hepatitis B virus-related hepatocellular carcinoma. Nat Genet. 2013;45:72-75.
67. Kumar V, Kato N, Urabe Y, Takahashi A, Muroyama R, Hosono N, Otsuka M, Tateishi R, Omata M, Nakagawa H, Koike K, Kamatani N, Kubo M, et al. Genome-wide association study identifies a susceptibility locus for HCV-induced hepatocellular carcinoma. Nat Genet. 2011;43:455-458.
68. Miki D, Ochi H, Hayes CN, Abe H, Yoshima T, Aikata H, Ikeda K, Kumada H, Toyota J, Morizono T, Tsunoda T, Kubo M, Nakamura Y, et al. Variation in the DEPDC5 locus is associated with progression to hepatocellular carcinoma in chronic hepatitis C virus carriers. Nat Genet. 2011;43:797-800.
69. Heid IM, Jackson AU, Randall JC, Winkler TW, Qi L, Steinthorsdottir V, Thorleifsson G, Zillikens MC, Speliotes EK, Magi R, Workalemahu T, White CC, Bouatia-Naji N, et al. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet. 2010;42:949-960.
70. Yamauchi T, Hara K, Maeda S, Yasuda K, Takahashi A, Horikoshi M, Nakamura M, Fujita H, Grarup N, Cauchi S, Ng DP, Ma RC, Tsunoda T, et al. A genome-wide association study in the Japanese population identifies susceptibility loci for type 2 diabetes at UBE2E2 and C2CD4A-C2CD4B. Nat Genet. 2010;42:864-868.
71. Kooner JS, Saleheen D, Sim X, Sehmi J, Zhang W, Frossard P, Been LF, Chia KS, Dimas AS, Hassanali N, Jafar T, Jowett JB, Li X, et al. Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat Genet. 2011;43:984-989.
72. Maher B. Personal genomes: The case of the missing heritability. Nature. 2008;456:18-21.
73. Walters RG, Jacquemont S, Valsesia A, de Smith AJ, Martinet D, Andersson J, Falchi M, Chen F, Andrieux J, Lobbens S, Delobel B, Stutzmann F, El-Sayed Moustafa JS, et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010;463:671-675.
74. Kilpelainen TO, Qi L, Brage S, Sharp SJ, Sonestedt E, Demerath E, Ahmad T, Mora S, Kaakinen M, Sandholt CH, Holzapfel C, Autenrieth CS, Hypponen E, et al. Physical activity attenuates the influence of FTO variants on obesity risk: a meta-analysis of 218,166 adults and 19,268 children. PLoS Med. 2011;8:e1001116.
75. Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet. 2013;93:779-797.
76. Meigs JB, Shrader P, Sullivan LM, McAteer JB, Fox CS, Dupuis J, Manning AK, Florez JC, Wilson PW, D’Agostino RB SR, Cupples LA. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med. 2008;359:2208-2219.
77. Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Lango Allen H, Lindgren CM, Luan J, Magi R, Randall JC, Vedantam S, Winkler TW, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937-948.