
INTRODUCTION
Breast cancer (BC) is the most common malignancy in women worldwide [1]. Recent advances in therapeutic strategies have improved BC-specific mortality and morbidity, but still only one-quarter of metastatic BC patients survive until 5 years after BC diagnosis [2].
In the era of next-generation sequencing (NGS), numerous genetic alterations causing BC have been discovered. The Cancer Genome Atlas (TCGA) consortium performed comprehensive genetic analysis of BCs [3]. They showed that TP53, PIK3CA, and GATA3 were the genes most commonly mutated, and that genetic alterations differed according to BC subtype (luminal A, B, basal-like, or HER2-enriched). The International Cancer Genome Consortium (ICGC) reported that 93 protein coding cancer genes carried driver mutations [4]. Similar to TCGA results, genetic alterations differed according to BC subtype; TP53, PTEN, and RB1 were the genes most frequently mutated in estrogen receptor (ER)-negative BC, whereas PIK3CA, CCND1, and GATA3 were rarely mutated in ER-negative BC.
Many clinical trials based on these mutated genes have been proposed and are on-going [5]. PIK3CA is considered a targetable potential driver of BC. PI3Kα and PIK3δ inhibitors and AKT inhibitors are being used to treat ER-positive BC patients harboring PIK3CA mutations in clinical trials [6–8]. Recently, everolimus, an mTOR inhibitor, was approved for postmenopausal ER-positive metastatic BCs [9, 10]. An additional biomarker study showed that BC patients with mutated PIK3CA derived clinical benefit from everolimus; however, BC patients with wild-type PIK3CA also responded to everolimus [11].
Here, we identified gene alterations in MBC using whole-exome and whole-transcriptome sequencing. We evaluated mutation profiles and expression patterns and analyzed the relationship between genetic alterations and expression of specific genes and pathways. Because we performed our large-scale genetic studies using BC surgical specimens, our findings, in addition to describing genetic alterations in advanced BC, could help establish treatment strategies for refractory BC. We conclude by proposing an optimal treatment plan for MBC BCs.
RESULTS
Samples and clinical data
We enrolled 54 patients with metastatic BC. Of these 54 patients, RNA sequencing was performed in 37. RNA-Seq was not performed for 17 samples due to RNA extraction failure. The characteristics of the 37 patients are described in Table 1. The median age of enrolled patients was 45.1 years, and 35.1% patients had TNBC. Fourteen of 37 patients (37.8%) had basal-like subtype BC. Five patients were tested for the BRCA1/2 mutation, and a germline BRCA1 and/or BRCA2 mutation was detected in three patients. Visceral metastasis was found in 15 patients, eight patients had brain metastasis, and the others had liver metastasis. All specimens were from biopsy from metastatic BC not archival tissue. Most common biopsy site was breast main mass (32.4%). Patients with metastatic BC received more than three palliative treatments on average. Thirty-six of 37 patients had received anthracycline-containing cytotoxic chemotherapy, and 31 patients were treated with taxane chemotherapy. All ER-positive BCs were treated with tamoxifen or a non-steroidal aromatase inhibitor. Anti-HER2 treatment was administered in all patients with HER2-positive BCs.
Table 1: Clinicopathological characteristics of metastatic breast cancer (N = 37)
N = 37 (%) |
|
|---|---|
Age (median) |
45.1 ± 11.0 |
Range |
26.5–75.7 |
< 40 years old |
15 (40.5) |
≥ 40 years old |
22 (59.5) |
Histology |
|
Invasive ductal carcinoma |
34 (91.9) |
Other |
3 (8.1) |
Subtype |
|
ER+HER2− |
12 (32.4) |
ER+HER2+ |
5 (13.5) |
ER-HER2− |
13 (35.1) |
ER-HER2+ |
7 (18.9) |
Intrinsic subtype |
|
Luminal A |
7 (18.9) |
Luminal B |
6 (16.2) |
Basal-like |
14 (37.8) |
Normal-like |
2 (5.4) |
HER2-enriched |
8 (21.6) |
BRCA1/2 |
|
Wild-type |
2 (5.4) |
Mutated |
3 (8.1) |
Not tested |
32 (86.5) |
Cancer status |
|
Recurrent |
27 (73.0) |
Initially metastatic |
10 (27.0) |
Visceral metastasis |
|
Yes |
15 (40.5) |
Liver metastasis |
7 (18.9) |
Brain metastasis |
8 (21.6) |
No |
22 (59.5) |
Biopsy site |
|
Breast |
12 (32.4) |
Lymph node |
7 (18.9) |
Pleura |
7 (18.9) |
Liver |
3 (8.1) |
Lung |
2 (5.4) |
Other |
6 (16.2) |
Chemotherapy agents (average 3.24) |
|
1 |
8 (21.6) |
2 |
11 (29.7) |
3 |
4 (10.8) |
≥ 4 |
14 (37.8) |
Chemotherapeutic regimen |
|
Anthracycline |
36 (97.3) |
Taxane |
31 (83.8) |
Both anthracycline and taxane |
27 (73.0) |
Hormone therapy (N = 17) |
|
Yes |
17 (100.0) |
No |
0 (0.0) |
HER2-targeted therapy (N = 12) |
|
Yes |
12 (100.0) |
No |
0 (0.0) |
The time elapsed between diagnosis with metastatic breast cancer and RNA-Seq differed according to breast cancer subtype (Table 2). For ER-HER2+ BC, mean time to RNA-Seq was 29.3 months (range 5.5–69.7 months), whereas in ER-HER2- BC, the corresponding time was 4.3 months (range 0.0–36.7 months).
Table 2: Previous chemotherapy and time to biopsy according to subtype
Subtype |
No. of previous chemotherapy agents |
Time to biopsy after metastasis |
|---|---|---|
ER+HER2− |
3.5 (range 1–6) |
13.6 months (range 0.1–126.0) |
ER+HER2+ |
4.4 (range 1–11) |
18.8 months (range 2.4–33.2) |
ER-HER2− |
2.5 (range 1–6) |
4.3 months (range 0.0–36.7) |
ER-HER2+ |
3.4 (range 1–9) |
29.3 months (range 5.5–69.7) |
Significantly mutated genes and mRNA expression in metastatic breast cancer
Overall, 34 tumor samples from 37 patients were subjected to whole-exome sequencing, resulting in identification of 3,278 somatic mutations comprising 3,069 point mutations (single nucleotide variants; SNVs) and 209 insertion/deletions. Among the point mutations, 44 were silent mutations, 2,830 were non-synonymous mutations, 184 were stop-gain, and 11 were stop-loss mutations. In addition, 136 frameshift deletions and 73 insertions were detected.
TP53 was the most frequently mutated gene in metastatic BC (64.7%, 14 SNVs, and 8 frameshift insertions and deletions (indels)), followed by MUC4 (38.2%) and PIK3CA (29.4%). Frameshift mutations were most commonly observed in ZNF717 (26.5%) (Figure 1A and 1B).
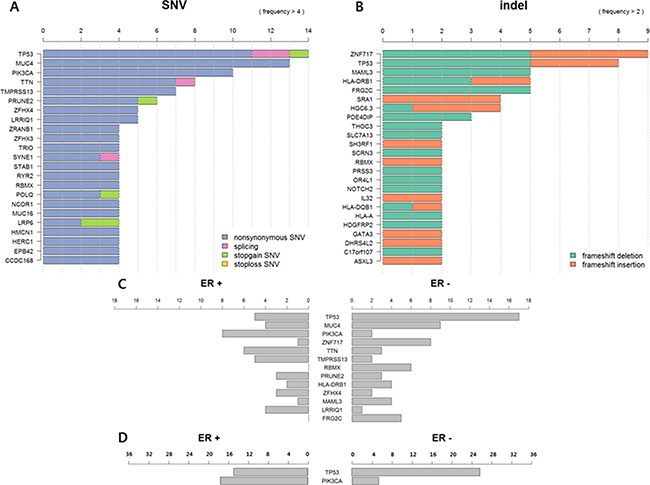
Figure 1: (A) Frequency of single nucleotide variants (SNVs) in metastatic breast cancer (BC) (N = 34). (B) Frequency of frame shift insertion/deletions in metastatic BC (N = 34). (C) Somatic mutation profile according to ER status in metastatic BC (N = 34). (D) TP53 and PIK3CA mutation profile according to ER status in metastatic BC (N = 63).
Somatic mutations differed according to ER status. TP53 was more frequently mutated in ER-negative BCs than ER-positive BCs (p = 0.126) in contrast to PIK3CA (p = 0.019). TTN, TMPRESS13, and LRRIQ1 were also more frequently mutated in ER-positive BCs, and ZNF717, RBNX, and FRG2C were more frequently mutated in ER-negative BCs, but there were no significant differences between the two groups (Figure 1C).
We additionally performed targeted deep sequencing of 29 metastatic BC samples to detect TP53 and PIK3CA mutations. Then, we reanalyzed 63 sequences. Of these 63 BCs, 36 (57.1%) were ER-positive (Supplementary Table 3). In this analysis, TP53 mutation was more frequently detected in ER-negative BC (ER-positive vs. ER-negative: 41.7% vs. 92.6% p < 0.001) and PIK3CA mutation was more common in ER-positive BC (ER-positive vs. ER-negative: 47.2% vs. 18.5%, p = 0.018) (Figure 1D).
Gene expression patterns also differed according to ER status. Among 22,072 genes, ERBB4, GATA3, FOXA1, and another 434 genes were more highly expressed in ER-positive BC than ER-negative BC (false discovery rate (FDR) p < 0.05 respectively). In contrast, KRT16, S100A2, RASAL1, and another 282 genes were more highly up-regulated in ER-negative BC than ER-positive BC (Supplementary Figure 2 and Supplementary Table 1).
The relationship between somatic mutations and gene expression
Because DNA is ultimately transcribed to messenger RNA followed by protein translation, we analyzed the association between somatic mutation and gene expression using Fisher’s exact test and the log-rank test. We focused on the two most commonly mutated genes, TP53 and PIK3CA.
Mutation of TP53 affected the level of gene expression (Figure 2A). Frameshift indels and stop-gain mutations of TP53 decreased gene expression compared to nonsynonymous mutations. In addition, high expression of CHEK2 and SNORA61 and low expression of LOC100499489 were detected in TP53-mutated BC (Figure 2B and Supplementary Figure 3). SNVs of TP53 (only nonsynonymous mutations, not frameshift indels or splicing variants) were associated with low expression of APBB2 and PPP1R3C and high expression of TNFRSF13C, but there were no statistically significant differences (Figure 2C).
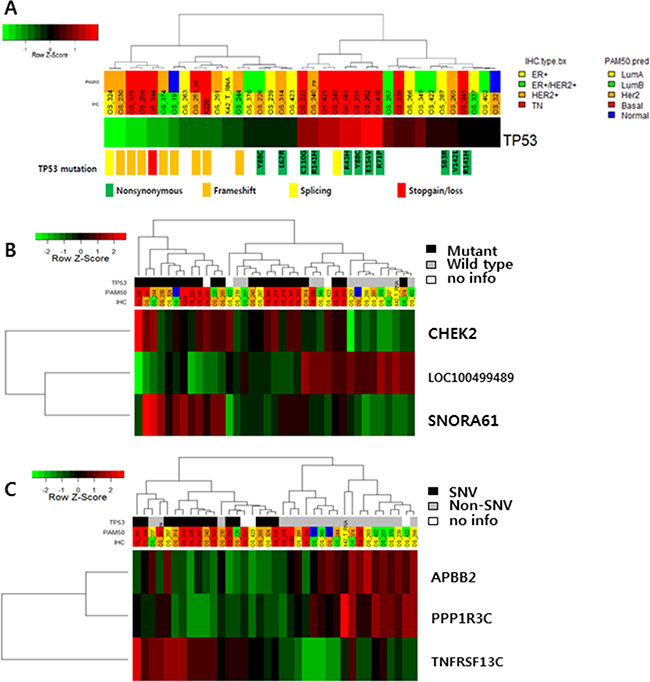
Figure 2: (A) TP53 expression according to TP53 mutation status. (B) Gene expression according to TP53 mutation status (TOP3 genes: CHEK2, SNORA61 and LOC100499489). (C) APBB2, PPP1R, and TNFRSF13C expression according to TP53 single nucleotide variants.
PIK3CA mutations were all nonsynonymous mutations. Nine of 10 mutation occurred at known hotspots: E545, H1047, and G1049. There was no relationship between mutation of PIK3CA and gene expression. We also analyzed PIK3CB, PIK3CD, PIK3CG, and PTEN, but did not find a correlation between genetic mutation of these genes and gene expression (Figure 3A). In one BC case with PIK3CA mutation, high expression of CALM1, SLC4A8, and NRK was observed (Figure 3B and Supplementary Figure 4). In pathway analysis, low scores for glyoxylate and dicarboxylate metabolism, drug metabolism, and RNA polymerase were associated with mutation of PIK3CA (Figure 3C and Supplementary Table 2).
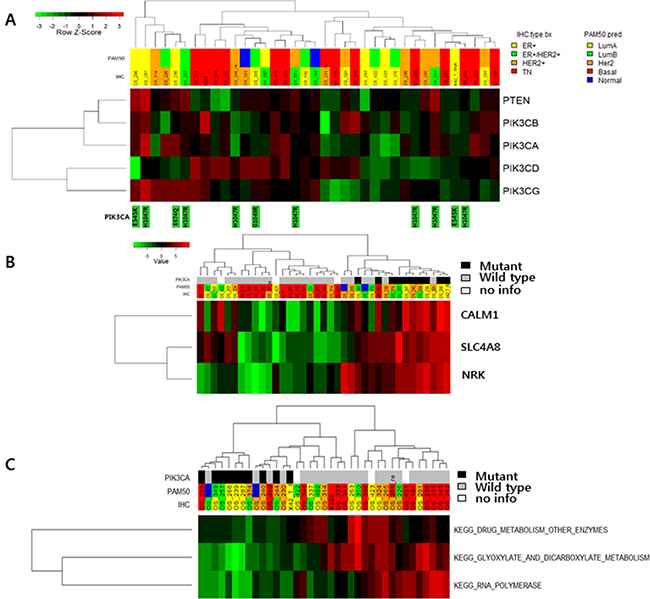
Figure 3: (A) Expression of PIK3CA pathway-associated genes according to PIK3CA mutation status. (B) Gene expression according to PIK3CA mutation status (TOP3 genes: CHEK2, SNORA61 and LOC100499489). (C) Pathway gene expression according to PIK3CA mutation status.
The relationship between genetic alterations and clinical outcomes
For analyzing the effect of genetic alterations to clinical outcome, we divided metastatic BC into two subtypes, ER-positive and ER-negative. According to subtypes, we analyzed the relationship between genetic alterations and overall survival.
TP53 mutation was related to shorter OS in ER-positive BC in contrast to ER- negative BC. In ER-positive BC, the median OS of TP53-mutated BC was 32.6 months compared to 88.5 months in wild type TP53 (median OS (wild type vs. mutated): 88.5 ± 54.4 vs. 32.6 ± 10.7 (months), p = 0.002). In contrast, TP53 mutation in ER-negative BC had longer OS compared to wild type TP53 (median OS (wild type vs. mutated): 0.1 vs. 32.6 ± 8.2 (months), p = 0.026) (Figure 4A and 4B). However, there were no significant differences in OS between those with SNVs and frameshift indel mutations.
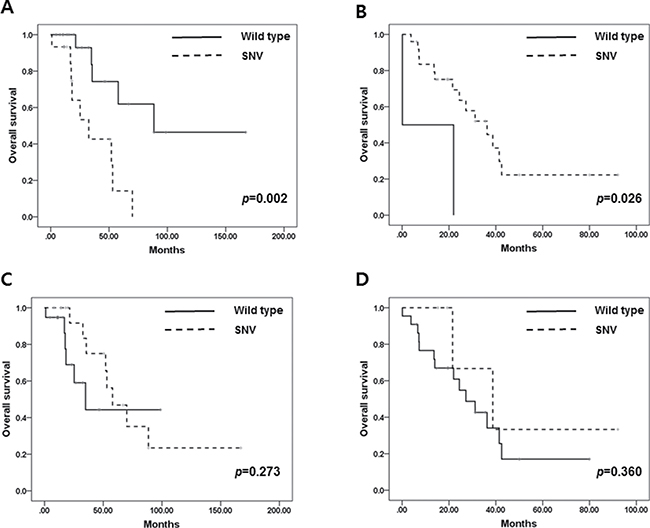
Figure 4: (A) Overall survival in ER-positive BC according to TP53 mutation. (B) Overall survival in ER-negative BC according to TP53 mutation. (C) Overall survival in ER-positive BC according to PIK3CA mutation. (D) Overall survival in ER-negative BC according to PIK3CA mutation.
PIK3CA mutation did not affect OS in metastatic BC regardless of ER status (median OS (wild-type vs. mutated): 34.9 ± 11.0 vs. 57.9 ± 12.1 (months), p = 0.269 in ER+ BC and median OS: 27.2 ± 5.9 vs. 38.7 ± 14.0 (months), p = 0.558 in ER- BC) (Figure 4C and 4D).
DISCUSSION
We explored genome-wide genetic alterations in metastatic BC in this study and confirmed that TP53 was the most frequently mutated gene in ER-negative metastatic BCs, while PIK3CA was the most frequently mutated gene in ER-positive metastatic BC.
TP53 mutation, the most common mutation in triple-negative breast cancer [3, 4], was also frequently detected in the metastatic setting. SNVs of TP53 were commonly observed in TNBC and increased TP53 mRNA expression. In contrast, TP53 frameshift indels were more frequently detected in HER2-positive BCs and decreased mRNA expression. Additional gene expression and pathway analysis revealed that SNVs and other types of TP53 mutations were diversely related to the expression patterns of other genes (Figure 2 and Supplementary Figure 3). Accordingly, TP53 mutation type affects mutational profile. In a previous study, we reported a relationship between TP53 mutation profile and expression in TNBCs [12]. Although statistical significance was not reached in the current study because of our small sample size, our results suggest that the TP53 mutation profile might impact all subtypes of BCs, as well as TNBCs.
We observed CHEK2 overexpression in TP53-mutated BCs. CHEK2, an inducer of the TP53 gene in response to DNA damage, acts as a tumor suppressor. TP53 mutation causes p53 dysfunction and DNA repair system malfunction. Therefore, CHEK2 overexpression may induce CHEK2-mediated DNA repair system activation as a compensatory mechanism in TP53-mutated BC. TNFRSF13C, also known as B-cell activating factor receptor (BAFFR), was especially activated in metastatic BC with TP53 SNVs. TNFRSF13C mutations have been studied in the context of lymphoid malignancy, with higher expression predicting better prognosis. Further studies to clarify the relationship between TP53 and TNFRSF13C are warranted.
SNVs were detected only in four loci in PIK3CA: three hotspots and one rare locus. Similar to previous comprehensive genetic studies of BCs, PIK3CA mutations were detected in five ER-positive cases, three ER-positive and HER2-positive cases, one HER2-positive case, and one TNBC case. No correlation between mutation and mRNA expression was observed for PIK3CA or PTEN. Additional analysis showed that calmodulin 1(CALM1) was highly expressed in PIK3CA-mutated BCs. Calmodulin, a regulator of calcium metabolism, is thought to be a regulator of AKT activity that indicates poor prognosis [13].
There are several current clinical trials targeting PIK3CA. BCs with PIK3CA mutation appear to have a poorer prognosis than wild-type BCs, and PI3K inhibitors appear to have a clinical benefit regardless of PIK3CA mutation status [6]. We propose that mutation of PIK3CA alters the activity of other genes involved in tumor aggressiveness, and that inhibiting PIK3CA modulates these altered pathways. This would explain why treatment with PIK3CA inhibitors improves the outcomes of BC patients with PIK3CA mutations as well as those patients without PIK3CA mutations. Three pathways related to PIK3CA might be targets of PIK3CA inhibitors; further validation studies are warranted.
We reviewed the TCGA dataset and found 20 metastatic BCs in the entire TCGA breast cancer cohort (n = 1046). The most common mutated gene in TCGA cohort was PIK3CA followed by TP53. Up to 30% of ER-positive BC had PIK3CA mutation (271/537, 53.5%) and 16.8% did TP53 mutation. Of ER-negative BC, TP53 was the most commonly mutated gene (154/238, 63.0%). PIK3CA was detected in 12.6% of ER-negative BC (30/238) (Supplementary Figure 5A). Among metastatic BCs in TCGA cohort, 3 of 4 ER-negative BC had TP53 mutation. Five of 16 ER-positive BC had PIK3CA mutation (31.3%) and 4 had TP53 mutation (25.0%) (Supplementary 5B). Metastatic BCs in our cohorts had a similar mutation profile to the profile observed in the TCGA cohort. Compared with early BC, metastatic BC more frequently had TP53 mutations in ER-positive BC. In ER-negative BC, the frequency of TP53 mutation did not vary between early and metastatic BC. PIK3CA mutation was detected at similar rates in early and metastatic BC. Considering TCGA cohort consisting of mainly early BCs, our study would give the novel genomic information of metastatic BCs to solve medical unmet need.
In conclusion, mutation of TP53, the most frequent genetic alteration in ER-negative BC, affected gene expression levels. Moreover, the type of TP53 mutation had a differential influence on clinical outcomes according to ER status. In contrast, there was no association between PIK3CA mutation and expression, or of other related genes, namely PIK3CB, PIK3CD, PIK3CG, and PTEN. Mutation of PIK3CA might alter calmodulin expression and other genetic pathways. Further functional validation studies are warranted.
MATERIALS AND METHODS
Patients
This study involved prospective explorative analysis of patients with metastatic BC at Samsung Medical Center as an establishing genomic platform for precision medicine in the era of NGS. Women diagnosed with stage IV BC or recurrent BC by diagnostic examination and staging work-up (breast magnetic resonance imaging [MRI], chest computed tomography [CT] scan, abdominal CT scan, bone scan, and/or positron emission tomography [PET]-CT scans if indicated) were included.
All patients provided written informed consent, and study approval was obtained from the Institutional Review Board of Samsung Medical Center, Seoul, Korea (IRB No: SMC 2012-08-065).
Immunohistochemical (IHC) staining
Two experienced pathologists reviewed all specimens to determine IHC staining for ER, progesterone receptor (PgR), and HER2. ER and PgR positivity were defined using Allred scores ranging from 3 to 8 based on IHC using antibodies to ER (Immunotech, Marseille, France) and PgR (Novocastra Laboratories Ltd., Newcastle upon Tyne, UK). HER2 status was evaluated using a specific antibody (Dako, Glostrop, Denmark) and/or silver in situ hybridization (SISH). Grades 0 and 1 for HER2, as assessed by IHC, were defined as a negative result, and grade 3 was defined as a positive result. Amplification of HER2 rated as 2+ by IHC was confirmed by SISH. Ki67 IHC analyses were performed independently using semi-quantitative and quantitative methods (Dako). Triple negativity was defined as a lack of expression of ER, PgR, and HER2.
DNA and RNA extraction
Unstained sections (4 mm) of tumors consisting of over 75% malignant cells were dissected under microscopy by comparison with an H&E-stained slide, and genomic DNA was extracted using a Qiagen DNA FFPE Tissue kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. After extraction, DNA concentration and 260/280- and 260/230-nm ratios were measured by spectrophotometry (ND1000, NanoDrop Technologies, ThermoFisher Scientific, MA, USA). Each sample was then quantified using a Qubit fluorometer (Life Technologies, Carlsbad, CA, USA). Libraries were prepared for samples with a genomic DNA total yield > 10 ng.
Areas containing representative invasive breast carcinoma were outlined on the slide. Total RNA was then extracted using a High Pure RNA Paraffin kit (Roche Diagnostic, Mannheim, Germany), and the RNA concentration and 260/280- and 260/230-nm ratios were measured using a NanoDrop ND1000 spectrophotometer (NanoDrop Technologies). Samples were concentrated by a SpeedVacTM concentrator (Thermo Scientific™, Waltham, MA, USA). After concentration, these with less than 1 g/L of total were excluded from downstream analysis.
Whole-exome sequencing (Table 3)
Table 3: Summary of whole-exome sequencing (N = 34)
Min. |
1st Qu. |
Median |
Mean |
3rd Qu. |
Max. |
|
|---|---|---|---|---|---|---|
TotalRead |
47,430,000 |
79,760,000 |
107,300,000 |
104,700,000 |
127,500,000 |
166,900,000 |
TotalRead_T |
47,430,000 |
110,100,000 |
119,300,000 |
122,300,000 |
132,900,000 |
166,900,000 |
TotalRead_N |
50,970,000 |
71,080,000 |
80,250,000 |
87,060,000 |
97,350,000 |
146,900,000 |
Coverage |
47.41 |
92.34 |
126.4 |
124.3 |
150.9 |
231.1 |
Coverage_T |
47.41 |
128.8 |
141.7 |
143.5 |
163.7 |
231.1 |
Coverage_N |
48.83 |
87.85 |
93.66 |
105.1 |
110.3 |
193.8 |
PCT_TARGET_BASES_50X |
0.363 |
0.7602 |
0.8365 |
0.7944 |
0.87 |
0.939 |
PCT_TARGET_BASES_50X_T |
0.363 |
0.8436 |
0.8655 |
0.8404 |
0.8835 |
0.939 |
PCT_TARGET_BASES_50X_N |
0.42 |
0.7117 |
0.7658 |
0.7484 |
0.8211 |
0.8733 |
PCT_TARGET_BASES_100X |
0.06776 |
0.3724 |
0.5346 |
0.4986 |
0.6341 |
0.7554 |
PCT_TARGET_BASES_100X_T |
0.07207 |
0.5511 |
0.6067 |
0.5836 |
0.6679 |
0.7554 |
PCT_TARGET_BASES_100X_N |
0.06776 |
0.3427 |
0.38 |
0.4136 |
0.4854 |
0.7329 |
Poor quality reads were filtered out and aligned to the human reference genome (hg19) using the Burrows-Wheeler Alignment tool (BWA, version 0.7.5a) [14]. To convert Sequence Alignment and Mapping (SAM) files into Binary Alignment and Mapping (BAM) files, we used SAMtools (version 0.1.19) [15]. Polymerase chain reaction (PCR) duplicates were removed from the BAM files by Picard (version 1.93, http://picard.sourceforge.net/) and SAMtools before variant calling. The Genome Analysis Toolkit (GATK, version 2.4.7) [16] was used to recalibrate base quality and optimize local realignment. Single nucleotide variants (SNVs) and indels were called using MuTect (version 1.1.4) [17] and Varscan2 (version 2.3.5) [18] with default parameter settings. Copy number variations were detected with CONTRA (version 2.0.4) [19]. Variants were annotated using ANNOVAR, with gene, chromosomal information, exonic function (synonymous, nonsynonymous, stop gain, non-frameshift, or frameshift indel), amino acid changes, and allele frequencies extracted from public databases such as the 1000 Genomes Project (2012 February version) and dbSNP (version 132).
Variants located in exonic regions with sufficient coverage (minimum depth of coverage ≥ 8) and variant allele frequency (VAF ≥ 0.1) were chosen for further statistical analyses. Synonymous variants were filtered out. Read alignments were manually investigated using the Integrative Genomic Viewer (http://www.broadinstitute.org/igv/).
Fisher’s exact test was used to analyze mutations and polymorphic variants separately to discover variants enriched in patients with a favorable outcome. P-values < 0.05 were considered significantly different. R version 3.0.2 (http://www.R-project.org/) and R package (ggplot2) were used for all statistical analyses and generation of heat maps and plots.
RNA-Seq analysis and normalization (Table 4)
Table 4: Summary of RNA-Seq (N = 37)
Min. |
1st Qu. |
Median |
Mean |
3rd Qu. |
Max. |
|
|---|---|---|---|---|---|---|
PF_ALIGNED_BASES |
3,426,000,000 |
4,818,000,000 |
5,413,000,000 |
5,377,000,000 |
5,814,000,000 |
6,976,000,000 |
CODING_BASES |
1,202,000,000 |
2,220,000,000 |
2,624,000,000 |
2,574,000,000 |
3,008,000,000 |
3,350,000,000 |
PCT_CODING_BASES |
0.226 |
0.4499 |
0.5045 |
0.4808 |
0.5289 |
0.599 |
PCT_UTR_BASES |
0.2149 |
0.2881 |
0.3209 |
0.3194 |
0.3483 |
0.415 |
PCT_INTRONIC_BASES |
0.02611 |
0.03425 |
0.04298 |
0.0453 |
0.05247 |
0.08502 |
PCT_INTERGENIC_BASES |
0.0573 |
0.09025 |
0.1271 |
0.1545 |
0.1617 |
0.5088 |
PCT_MRNA_BASES |
0.4408 |
0.7687 |
0.8373 |
0.8002 |
0.8652 |
0.9136 |
MEDIAN_CV_COVERAGE |
0.494 |
0.5271 |
0.5776 |
0.5939 |
0.6472 |
0.753 |
After trimming poor-quality bases from FASTQ files obtained from whole-transcriptome sequencing, we aligned the reads to the human reference genome hg19 with Tophat (version 2.0.6) [20] and performed reference-guided assembly of transcripts with Cufflinks (version 2.1.1) [21]. Alignment quality was verified with SAMtools (version 0.1.19). Transcript abundance was estimated using a count-based method with htseq-count. Gene counts were used as the input for TMM (Trimmed Mean of M values) normalization by the R package edgeR [22], and normalized counts were transformed to log2-counts per million (logCPM) by applying voom from the R package limma [23] to account for higher variability at low expression levels. Genes with zero read counts across all samples were removed for a more powerful statistical test. Pathway analysis was performed using GSVA [24] (Supplementary Figure 1).
Targeted deep sequencing of TP53 and PIK3CA
We used CancerScanTM to detect TP53 and PIK3CA mutation. After enriched exome libraries were multiplexed, the libraries were sequenced on a HiSeq 2500 sequencing platform (Illumina). Briefly, a paired-end DNA sequencing library was prepared through gDNA shearing, end-repair, A-tailing, paired-end adapter ligation, and amplification. After hybridization of the library with bait sequences for 27 hours, the captured library was purified and amplified with an index barcode tag, and the library quality and quantity were assessed. Sequencing of the exome library was performed using the 100-bp paired-end mode of the TruSeq Rapid PE ClusterKit and TruSeq Rapid SBS Kit (Illumina).
Intrinsic subtyping
We performed intrinsic subtyping with log-scaled normalized expression values using the 50-gene Prediction Analysis of Microarray (PAM50) subtype predictor as described by Parker et al. [25]. The PAM50 subtype predictor classified tumors into the following groups: luminal A, luminal B, HER2-enriched, basal-like, and normal-like (Supplementary Figure 1).
Survival analysis
We evaluated the association between gene expression and overall survival (OS) using the R package RcmdrPlugin.survival. OS was defined as the time elapsed between the date of stage IV breast cancer diagnosis and the date of death. For each gene, patients were grouped based on the normalized expression value of the gene, with the top 50% and the bottom 50% representing high and low expression groups, respectively. Survival curves for the two groups were estimated with the Kaplan-Meier method, and the log-rank test was used to compare overall survival curves between the two groups (p < 0.05). After pathway analysis with GSVA, Fisher’s exact test was used to identify pathways that were enriched with genes significantly associated with overall survival (p < 0.05).
ACKNOWLEDGMENTS AND FUNDING
J-K performed statistical analyses of genetic and clinical data and drafted and revised the manuscript. EL and KP handled whole-exome and whole-transcriptome data analysis. HHJ carried out sample preparation. J-K and HHJ participated in interpreting the genetic and clinical data. JSA, YHP and Y-I participated in sample preparation and coordination for clinical data analysis. YHP conceived and designed this study. All authors read and approved the final manuscript. The authors have no conflicts of interest to declare.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Ferlay J SI EM, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray, F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. Lyon, France: International Agency for Research on Cancer. 2013.
2. Kohler BA, Sherman RL, Howlader N, Jemal A, Ryerson AB, Henry KA, Boscoe FP, Cronin KA, Lake A, Noone AM, Henley SJ, Eheman CR, Anderson RN, et al. Annual Report to the Nation on the Status of Cancer, 1975–2011, Featuring Incidence of Breast Cancer Subtypes by Race/Ethnicity, Poverty, and State. J Natl Cancer Inst. 2015; 107:djv048.
3. Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70.
4. Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, Van Loo P, Ju YS, Smid M, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016; 534:47–54.
5. Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, Fu S, Falchook GS, Hong DS, Garrido-Laguna I, Luthra R, Lee JJ, Lu KH, et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012; 30:777–782.
6. Schmid P, Pinder SE, Wheatley D, Macaskill J, Zammit C, Hu J, Price R, Bundred N, Hadad S, Shia A, Sarker SJ, Lim L, Gazinska P, et al. Phase II Randomized Preoperative Window-of-Opportunity Study of the PI3K Inhibitor Pictilisib Plus Anastrozole Compared With Anastrozole Alone in Patients With Estrogen Receptor-Positive Breast Cancer. J Clin Oncol. 2016; 34:1987–1994.
7. Mayer IA, Abramson VG, Isakoff SJ, Forero A, Balko JM, Kuba MG, Sanders ME, Yap JT, Van den Abbeele AD, Li Y, Cantley LC, Winer E, Arteaga CL. Stand up to cancer phase Ib study of pan-phosphoinositide-3-kinase inhibitor buparlisib with letrozole in estrogen receptor-positive/human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol. 2014; 32:1202–1209.
8. Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, Baird RD, Delgado L, Taylor A, Lupinacci L, Riisnaes R, Pope LL, Heaton SP, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol. 2011; 29:4688–4695.
9. Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012; 366:520–529.
10. Mohseni M, Park BH. PIK3CA and KRAS mutations predict for response to everolimus therapy: now that’s RAD001. J Clin Invest. 2010; 120:2655–2658.
11. Hortobagyi GN, Chen D, Piccart M, Rugo HS, Burris HA 3rd, Pritchard KI, Campone M, Noguchi S, Perez AT, Deleu I, Shtivelband M, Masuda N, Dakhil S, et al. Correlative Analysis of Genetic Alterations and Everolimus Benefit in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results From BOLERO-2. J Clin Oncol. 2016; 34:419–426.
12. Kim JY, Park K, Jung HH, Lee E, Cho EY, Lee KH, Bae SY, Lee SK, Kim SW, Lee JE, Nam SJ, Ahn JS, Im YH, et al. Association Between Mutation and Expression of TP53 as a Potential Prognostic Marker of Triple-Negative Breast Cancer. Cancer Res Treat. 2016; 48:1338–1350.
13. Coticchia CM, Revankar CM, Deb TB, Dickson RB, Johnson MD. Calmodulin modulates Akt activity in human breast cancer cell lines. Breast Cancer Res Treat. 2009; 115:545–560.
14. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010; 26:589–595.
15. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011; 27:2987–2993.
16. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011; 43:491–498.
17. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013; 31:213–219.
18. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012; 22:568–576.
19. Li J, Lupat R, Amarasinghe KC, Thompson ER, Doyle MA, Ryland GL, Tothill RW, Halgamuge SK, Campbell IG, Gorringe KL. CONTRA: copy number analysis for targeted resequencing. Bioinformatics. 2012; 28:1307–1313.
20. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36.
21. Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013; 31:46–53.
22. McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012; 40:4288–4297.
23. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47.
24. Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013; 14:7.
25. Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, Quackenbush JF, Stijleman IJ, Palazzo J, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009; 27:1160–1167.