
INTRODUCTION
Members of the tumor necrosis factor transmembrane cytokine superfamily, such as TNFα and Fas ligand (FasL), play crucial roles in inflammation and immunity [1]. TRAIL is a member of this superfamily with the ability to selectively trigger cancer cell death, but does not motive cytotoxicity to most normal cells [2]. Five members of the death receptor family have been established that can bind TRAIL. The death receptors, DR4 (TRAIL-R1) and DR5 (TRAIL-R2), involve two cysteine-rich extracellular TRAIL-mandatory domains and a cytoplasmic death domain, which are essential for transmitting a cytotoxic signal [3, 4]. The decoy receptors, DcR1 (TRAIL-R3), DcR2 (TRAIL-R4), and osteoprotegerin (TRAIL-R5), can also bind TRAIL, but they do not transmit apoptotic signals as a result of a nonfunctional death domain [5, 6]. Finally, TRAIL initiates apoptosis upon the binding of TRAIL receptors, which promotes the recruitment of the accessory molecule FAS-associated death domain protein and ultimately, procaspase-8, to the formation of death-inducing signaling complex, leading to subsequent effector caspases (caspase-8, -9, -10, and -3) [7, 8]. However, the downregulation of DR, upregulation of anti-apoptotic protein, and ultimately, upregulation of inhibitors of apoptosis proteins contributes resistance to TRAIL-mediated apoptosis in many cancer cells [9–13]. Many studies have demonstrated that the related mechanisms of association exist between TRAIL and numerous agents [14–23]. Accordingly, combination treatment with TRAIL sensitizers is one direction to overcoming TRAIL resistance.
The peroxisome proliferator-activator receptors are members of ligand-stimulated nuclear receptors family that include PPARα, PPARδ, and PPARγ [24]. PPARγ is mainly exposed in adipose tissue and plays eventual roles in lipid metabolism, adipocyte differentiation, and progressing insulin sensitivity [25]. Notwithstanding, information on the molecular mechanism of PPARγ in tumorigenesis is fractioning. Many studies have revealed that PPARγ acts as a tumor inhibitor, because it is mainly exposed in prostate, breast, and colonic epithelium and the appearance of cultured cancer cell lines suppositious of these tissue types with ligands of PPARγ restrain cellular proliferation [26–31].
Fortunately, many studies have revealed that chemotherapeutic drugs can lead to increased apoptosis induction in TRAIL-resistant tumor cells [32, 33]. The thiazolidinedione (TZD) groups of antidiabetic drugs, such as troglitazone are used in the cure of type II diabetes to reduce blood glucose levels and improve the sensitivity of numerous tissues to insulin [34]. The ability of TZDs to inhibit a number of cancers has been associated with their ability to suppress growth and stimulate apoptosis in a PPARγ-dependent manner [35, 36]. One recent study has shown that the activation of PPARγ induces autophagy in breast cancer cells [37].
Autophagy has been characterized as a particular homeostatic and adaptive system that serves to convey cytoplasmic components and organelles to the lysosomes for digestion [38, 39]. The autophagy process starts with the formation of double-membrane vesicles conversant as autophagosomes that engulf cytoplasmic constituents including organelles proceeding from a maturation system upon fusion with lysosomes and finally, dilapidation and recycling of the cargo [40, 41]. Autophagosome formation is coordinated by the Atg12-Atg5-Atg16 complex and conversion of LC3-I-phospholipid conjugates LC3-II, which is used extensively as an autophagy marker [42, 43]. P62 is a well-establish autophagy marker that is organized into autophagosomes by exactly interacting with LC3 and is comfortably degraded by autophagy. Autophagy and apoptosis coordinately govern cell survival and cell death, and appear simultaneously in cancers [44, 45]. Chemotherapeutic agents frequently activate autophagy in cancer cells. Therefore, activation of autophagy in cancer cells has been revealed to play an extensive induction as a cell death mechanism by tumor development, where apoptosis is limited [46].
Though the effect of the antidiabetic drugs such as troglitazone and the synergistic outcome of troglitazone combined with TRAIL are well-establish; however, the molecular mechanisms involved in these effects are presently unknown. Therefore, the objective of this research was to supervise the molecular mechanisms underlying the anticancer effect of troglitazone and the synergistic outcome of troglitazone combined with TRAIL in lung A549 cells.
RESULTS
Troglitazone sensitizes TRAIL-mediated apoptosis in human lung adenocarcinoma cells
To understand the effect of troglitazone on TRAIL-mediated apoptosis, lung adenocarcinoma cells were pre-incubated with different concentrations of troglitazone for 12 h and exposed to TRAIL for 2 h. We photographed adenocarcinoma cells under a light microscope to visualize the morphological changes. Treatment of TRAIL or troglitazone alone did not or only slightly induced cell death (Figure 1) and did not morphological change was identified compared with that in control, suggesting that A549 cells were highly resistant to TRAIL-induced apoptosis. Nevertheless, co-treatment of TRAIL with serial concentrations of troglitazone significantly reduced the cell viability compared with troglitazone or TRAIL alone (Figure 1A–1D). Furthermore, troglitazone and TRAIL co-treatment also decreased the cell viability and significantly sensitized apoptosis in Calu-3, HCC-15 cells (Figure 1E–1H). These data suggested that troglitazone sensitized TRAIL-mediated apoptosis in lung adenocarcinoma cells.
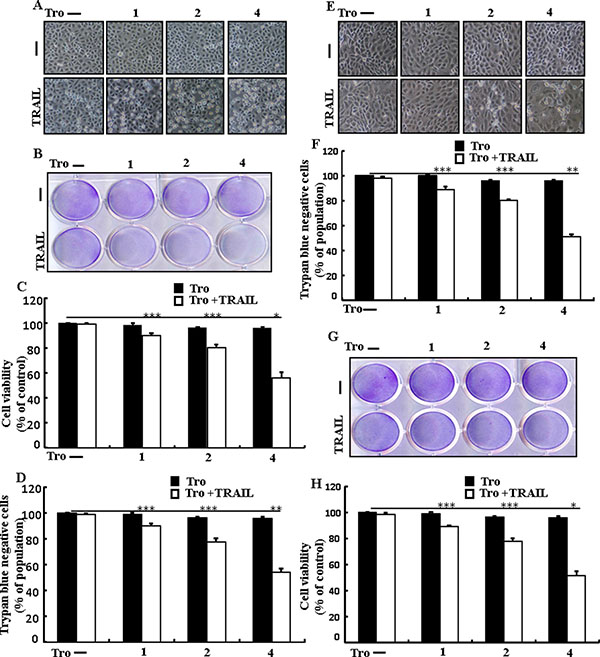
Figure 1: Troglitazone sensitizes TRAIL-mediated apoptosis in human lung adenocarcinoma cells. A549, HCC-15 and Calu-3 cells were pre-incubated with troglitazone at different doses (0, 1, 2, and 4 μM) for 12 h and exposed to TRAIL protein 200 ng/ml for 2 h. (A and E) Cell morphology photographed using light microscope in A549 and Calu-3 Cells (×100); (B and G) Cell viability was measured with crystal violet assay in A549 and HCC-15 Cells; (C and H) Bar graph indicating the average density of crystal violet in A549 and HCC-15 Cells; (D and F) Cell viability was measured with trypan blue dye exclusion assays in A549 and Calu-3 Cells. *p < 0.05 **p < 0.01, ***p < 0.001: represent significant differences between control and each treatment group; Tro: Troglitazone; TRAIL: Tumor necrosis factor (TNF)-related apoptosis-inducing ligand.
Troglitazone induces autophagy and sensitized apoptosis mediated by TRAIL
To understand the effect of troglitazone on autophagy flux. All the cell lysates were included to western blot analysis. As displayed in Figure 2A, the protein expression levels of DR4 and DR5, were unchanged by troglitazone at varying concentrations. P62 is a well-establish autophagy marker that is organized into autophagosomes by exactly interacting with LC3 and is comfortably degraded by autophagy. Inhibiting autophagy results in prompt accumulation of cellular p62, on the contrary decreased p62 levels are amalgamated with activating autophagy. However, LC3-II was significantly increased and p62 was decreased after troglitazone treatment in a dose-dependent manner (Figure 2B). Immunocytochemistry results also supported that various concentrations of troglitazone decreased p62 protein levels (Figure 2C). A TEM assay suggested that numerous autophagic vacuoles and empty vacuoles were appeared in the cells treated with troglitazone (Figure 2D). The combined treatment of troglitazone and TRAIL enhanced intracellular apoptosis indicators Ac-cas3 and Ac-cas8 expression levels compare with the single treatment with TRAIL or troglitazone (Figure 2E). These results suggested that troglitazone could induce autophagy in A549 cells.
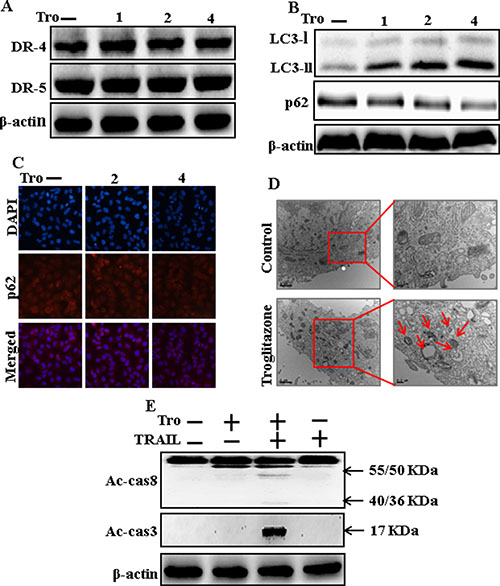
Figure 2: Troglitazone induces autophagy and sensitized apoptosis mediated by TRAIL. A549 cells were pre-incubated with troglitazone at varying doses (0, 1, 2, and 4 μM) for 12 h. (A and B) Western blot for DR-4, DR-5, LC3-II, and p62 proteins was analyzed from A549 cells; (C) Cells were immunostained with p62 antibody (red) and observed in fluorescent view; (D) TEM shows the ultrastructure of cells treated with troglitazone for 12 h. Arrows indicate autophagosomes, together with residual digested material and empty vacuoles; (E) Western blot for Ac-cas3 and Ac-cas8 expression levels was conducted with A549 cells. Cells were pre-incubated with troglitazone for 12 h and exposed to TRAIL protein for an additional 1 h. β-actin was used as the loading control. Tro: Troglitazone; TRAIL: Tumor necrosis factor (TNF)-related apoptosis-inducing ligand; Ac-cas3: Activated caspase 3; Ac-cas8: Activated caspase 8.
Troglitazone enhancement of TRAIL-induced apoptosis is blocked by inhibition of autophagy
Chloroquine was used to investigate the effect of troglitazone on TRAIL-induced apoptosis. A549 cells were pre-incubated with the indicated troglitazone concentrations for 12 h and exposed to TRAIL for 2h. A549 cells were also pre-incubated with autophagy inhibitor chloroquine for 1 h followed by troglitazone. Co-treatment of troglitazone, chloroquine, and TRAIL blocked cell death. However, Cell morphology results also supported that chloroquine enclosed the cell death effect compared to treatment with troglitazone and TRAIL (Figure 3A). Co-treatment of troglitazone, TRAIL, and chloroquine strongly increased cell viability in human lung adenocarcinoma A549 cells with significantly decreased cell death (Figure 3B–3D). These data suggested that chloroquine could promote troglitazone-mediated cancer cell survival induced by TRAIL.
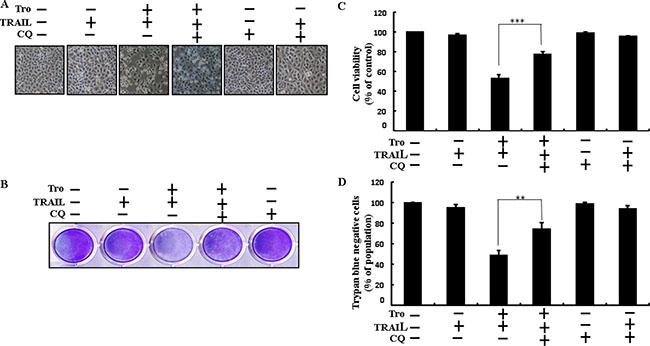
Figure 3: Troglitazone enhancement of TRAIL-induced apoptosis is blocked by inhibition of autophagy. Cells were pre-incubated with the indicated troglitazone doses for 12 h and exposed to TRAIL protein for an additional 2h. Additional cells were also pre-incubated with autophagy inhibitor chloroquine for 1 h followed by troglitazone treatment. (A) Cell morphology photographed using light microscope (×100); (B) Cell viability was measured with crystal violet assay; (C) Bar graph indicating average density of crystal violet; (D) Cell viability was measured with trypan blue dye exclusion assays. **p < 0.01, ***p < 0.001: represent significant differences between control and each treatment group; Tro: Troglitazone; TRAIL: Tumor necrosis factor (TNF)-related apoptosis-inducing ligand; CQ: Chloroquine.
Inhibition of autophagy blocks TRAIL-mediated apoptosis by troglitazone through activation of autophagy flux
We determine the effect of troglitazone on TRAIL induction of the apoptotic pathway by activating autophagy flux with pharmacological autophagy inhibitor chloroquine. All the cell lysates were included to western blot analysis. The expression levels of DR4 and DR5 were unchanged by troglitazone or chloroquine alone or by combined treatment with troglitazone and chloroquine in A549 cells (Figure 4A). Autophagy induction was further adopted by the observation of autophagic flux using chloroquine. Autophagy inhibitor Chloroquine caused impressed accumulation of membrane-bound LC3-II levels, with decreasing p62 (Figure 4B). Immunocytochemistry results also supported that troglitazone treatment decreased the p62 protein level compared with chloroquine or by treatment with both troglitazone and chloroquine (Figure 4C). The combined treatment of troglitazone and TRAIL enhanced intracellular apoptosis indicators Ac-cas3 and Ac-cas8 expression levels. However, co-treatment of troglitazone, TRAIL, and chloroquine enclosed the increase in expression level of Ac-cas3 and Ac-cas8 (Figure 4D). These results suggested that troglitazone-mediated enhancement of the TRAIL-induced apoptosis could be blocked by chloroquine via activation of autophagy flux.
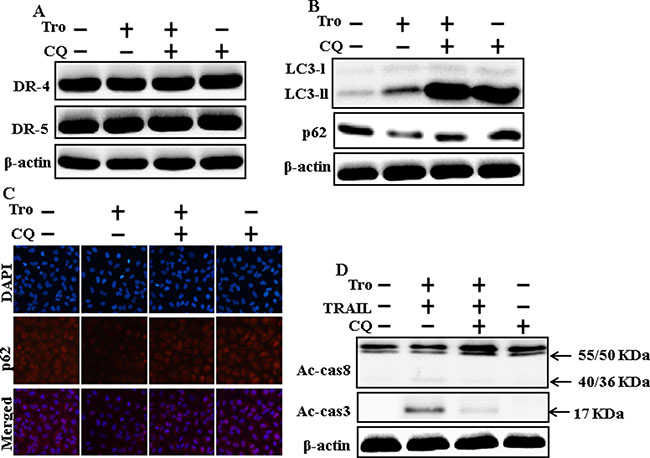
Figure 4: Inhibition of autophagy blocks TRAIL-mediated apoptosis by troglitazone via activation of autophagy flux. A549 cells were pre-incubated with chloroquine for 1h followed by indicated troglitazone doses for 12 h. (A and B) Western blot for DR-4, DR-5, LC3-II, and p62 proteins was analyzed from A549 cells; (C) Cells were immunostained with p62 antibody (red) and observed in fluorescent view; (D) Western blot for Ac-cas3 and Ac-cas8 expression levels was conducted with A549 cells. Cells were pre-incubated with the indicated troglitazone concentrations for 12 h and exposed to TRAIL protein for an additional 1h. Additional cells were pre-incubated with autophagy inhibitor chloroquine for 1 h, followed by troglitazone treatment. β-actin was used as the loading control. Tro: Troglitazone; Tumor necrosis factor (TNF)-related apoptosis-inducing ligand; Ac-cas3: Activated caspase 3; Ac-cas8: Activated caspase 8; CQ: Chloroquine.
Troglitazone enhanced TRAIL-induced apoptosis is blocked by genetic inhibition of autophagy
Genetic autophagy inhibitor ATG5 siRNA used to determine the effect of troglitazone on TRAIL-induced apoptosis. A549 cells were pre-incubated with ATG5 siRNA or NC for 24 h and then exposed to indicate troglitazone doses for 12 h with or without TRAIL for 2 h. Co-treatment of troglitazone, ATG5 siRNA, and TRAIL blocked cell death. However, Cell morphology results also supported that ATG5 siRNA blocked cell death effect compared to troglitazone, TRAIL, and negative control siRNA treatment (Figure 5A). Co-treatment with troglitazone, TRAIL, and ATG5 siRNA strongly increased cell viability in human lung adenocarcinoma A549 cells with significantly decreased cell death (Figure 5B–5D). These results suggested that ATG5 siRNA could promote troglitazone-mediated cancer cell survival induced by TRAIL.
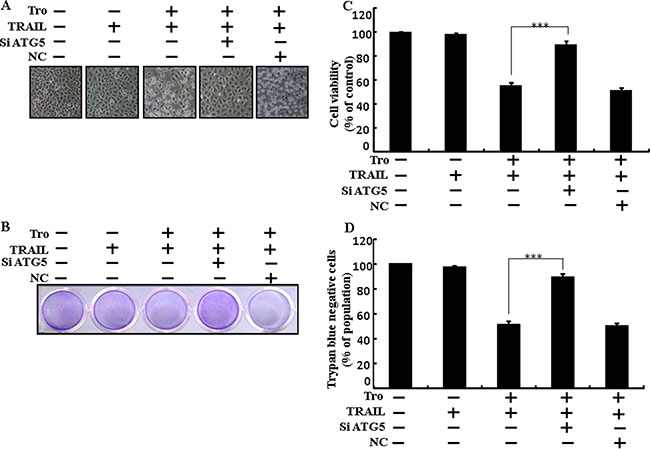
Figure 5: Troglitazone enhanced TRAIL-induced apoptosis is blocked by genetic inhibition of autophagy. A549 cells were pre-incubated with ATG5 siRNA or negative control siRNA for 24 h and then exposed to indicated troglitazone doses for 12 h with or without TRAIL protein for an additional 2 h. (A) Cell morphology photographed using light microscope (×100); (B) Cell viability was measured with crystal violet assay; (C) Bar graph indicating average density of crystal violet; (D) Cell viability was measured with trypan blue dye exclusion assays. ***p < 0.001: represent significant differences between control and each treatment group. Tro: Troglitazone; TRAIL: Tumor necrosis factor (TNF)-related apoptosis-inducing ligand; siATG5: ATG5 small interfering RNA; NC: Negative control.
Genetic inhibition of autophagy blocks TRAIL-induced apoptosis by troglitazone through activation of autophagy flux
To understand the effect of troglitazone-induced TRAIL-mediated apoptotic pathway by activating autophagy flux with genetic autophagy inhibition by ATG5 siRNA. All the cell lysates were included to western blot analysis. The expression levels of DR4 and DR5 were unchanged by troglitazone alone or by combined treatment with ATG5 siRNA or NC siRNA in A549 cells (Figure 6A). To address the induction of autophagy, cells were transfected with siRNA directed in opposition to autophagy protein 5 (Atg5) to block autophagic vesicle composition, and silencing of ATG5 was confirmed. Knockdown of ATG5 markedly decreased the troglitazone-induced LC3-II protein level (Figure 6B). Immunocytochemistry results also suggested this p62 protein level in A549 cells (Figure 6C). Co-treatment of troglitazone, NC siRNA, and TRAIL enhanced intracellular apoptosis indicators Ac-cas3 and Ac-cas8. However, co-treatment with troglitazone, ATG5 siRNA, and TRAIL enclosed the increase in Ac-cas3 and Ac-cas8 expression levels (Figure 6D). These results suggested that troglitazone-mediated enhancement of the TRAIL-induced apoptosis could be blocked by genetic inhibition of autophagy via activation of autophagy flux.
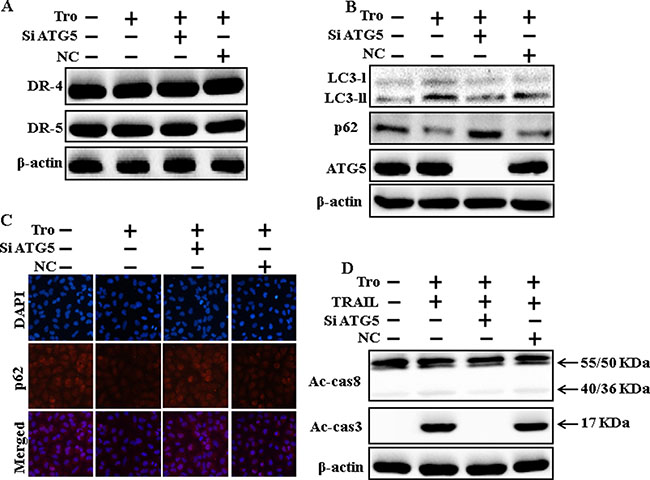
Figure 6: Genetic inhibition of autophagy blocks TRAIL-induced apoptosis by troglitazone via activation of autophagy flux. A549 cells were pre-incubated with ATG5siRNA or negative control siRNA for 24 h, and then exposed to indicated troglitazone doses for 12 h. (A and B) Western blot for DR-4, DR-5, LC3-II, p62 and ATG5 proteins was analyzed from A549 cells; (C) Cells were immunostained with p62 antibody (red) and observed in fluorescent view; (D) Western blot for Ac-cas3 and Ac-cas8 expression levels was conducted. A549 cells were pre-incubated with ATG5siRNA or negative control siRNA for 24 h, and then exposed to indicated troglitazone doses for 12 h with or without TRAIL protein for an additional 1 h. β-actin was used as the loading control. Tro: Troglitazone; TRAIL: Tumor necrosis factor (TNF)-related apoptosis-inducing ligand; Ac-cas3: Activated caspase 3; Ac-cas8: Activated caspase 8; siATG5: ATG5 small interfering RNA; NC: Negative control.
Effects of troglitazone are induced by PPARγ activation in A549 cells
To understand the effects of troglitazone are induced by PPARγ activation in A549 cells. Pretreatment of troglitazone induced activation of PPARγ in a dose-dependent manner (Figure 7A and 7B). Western blot result demonstrated that the conversion rate of LC3-I to LC3-II and PPARγ was suppressed in the presence of GW9662 (Figure 7C). Morphological image and crystal violet staining results display that co-treatment with troglitazone, GW9662, and TRAIL enclosed the cell death effect compared to treatment with troglitazone and TRAIL (Figure 7D–7F). These data suggested that troglitazone-induced autophagy flux was dependent on the activation of PPARγ in A549 cells.
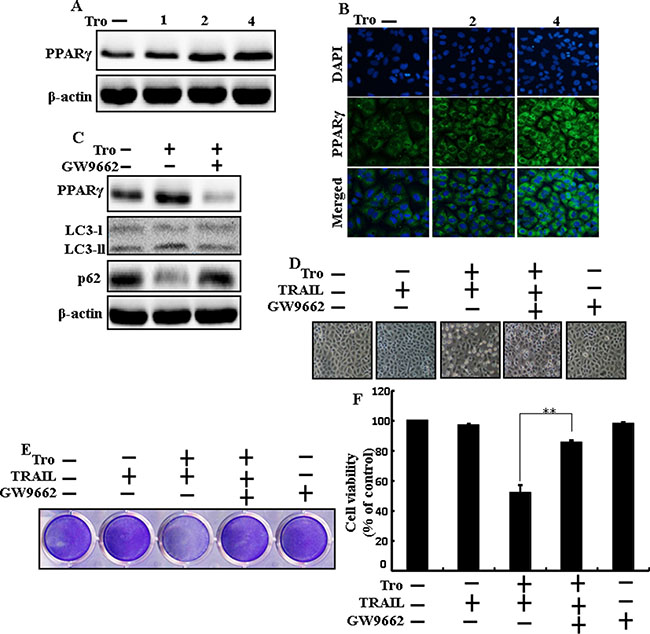
Figure 7: Effects of troglitazone are induced by PPARγ activation in A549 cells. Lung adenocarcinoma cells were pre-incubated with different doses of troglitazone (0, 1, 2, and 4 μM) for 12 h and exposed to TRAIL protein for an additional 2 h. Additional cells were pretreated with GW9662 for 1 h followed by treatment with troglitazone. After that, (A and C) Western blot for PPARγ, LC3-II, and p62 proteins was analyzed from A549 cells; (B) Cells were immunostained with PPARγ antibody (green) and observed in fluorescent view; (D) Cell morphology photographed using light microscope (×100); (E) Cell viability was measured with crystal violet assay; (F) Bar graph indicating average density of crystal violet. β-actin was used as the loading control. **p < 0.01: represent significant differences between control and each treatment group; Tro: Troglitazone; TRAIL: Tumor necrosis factor (TNF)-related apoptosis-inducing ligand.
DISCUSSION
The purpose of this study was to determine the effect of troglitazone with or without TRAIL on lung adenocarcinoma A549 cells. Our results demonstrated that PPARγ activation by troglitazone enhances TRAIL-mediated tumor cell death in A549 cells via regulation of autophagy flux.
TRAIL is a member of cytokine superfamily with the ability to selectively trigger cancer cell death but does not motive cytotoxicity to normal cells. The therapeutic probability of TRAIL is being demonstrated in various clinical trials [47]. The PPARγ ligands include naturally appearing fatty acids and members of the thiazolidinedione (TZD) group, such as troglitazone and rosiglitazone, and it has been reported extensively that PPARγ ligands inhibit cellular proliferation and selectively induce apoptosis [28, 48–54]. Antidiabetic drugs troglitazone and other TZDs have been investigated to suppress growth and to stimulate apoptosis and autophagy in a number of tumorigenic models and PPARγ has been recommended to be involved [55, 56]. Autophagy flux is the whole process through which cytoplasmic elements are initiated to lysosomes for degradation [57, 58]. Therefore, autophagy is outgoing as an important destination for the interruption and treatment of tumors [59], and targeting the autophagic pathways yields an innovative strategy for drug detection and new goals for drug improvement in cancer treatment [60].
Recent evidence has shown that considerable extent of cancer cell lines, including A549 cells were highly resistant to TRAIL-induced apoptosis [61]. In our present study, we also observed that Treatment of TRAIL or troglitazone alone did not or only slightly induced-apoptosis (Figure 1) and did not morphological change was identified compared with the control. We suggested that troglitazone and TRAIL co-treatment motives a significant induction of cancer cell death in lung A549 cells that are highly resistant to either agent alone. Some reports have demonstrated that troglitazone treatment inhibited cancer cell proliferation and induction of autophagy [37]. However, our western blot and immunocytochemistry results suggested LC3-II was increased and p62 was decreased after troglitazone treatment in a dose- dependent manner, though co-treatment of troglitazone with TRAIL enhanced intracellular apoptosis indicators Ac-cas3 and Ac-cas8 expression levels compared to treatment with troglitazone or TRAIL alone (Figure 2). Our results also suggested that specific pharmacological inhibitor chloroquine promoted the survival of lung adenocarcinoma A549 cells (Figure 3 and Figure 4). In addition, genetic autophagy inhibitor blocked troglitazone mediated apoptosis of A549 cells induced by TRAIL (Figure 5 and Figure 6). The ability of troglitazone to suppress a number of tumors has been attributed to its potential to inhibit growth and trigger apoptosis in a PPARγ-dependent manner [35, 62]. Recently revealed that PPARγ activation depend on autophagy, since GW9662 was able to prevent the upregulation of Beclin-1 as well as the accumulation of LC3 and MDC labeled vacuoles in breast cancer cells [63]. Our results demonstrated that Pretreatment of troglitazone induced activation of PPARγ in a dose-dependent manner. Western blot result suggested that the conversion rate of LC3-I to LC3-II and PPARγ was suppressed in the presence of GW9662 (Figure 7).
In summary, PPARγ activation by troglitazone sensitizes TRAIL-induced tumor cell death in A549 cells via autophagy flux. Combined treatment of troglitazone with TRAIL might be an adequate therapeutic technique to carefully treat some TRAIL-resistant cancers, including lung adenocarcinoma cells.
MATERIALS AND METHODS
Cell culture
Cancer cells originating from human lung (A549, HCC-15 and Calu-3) tumors were obtained from the American Type Culture Collection (Global Bioresource Center, Manassas, VA, USA). Cells were maintained in RPMI-1640 (Gibco BRL, Grand Island, NY, USA) medium containing 10% fetal bovine serum and 100 μg/ml penicillin-streptomycin. Cells were maintained at 37°C and 5% CO2 in humidified incubator.
Reagents
Recombinant troglitazone, chloroquine (20 μM) and GW9662 (10 μM) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Recombinant TRAIL (200 ng/ml) was purchased from Abfrontier (Geumcheon-gu, Seoul, South Korea).
Cell viability assay
A549, HCC-15 and Calu-3 cells were plated at 1.0 × 104 cells/well in 12-well plates and incubated at 37°C for 24 h. The A549 cells were pretreated with troglitazone in a dose-dependent manner (0, 1, 2, and 4 μM). After pretreated with different doses of troglitazone for 12 h and were treated with TRAIL protein for an additional 2 h. Additional cells were also pretreated with chloroquine (20 μM) and GW9662 (10 μM) for 1 h, followed by troglitazone treatment. Cell morphology was examined by photographs taken under inverted microscopy (Nikon, Japan). Cell viability was determined applying crystal violet staining method as previously described [64].
Trypan blue exclusion assay
The number of cell viability was examined by trypan blue dye exclusion assay (Sigma-Aldrich) using a hemocytometer. The result was mainly expressed as the percentage of viable cells compared with that of vehicle-treated controls.
Western blot assay
A549 cell lysates were prepared by harvesting, washing in cold PBS, resuspending in lysis buffer followed by sonication. Proteins (35 μg) were resolved by 10%–15% SDS gels and transferred to a nitrocellulose membrane, and analyzed by western blotting as described previously [65]. The following antibodies were used for immunoblotting: LC3(Novus Biologicals, Littleton, CO, USA), DR-4, DR-5, and β-actin Sigma-Aldrich (St. Louis, MO, USA), p62 (Millipore Corp., Milford, MA, USA), ATG5, cleaved caspase-3 (Cell Signaling Technology, Danvers, MA, USA), cleaved caspase-8 (BD pharmingen, USA), PPARγ Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Immunocytochemistry
A549 cell lines cultured on glass coverslips positioned on a 24-well plate. The cells were washed with PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. Following this, Cells were then washed twice with ice-cold PBS, blocked with 5% FBS inTris-buffered saline with Tween, and incubated with monoclonal antibodies against p62, PPARγ at room temperature for 24 h. Unbound antibody was removed with PBS wash ( three times) and Cells were then incubated again with secondary antibody at room temperature for 2 h in the dark. Finally, cells were mounted with DakoCytomation fluorescent mounting medium and visualized via a fluorescence microscopy.
TEM (transmission electron microscopy) analysis
TEM samples were analyzed by Transmission Electron Microscope (JEM-2010, JEOL) installed in the Center for University-Wide Research Facilities (CURF) at Chonbuk National University. After fixation of A549 cell samples in 2% glutaraldehyde and 2% paraformaldehyde in 0.05 sodium cacodylate buffer, specimens were post fixed in 1% osmium tetroxide, dehydrated in graded ethanol and propylene oxide. A549 cells were embedded in Epoxy resin. Ultrathin sections were cut on an LKB-III ultratome and were stained with 0.5% uranyl acetate and lead citrate. The images were taken on a Hitachi H7650 electron microscope at an accelerating voltage of 100 kV.
RNA interference
A549 cells were transfected with ATG5-specific small interfering RNA (siRNA; oligo ID HSS114103; Invitrogen, Carlsbad, CA, USA) using Lipofectamine 2000 according to the manufacturer’s instructions. After 36-h post transfection, the knockdown efficiency at protein level was observed by immunoblotting and cell viability test. Nonspecific siRNA was used as a negative control.
Statistical analysis
All data are expressed as means ± standard deviation (SD) and were compared using the Student’s t-test, analysis of variance and the ANOVA Duncan test using SAS statistical package (SAS Institute, Cary, NC, USA). Statistical significance was indicated by a P value less than 0.05 (*), 0.01 (**), or 0.001 (***).
ACKNOWLEDGMENTS AND FUNDING
This study was supported by a grant from the National Research Foundation of Korea (NRF), funded by the Korean government (MISP) (2013R1A4A1069486).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Ashkenazi A, Dixit VM. Apoptosis control by death and decoy receptors. Current opinion in cell biology. 1999; 11:255–260.
2. Almasan A, Ashkenazi A. Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy. Cytokine & growth factor reviews. 2003; 14:337–348.
3. Pan G, O'Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, Dixit VM. The receptor for the cytotoxic ligand TRAIL. Science (New York, NY). 1997; 276:111–113.
4. Walczak H, Degli-Esposti MA, Johnson RS, Smolak PJ, Waugh JY, Boiani N, Timour MS, Gerhart MJ, Schooley KA, Smith CA, Goodwin RG, Rauch CT. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. The EMBO journal. 1997; 16:5386–5397.
5. Degli-Esposti MA, Smolak PJ, Walczak H, Waugh J, Huang CP, DuBose RF, Goodwin RG, Smith CA. Cloning and characterization of TRAIL-R3, a novel member of the emerging TRAIL receptor family. The Journal of experimental medicine. 1997; 186:1165–1170.
6. Degli-Esposti MA, Dougall WC, Smolak PJ, Waugh JY, Smith CA, Goodwin RG. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity. 1997; 7:813–820.
7. Gonzalvez F, Ashkenazi A. New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene. 2010; 29:4752–4765.
8. Johnstone RW, Frew AJ, Smyth MJ. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nature reviews Cancer. 2008; 8:782–798.
9. Jin Z, McDonald ER, 3rd, Dicker DT, El-Deiry WS. Deficient tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor transport to the cell surface in human colon cancer cells selected for resistance to TRAIL-induced apoptosis. The Journal of biological chemistry. 2004; 279:35829–35839.
10. Kelly MM, Hoel BD, Voelkel-Johnson C. Doxorubicin pretreatment sensitizes prostate cancer cell lines to TRAIL induced apoptosis which correlates with the loss of c-FLIP expression. Cancer biology & therapy. 2002; 1:520–527.
11. Ng CP, Zisman A, Bonavida B. Synergy is achieved by complementation with Apo2L/TRAIL and actinomycin D in Apo2L/TRAIL-mediated apoptosis of prostate cancer cells: role of XIAP in resistance. The Prostate. 2002; 53:286–299.
12. Walczak H, Bouchon A, Stahl H, Krammer PH. Tumor necrosis factor-related apoptosis-inducing ligand retains its apoptosis-inducing capacity on Bcl-2- or Bcl-xL-overexpressing chemotherapy-resistant tumor cells. Cancer research. 2000; 60:3051–3057.
13. Zhang Y, Zhang B. TRAIL resistance of breast cancer cells is associated with constitutive endocytosis of death receptors 4 and 5. Molecular cancer research. 2008; 6:1861–1871.
14. Jung KJ, Min KJ, Bae JH, Kwon TK. Carnosic acid sensitized TRAIL-mediated apoptosis through down-regulation of c-FLIP and Bcl-2 expression at the post translational levels and CHOP-dependent up-regulation of DR5, Bim, and PUMA expression in human carcinoma caki cells. Oncotarget. 2015; 6:1556–1568. doi: 10.18632/oncotarget.2727.
15. Cao W, Li X, Zheng S, Zheng W, Wong YS, Chen T. Selenocysteine derivative overcomes TRAIL resistance in melanoma cells: evidence for ROS-dependent synergism and signaling crosstalk. Oncotarget. 2014; 5:7431–7445. doi: 10.18632/oncotarget.2008.
16. Chen JJ, Bozza WP, Di X, Zhang Y, Hallett W, Zhang B. H-Ras regulation of TRAIL death receptor mediated apoptosis. Oncotarget. 2014; 5:5125–5137. doi: 10.18632/oncotarget.2091.
17. Shin EA, Sohn EJ, Won G, Choi JU, Jeong M, Kim B, Kim MJ, Kim SH. Upregulation of microRNA135a-3p and death receptor 5 plays a critical role in Tanshinone I sensitized prostate cancer cells to TRAIL induced apoptosis. Oncotarget. 2014; 5:5624–5636. doi: 10.18632/oncotarget.2152.
18. Di X, Zhang G, Zhang Y, Takeda K, Rivera Rosado LA, Zhang B. Accumulation of autophagosomes in breast cancer cells induces TRAIL resistance through downregulation of surface expression of death receptors 4 and 5. Oncotarget. 2013; 4:1349–1364. doi: 10.18632/oncotarget.1174.
19. Saturno G, Valenti M, De Haven Brandon A, Thomas GV, Eccles S, Clarke PA, Workman P. Combining trail with PI3 kinase or HSP90 inhibitors enhances apoptosis in colorectal cancer cells via suppression of survival signaling. Oncotarget. 2013; 4:1185–1198. doi: 10.18632/oncotarget.1162.
20. Chen JJ, Mikelis CM, Zhang Y, Gutkind JS, Zhang B. TRAIL induces apoptosis in oral squamous carcinoma cells--a crosstalk with oncogenic Ras regulated cell surface expression of death receptor 5. Oncotarget. 2013; 4:206–217. doi: 10.18632/oncotarget.813.
21. Chen JJ, Shen HC, Rivera Rosado LA, Zhang Y, Di X, Zhang B. Mislocalization of death receptors correlates with cellular resistance to their cognate ligands in human breast cancer cells. Oncotarget. 2012; 3:833–842. doi: 10.18632/oncotarget.542.
22. Fulda S. Novel insights into the synergistic interaction of Bortezomib and TRAIL: tBid provides the link. Oncotarget. 2011; 2:418–421. doi: 10.18632/oncotarget.277.
23. Eberhard Y, Gronda M, Hurren R, Datti A, MacLean N, Ketela T, Moffat J, Wrana JL, Schimmer AD. Inhibition of SREBP1 sensitizes cells to death ligands. Oncotarget. 2011; 2:186–196. doi: 10.18632/oncotarget.239.
24. Rosen ED, Spiegelman BM. PPARgamma : a nuclear regulator of metabolism, differentiation, and cell growth. The Journal of biological chemistry. 2001; 276:37731–37734.
25. Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994; 79:1147–1156.
26. Brockman JA, Gupta RA, Dubois RN. Activation of PPARgamma leads to inhibition of anchorage-independent growth of human colorectal cancer cells. Gastroenterology. 1998; 115:1049–1055.
27. Mueller E, Sarraf P, Tontonoz P, Evans RM, Martin KJ, Zhang M, Fletcher C, Singer S, Spiegelman BM. Terminal differentiation of human breast cancer through PPAR gamma. Molecular cell. 1998; 1:465–470.
28. Elstner E, Muller C, Koshizuka K, Williamson EA, Park D, Asou H, Shintaku P, Said JW, Heber D, Koeffler HP. Ligands for peroxisome proliferator-activated receptorgamma and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95:8806–8811.
29. Ricote M, Huang J, Fajas L, Li A, Welch J, Najib J, Witztum JL, Auwerx J, Palinski W, Glass CK. Expression of the peroxisome proliferator-activated receptor gamma (PPARgamma) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95:7614–7619.
30. Sarraf P, Mueller E, Jones D, King FJ, DeAngelo DJ, Partridge JB, Holden SA, Chen LB, Singer S, Fletcher C, Spiegelman BM. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nature medicine. 1998; 4:1046–1052.
31. Wang C, Fu M, D'Amico M, Albanese C, Zhou JN, Brownlee M, Lisanti MP, Chatterjee VK, Lazar MA, Pestell RG. Inhibition of cellular proliferation through IkappaB kinase-independent and peroxisome proliferator-activated receptor gamma-dependent repression of cyclin D1. Molecular and cellular biology. 2001; 21:3057–3070.
32. Singh TR, Shankar S, Chen X, Asim M, Srivastava RK. Synergistic interactions of chemotherapeutic drugs and tumor necrosis factor-related apoptosis-inducing ligand/Apo-2 ligand on apoptosis and on regression of breast carcinoma in vivo. Cancer research. 2003; 63:5390–5400.
33. Hesry V, Piquet-Pellorce C, Travert M, Donaghy L, Jegou B, Patard JJ, Guillaudeux T. Sensitivity of prostate cells to TRAIL-induced apoptosis increases with tumor progression: DR5 and caspase 8 are key players. The Prostate. 2006; 66:987–995.
34. Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, Burris TP, Rastinejad F. Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature. 2008; 456:350–356.
35. Elrod HA, Sun SY. PPARgamma and Apoptosis in Cancer. PPAR research. 2008; 2008:704165.
36. Kiss-Toth E, Roszer T. PPARgamma in Kidney Physiology and Pathophysiology. PPAR research. 2008; 2008:183108.
37. Zhou J, Zhang W, Liang B, Casimiro MC, Whitaker-Menezes D, Wang M, Lisanti MP, Lanza-Jacoby S, Pestell RG, Wang C. PPARgamma activation induces autophagy in breast cancer cells. The international journal of biochemistry & cell biology. 2009; 41:2334–2342.
38. Jiang P, Mizushima N. Autophagy and human diseases. Cell research. 2014; 24:69–79.
39. Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005; 120:237–248.
40. Mizushima N. Autophagy: process and function. Genes & development. 2007; 21:2861–2873.
41. Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008; 4:151–175.
42. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. The EMBO journal. 2000; 19:5720–5728.
43. Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005; 1:84–91.
44. Kim AD, Kang KA, Kim HS, Kim DH, Choi YH, Lee SJ, Kim HS, Hyun JW. A ginseng metabolite, compound K, induces autophagy and apoptosis via generation of reactive oxygen species and activation of JNK in human colon cancer cells. Cell death & disease. 2013; 4:e750.
45. Kim A, Yim NH, Ma JY. Samsoeum, a traditional herbal medicine, elicits apoptotic and autophagic cell death by inhibiting Akt/mTOR and activating the JNK pathway in cancer cells. BMC complementary and alternative medicine. 2013; 13:233.
46. Chen YJ, Chi CW, Su WC, Huang HL. Lapatinib induces autophagic cell death and inhibits growth of human hepatocellular carcinoma. Oncotarget. 2014; 5:4845–4854. doi: 10.18632/oncotarget.2045.
47. Mahalingam D, Szegezdi E, Keane M, de Jong S, Samali A. TRAIL receptor signalling and modulation: Are we on the right TRAIL? Cancer treatment reviews. 2009; 35:280–288.
48. Clay CE, Namen AM, Atsumi G, Willingham MC, High KP, Kute TE, Trimboli AJ, Fonteh AN, Dawson PA, Chilton FH. Influence of J series prostaglandins on apoptosis and tumorigenesis of breast cancer cells. Carcinogenesis. 1999; 20:1905–1911.
49. Nwankwo JO, Robbins ME. Peroxisome proliferator-activated receptor- gamma expression in human malignant and normal brain, breast and prostate-derived cells. Prostaglandins, leukotrienes, and essential fatty acids. 2001; 64:241–245.
50. Martelli ML, Iuliano R, Le Pera I, Sama I, Monaco C, Cammarota S, Kroll T, Chiariotti L, Santoro M, Fusco A. Inhibitory effects of peroxisome poliferator-activated receptor gamma on thyroid carcinoma cell growth. The Journal of clinical endocrinology and metabolism. 2002; 87:4728–4735.
51. Shimada T, Kojima K, Yoshiura K, Hiraishi H, Terano A. Characteristics of the peroxisome proliferator activated receptor gamma (PPARgamma) ligand induced apoptosis in colon cancer cells. Gut. 2002; 50:658–664.
52. Toyoda M, Takagi H, Horiguchi N, Kakizaki S, Sato K, Takayama H, Mori M. A ligand for peroxisome proliferator activated receptor gamma inhibits cell growth and induces apoptosis in human liver cancer cells. Gut. 2002; 50:563–567.
53. Wang YL, Frauwirth KA, Rangwala SM, Lazar MA, Thompson CB. Thiazolidinedione activation of peroxisome proliferator-activated receptor gamma can enhance mitochondrial potential and promote cell survival. The Journal of biological chemistry. 2002; 277:31781–31788.
54. Yoshizawa K, Cioca DP, Kawa S, Tanaka E, Kiyosawa K. Peroxisome proliferator-activated receptor gamma ligand troglitazone induces cell cycle arrest and apoptosis of hepatocellular carcinoma cell lines. Cancer. 2002; 95:2243–2251.
55. Kim JB, Wright HM, Wright M, Spiegelman BM. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95:4333–4337.
56. Krishnan A, Nair SA, Pillai MR. Biology of PPAR gamma in cancer: a critical review on existing lacunae. Current molecular medicine. 2007; 7:532–540.
57. Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell death and differentiation. 2005; 12 Suppl 2:1509–1518.
58. Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science (New York, NY). 2000; 290:1717–1721.
59. Hippert MM, O'Toole PS, Thorburn A. Autophagy in cancer: good, bad, or both? Cancer research. 2006; 66:9349–9351.
60. Liu B, Bao JK, Yang JM, Cheng Y. Targeting autophagic pathways for cancer drug discovery. Chinese journal of cancer. 2013; 32:113–120.
61. Jin CY, Park C, Hwang HJ, Kim GY, Choi BT, Kim WJ, Choi YH. Naringenin up-regulates the expression of death receptor 5 and enhances TRAIL-induced apoptosis in human lung cancer A549 cells. Molecular nutrition & food research. 2011; 55:300–309.
62. Campbell MJ, Carlberg C, Koeffler HP. A Role for the PPARgamma in Cancer Therapy. PPAR research. 2008; 2008:314974.
63. Rovito D, Giordano C, Plastina P, Barone I, De Amicis F, Mauro L, Rizza P, Lanzino M, Catalano S, Bonofiglio D, Ando S. Omega-3 DHA- and EPA-dopamine conjugates induce PPARgamma-dependent breast cancer cell death through autophagy and apoptosis. Biochimica et biophysica acta. 2015; 1850:2185–2195.
64. Nazim UM, Moon JH, Lee JH, Lee YJ, Seol JW, Eo SK, Lee JH, Park SY. Activation of autophagy flux by metformin downregulates cellular FLICE-like inhibitory protein and enhances TRAIL- induced apoptosis. Oncotarget. 2016; 7:23468–23481. doi: 10.18632/oncotarget.8048.
65. Lee JH, Moon JH, Kim SW, Jeong JK, Nazim UM, Lee YJ, Seol JW, Park SY. EGCG-mediated autophagy flux has a neuroprotection effect via a class III histone deacetylase in primary neuron cells. Oncotarget. 2015; 6:9701–9717. doi: 10.18632/oncotarget.3832.