
INTRODUCTION
In recent years, extensive effort has been made towards a better understanding of the oncogenic events that drive breast cancer initiation and progression. However, successful treatments remain a challenge in many cases due to heterogeneity of breast tumors. Gene amplification is one of the most common genetic abnormality events in breast cancer. Chromosome 20q is frequently amplified in a variety of cancers including prostate, ovarian, bladder, colorectal, pancreatic, and breast [1-6]. Amplification of 20q is also associated with poor clinical outcome [7] and serves as an indicator for cancer progression and metastasis [6,8]. In breast cancer, multiple genes localized to 20q have been identified as oncogenes, including Aurora-A kinase (AURKA), ZNF217 [9], UBE2C [10] and TPX2 [11,12]. AURKA is the most studied gene on 20q. High expression levels of AURKA indicate decreased survival in breast cancer patients [13] and is currently an anticancer target [14]. Another gene on 20q, ZNF217 was shown to be a marker for poor breast cancer prognostis [15,16] and its overexpression promotes epithelial-mesenchymal transition (EMT) and invasion [16]. However, the detailed and integral mechanism for how chromosome 20q affects tumorigenesis and tumor behavior is not clearly understood. Other genes on 20q are also likely to participate in tumorigenesis and/or metastasis, but their functions are yet to be defined.
Here we focus on the gene named ‘family with sequence similarity 83, member D’ (FAM83D), located on chromosome 20q. FAM83D was first identified as a protein with spindle localization in mitosis [17]. FAM83D interacts with the chromokinesin KID22 and is required for correct chromosome congression in metaphase [18]. The mitotic spindle is responsible for accurate distribution of sister chromatids in cell division. Functional aberration of the mitotic spindle may lead to errors in chromosome segregation and aneuploidy, which are often seen in advanced human cancers. Various microarray studies suggest that FAM83D expression is elevated in hepatoacellular carcinoma [19], ovarian cancer [20] and metastatic lung adenocarcinomas [21]. However, the function and mechanism of FAM83D in tumorigenesis has not yet been studied.
FBXW7 is a bona fide tumor suppressor that is inactivated by gene mutation or expression downregulation in numerous human malignancies, including breast cancer [22]. It is a member of the F-box family of proteins, which function as the substrate recognition components of the Skp-Cullin-F-box (SCF) E3 ubiquitin ligase [22]. The SCFFBXW7 complex targets several well-known onco-proteins for ubiquitin-mediated degradation in a phosphorylation-dependent manner, including c-Jun, c-Myc, Cyclin E, KLF15, Notch and mTOR [23-28].
In the present study, we investigated whether FAM83D plays a role in breast cancer initiation and progression. We showed that overexpression of FAM83D inactivates FBXW7 by downregulating FBXW7 protein expression, leading to up-regulation of FBXW7 downstream targets, which in turn results in elevated cell proliferation, migration and invasion.
RESULTS
Elevated expression of FAM83D in human breast cancers
We first revisited the CGH microarray data previously published on primary breast cancers [29-31] and cell lines [32] and refined 20q into 5 sub-amplicon regions, one containing FAM83D (Fig. 1A). Next we examined FAM83D expression levels in a panel of 20 widely used human breast cancer cell lines. As expected, we found that the level of FAM83D mRNA was elevated in most of the malignant cell lines by 1.5 to 4 fold, in comparison to levels in non-malignant cell lines MCF10A and 184A1 (Fig. 1B). Correspondingly, FAM83D protein levels are consistently increased in breast cancer cell lines (Fig. 1C). FAM83D expression was further assessed in three publicly available microarray datasets in the Gene Expression Omnibus (GEO) database (GSE10780 [33], GSE3744 [34], and GSE14548 [35]) that contain both normal and breast cancer samples. FAM83D expression levels were measured as log2 (probe intensities) using Affymetrix microarrays. In all three datasets, the levels of FAM80D mRNA in breast cancers were statistically significantly higher than those in normal breast tissues (Fig. 1D). These results indicate that the expression level of FAM83D is elevated in breast tumors.
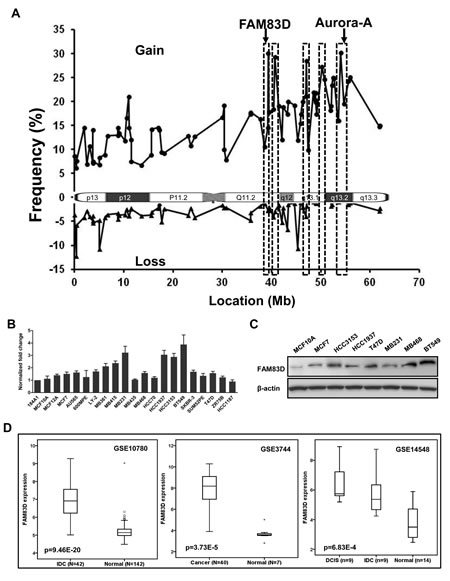
Figure 1: The expression of FAM83D is elevated in human breast cancers. (A) Genomic amplification on chromosome 20q was refined by integrative analysis of public copy number datasets for breast cancers, indicating that FAM83D is located at a peak of a sub-amplicon. (B) Expression profile of FAM83D in breast cancer cell lines. FAM83D mRNA levels relative to normal breast epithelial cell line 184A1 were determined by qRT-PCR. Gene expression was normalized to GAPDH. Data are presented as means ± Standard deviation. (C) Protein level of FAM83D in cultured breast cancer cell lines. (D) FAM83D mRNA expression levels are significantly elevated in breast tumors in comparison to normal breast tissues, using three public expression datasets. FAM83D expression is measured as log2 (probe intensities). The p-values were obtained from Mann-Whitney U or Kruskal-Wallis tests.
mRNA level of FAM83D is associated with clinical outcome of breast cancers
To investigate the clinical impact of elevated FAM83D expression in human breast cancer, we assessed the association between FAM83D mRNA levels and clinical outcome in four independent breast cancer cohorts [36-39] with clinical information (GEO database). To determine the prognostic impact of FAM83D expression in breast cancer, we categorized breast cancer patients into three groups based on FAM83D mRNA expression levels (low=bottom tertile, intermediate=middle tertile, and high=top tertile). Patients with tumors displaying high FAM83D expression levels had significantly shorter disease-free survival (DFS) compared to those with low FAM83D (p=0.0011, 0.0063, 1.93E-5 and 0.0021 in dataset GSE1456, GSE3494, GSE6532 and GSE20685, respectively) (Fig. 2). Moreover, in all four datasets, DFS curves for patients with intermediate levels of FAM83D was between those with high and low levels (Fig. 2), suggesting a dose-dependent effect of FAM83D expression on DFS.
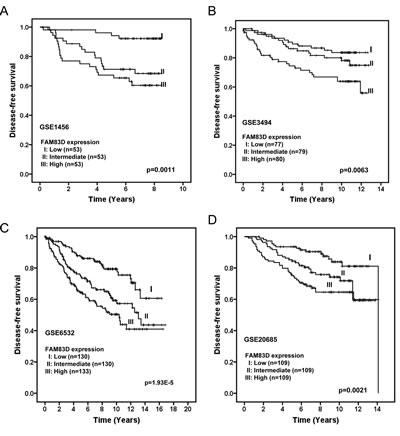
Figure 2: Elevated expression of FAM83D is associated with poor disease-free survival of breast cancer patients in four different cohorts (A) GSE1456, (B) GSE3494, (C) GSE6532, and (D) GSE20685. FAM83D expression is measured as log2 (probe intensities). The p-values were obtained from a long-rank test.
Overexpression of FAM83D promotes cell growth, migration and invasion
Since FAM83D is overexpressed in human tumors, we questioned whether FAM83D functions as an oncogene. To corroborate this hypothesis, we first tested whether overexpression of FAM83D in non-malignant MCF10A cells promotes cell growth. MCF10A cells have a low endogenous level of FAM83D (Fig. 1B). We generated MCF10A cells with stable expression of FLAG-tagged FAM83D gene (designated as MCF10A-FAM83D) (Fig. 3A). Overexpression of FAM83D strongly stimulates cell proliferation compared to control cells (Fig. 3B), indicating that FAM83D is potentially oncogenic.
Migration and invasion are hallmarks for cancer metastasis. To evaluate the functional role of FAM83D overexpression in cell motility in MCF10A cells, we performed an in vitro scratch healing assay and a Boyden chamber migration/invasion assay. As shown in Fig. 3C and 3D, FAM83D overexpression in MCF10A accelerated wound closure and migration through the uncoated membrane of the Boyden chamber compared to control cells. Moreover, MCF10A-FAM83D cells displayed a significant increase in the number of cells that migrated through the Matrigel-coated membrane in the Boyden chamber (invasion assay) (Fig. 3D). Consistent with these in vitro observations, we found that high level of FAM83D expression shortened the distant metastasis-free survival in breast cancer patients (Fig. 3E). Together, these results clearly suggest that FAM83D overexpression promotes cell motility.
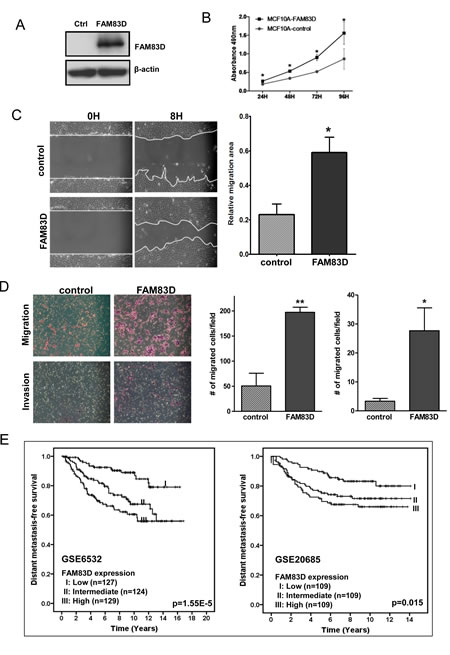
Figure 3: Ectopic expression of FAM83D enhances cell proliferation, migration and invasion. (A) MCF10A cells with FLAG-tagged FAM83D (MCF10A-FAM83D) were established with stable expression of FAM83D and confirmed by Western blot using an anti-Flag antibody. (B) Proliferation of MCF10A-FAM83D is significantly accelerated compared to normal MCF10A control cells measured by MTT assay. (C) MCF10A-FAM83D (FAM83D) closed the scratched area significantly faster than MCF10A control cells (control). The representative photographs are shown in left panel. Quantification of the data is presented as relative migration area in each cell line (right panel). (D) Migration and invasion ability of MCF10A-FAM83D cells (FAM83D) is significantly enhanced compared to MCF10A control cells (control) using the transwell assay. The representative photographs are shown in the left panel and the average number of migrated cells/field in the right panel. In b-d, data are presented as means ± standard deviation from three independent experiments, each performed in triplicate. * indicates p < 0.05 and ** indicates p < 0.01, which were obtained by a t-test. (E) High FAM83D mRNA levels reduce distant metastasis-free survival for breast cancer patients in two independent datasets (GSE6532 and GSE20685). The p-values in the graphs were obtained from a long-rank test.
FAM83D induces epithelial–mesenchymal transition in MCF10A cells
Given that ectopic expression of FAM83D increased cell motility and invasiveness, we next examined whether FAM83D overexpression could affect cell morphology. MCF10A-FAM83D cells exhibited a discohesive growth pattern and a spindle-shaped fibroblastic morphology, hallmarks of a mesenchymal phenotype, which was not observed in vector-only control cells (Fig. 4A). Consistently, the protein level of an epithelial marker E-cadherin is mildly suppressed in MCF10A-FAM83D cells (Fig. 4B). Moreover, immunofluorescent staining showed that the clear membrane distribution of E-cadherin MCF10A-control cells display, consistent with its normal function in cell-cell adhesion, was absent in MCF10A-FAM83D cells, and instead was enriched in the cytoplasm (Fig. 4C). Additionally, we found that Vimentin, a mesenchymal marker, dramatically increased in MCF10A-FAM83D cells (Fig. 4D). These findings suggest that FAM83D overexpression induces EMT.
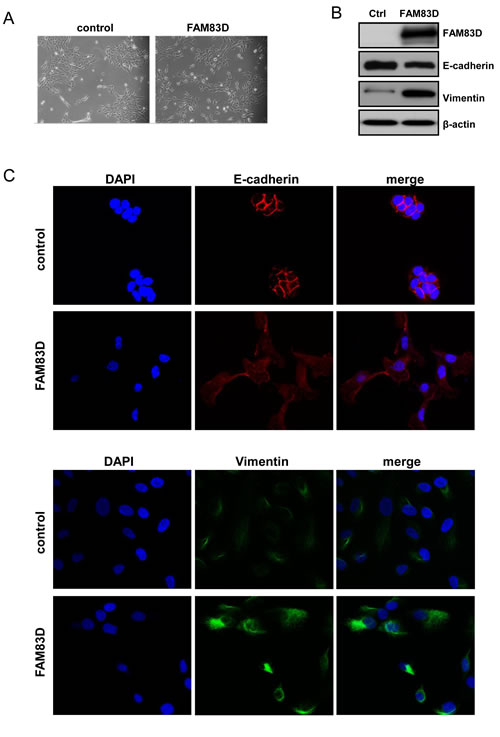
Figure 4: Overexpression of FAM83D in MCF10A cells leads to an epithelial-mesenchymal transition. (A) Representative micrographs of MCF10A-FAM83D and MCF10A control cells under bright field display significant differences in morphology. (B) Western analysis of E-cadherin and vimentin indicates EMT in MCF10A-FAM83D compared to MCF10A control cells. Loading control is β-actin. (C) Immunofluorescent staining of E-cadherin (red, upper panels) and vimentin (green, lower panels) demonstrates EMT in MCF10A-FAM83D compared to MCF10A control cells. DNA is stained by DAPI (blue).
Ablation of FAM83D inhibits tumor growth through apoptosis
Next we examined whether depletion of the high level of endogenous FAM83D proteins in BT549 cells (Fig. 1B) inhibited the cell proliferation and motility phenotypes. FAM83D was depleted using both stable (shFAM83D) and transient (siFAM83D) knockdown systems, which was verified at the protein level by Western blotting (Fig. 5A). Depletion of FAM83D dramatically inhibited BT549 cell proliferation (Fig. 5B). We observed many floating BT549-shFAM83D cells, implicating a potential apoptotic effect resulting from FAM83D depletion, which was confirmed by the Caspase3/7 apoptosis assay (Fig. 5C). Consistent with this result, we found a dramatic reduction in colony formation of BT549-shFAM83D cells (Fig. 5D). In addition, significantly fewer BT549-shFAM83D cells were able to migrate through Matrigel compared to controls (Fig. 5E), indicating that inhibition of FAM83D dramatically restrained the invasiveness of BT549 cells.
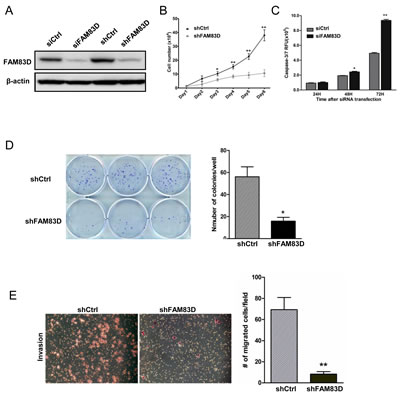
Figure 5: Biological consequences of FAM83D knockdown in BT549 cells. (A) FAM83D expression was analyzed by Western blotting using anti-FAM83D antibody in both stable shRNA and transient siRNA knockdown in BT549 cells. Loading was controlled by β-actin levels. (B) BT549 cells with stable FM83D knockdown (shFAM83D) show a significant reduction in proliferation in comparison to control cells (shCtrl). BT549-shFAM83D and control cells were seeded in six-well dish in triplicates and cell numbers were counted by hemocytometer at indicated time points. (C) Knockdown of FAM83D induces cell apoptosis in BT549 cells. BT549 cells were transfected with siFAM83D or control siRNA (siCtrl) in a 96-well plate in triplicates for the indicated times. Apoptosis was determined by a Caspase3/7 assay. (D) Knockdown of FAM83D reduces colony formation in BT549 cells. (E) Knockdown of FAM83D significantly decreases cell invasiveness. Representative photographs from the Matrigel-coated transwell invasion assay of BT549 shCtrl control cells and shFAM83D cells. Data are presented as means ± standard deviation from three independent experiments, each performed in triplicate. * indicates p<0.05 and ** indicates p< 0.01, which were obtained from t-test.
FAM83D downregulates tumor suppressor FBXW7 via interaction
To understand how FAM83D exerts its oncogenic effects, we used a yeast two-hybrid screening approach (data not shown) and discovered that FAM83D interacts with the F-box protein FBXW7. To confirm this result, 293T cells were co-transfected with FLAG-tagged FAM83D and HA-tagged FBXW7 constructs. Immunoprecipitation and subsequent immunoblot analyses revealed that HA-tagged FBXW7 co-immunoprecipitated with FLAG-tagged FAM83D, confirming a physical interaction between the two proteins (Fig. 6A). Moreover, we observed that HA-tagged FBXW7 co-immunoprecipitated with endogenous FAM83D (Fig. 6B), demonstrating that the physical interaction is not an artifact due to ectopic expression.
The physical interaction between FAM83D and FBXW7 suggested that FBXW7 could target FAM83D for ubiquitination and subsequent degradation. However, HCT116 and DLD1 FBXW7 knockout cells displayed no effect on FAM83D protein levels (Fig. 6C), implicating that FAM83D is not a downstream substrate of FBXW7.
We then addressed an alternative possibility in which FAM83D regulates FBXW7. Indeed, overexpression of FAM83D dramatically reduced endogenous FBXW7 protein levels (Fig. 6D), and was accompanied by increases in protein levels of FBXW7 substrates, including c-Myc, c-Jun and mTOR (Fig. 6D). Furthermore, FAM83D decreases the levels of FBXW7 proteins in a dose–dependent manner (Fig. 6E). Proteasome inhibition by addition of MG132 caused a significant increase in FBXW7 levels (Fig. 6F), suggesting that FAM83D promotes degradation of FBXW7.
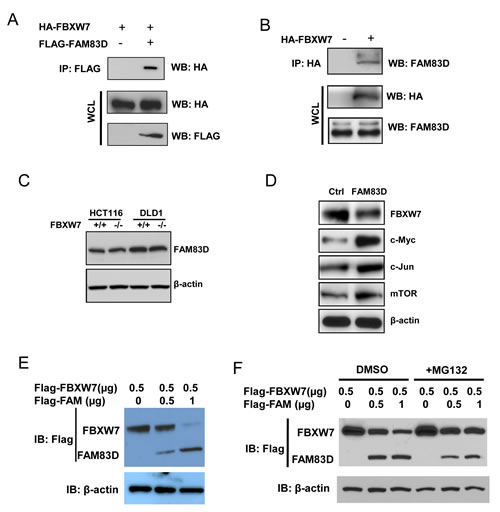
Figure 6: FAM83D downregulates FBXW7 via physical interaction. (A) FLAG-FAM83D is specifically co-immunoprecipitated with HA-FBXW7. (B) Ectopic FBXW7 (HA-FBXW7) binds to endogenous FAM83D in vivo. (C) FAM83D protein levels are not regulated by FBXW7. (D) Overexpression of FAM83D in MCF10A represses FBXW7 expression and leads to elevated expression of known FBXW7 targets, including c-Myc, c-Jun and mTOR. (E) Ectopic overexpression of FAM83D (FLAG-FAM83D) reduces FLAG-FBXW7 protein levels in a dose-dependent manner. Amounts of transfected plasmids are indicated above the blot. (F) Proteasome inhibition by MG132 suppresses reduction of FLAG-FBXW7 by FAM83D ectopic overexpression. In co-immunoprecipitated experiments, 5% of extracts used were shown as loading controls (WCL). In Western-blotting experiments, β-actin serves as the loading control. In a and b, “-“ indicates empty vector.
Inhibition of mTOR signaling suppresses FAM83D-induced oncogenic phenotypes
Since FAM83D directly regulates FBXW7 to control cell growth and motility, we tested whether inhibition of downstream substrates of FBXW7 can suppress oncogenic phenotypes induced by FAM83D overexpression. We focused on mTOR, since our previous study showed that mTOR inhibition by rapamycin suppresses EMT, invasion and stemness driven by loss of FBXW7 in colon cancer cells [40]. As shown in Fig. 7A, rapamycin treatment significantly decreased the closure rate of MCF10A-FAM83D cells in the scratch healing assay. Consistent with this result, rapamycin also significantly decreased migration (Fig. 7B) and invasion (Fig. 7C) of MCF10A-FAM83D cells in the Boyden chamber assay. These results indicate that FAM83D executes oncogenic functions via regulation of FBXW7, which can be attenuated by inhibition of mTOR signaling, an FBXW7-downstream target.
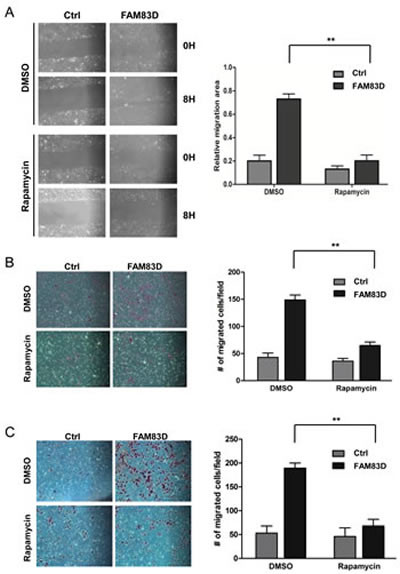
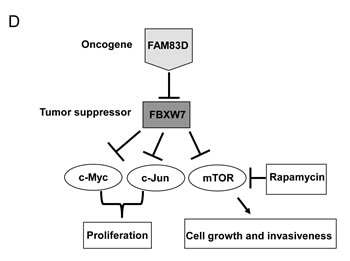
Figure 7: mTOR inhibition by rapamycin alleviates the enhanced migration and invasion caused by ectopic overexpression of FAM83D. (A) Inhibition of mTOR by rapamycin significantly suppresses cell motility induced by FAM83D ectopic overexpression in scratch-healing assay. The representative photographs are shown in left panel. Quantification of the data was presented as relative migration area in each cell line (right panel). (B and C) Enhanced cell migration and invasion caused by FAM83D ectopic overexpression were rescued by treatment of Rapamycin. Uncoated (B) or Matrigel-coated (C) transwell assays on MCF10A control cells and MCF10A-FAM83D cells treated with DMSO control and rapamycin. In a-c, data are presented as means ± Standard deviation from three independent experiments each performed in triplicates. ** indicates p<0.01, which were obtained from t-test. (D) Schematic illustration of the proposed oncogenic function of FAM83D in breast cancer development through inhibition of the FBXW7 tumor suppressor by physical interaction.
DISCUSSION
To our knowledge, this is the first study to report that FAM83D is an oncogene, based on several lines of evidence. First, we showed consistent overexpression of FAM83D mRNAs using a panel of breast cancer cell lines and primary breast cancers, and that high levels of FAM83D expression are associated with poor prognosis in breast cancer patients. Second, in vitro data provided direct evidence that FAM83D acts as an oncogene in breast cancer development by promoting cell growth, migration, and invasion. Third, overexpression of FAM83D induced EMT, a crucial mechanism regulating the initial steps in metastatic progression. Fourth, depletion of FAM83D induced cell death, reduced cell proliferation and colony formation, and abolished the invasive capability in aggressive and metastatic BT549 cells. Finally, we found that FAM83D downregulates the tumor suppressor gene FBXW7. These results led us to propose a model for how FAM83D executes its oncogenic function (Fig. 7D).
Mounting evidence shows that in epithelial cancers, including breast cancer, induction of EMT is a major event that provides mobility to cancer cells in order to generate metastases [41]. EMT is characterized by the loss of epithelial characteristics and acquisition of a mesenchymal phenotype, which confers the ability for cancer cells to invade adjacent tissue and migrate to distant sites [41]. In this study, we showed that ectopic expression of FAM83D in MCF10A cells displayed morphological and molecular characteristics in common with EMT, including the associated stimulatory effects on in vitro migration and invasion. These findings provide a mechanistic framework to explain the clinical observations that breast cancer patients with high levels of FAM83D in cancer tissue samples have significantly shorter disease-free survival. Silencing of FAM83D induced cell death, suggesting FAM83D is a potential target for cancer therapy to improve patients’ prognosis, especially for cancers with overexpression of FAM83D.
FBXW7 is a tumor suppressor that controls the protein levels of many well-known oncogenes and is frequently mutated in many cancers [22]. However, little is known about the regulation of FBXW7 itself. We found that FAM83D physically interacts with and downregulates FBXW7 protein levels. Consequently, the levels of FBXW7-targeted oncoproteins, such as c-Myc, C-Jun and mTOR, are increased upon overexpression of FAM83D, which in turn leads to the increase in cell proliferation and motility. Surprisingly, inhibition of mTOR alone by rapamycin blocks FAM83D-induced oncogenic characteristics in cells, clearly suggesting that FAM83D executes its oncogenic function at least partially due to mTOR activation through FBXW7 downregulation. However, further experiments are necessary to explore the possible involvement of other oncogenic pathways.
Several mechanisms have been reported for inactivation of FBXW7 in human cancer including mutation, deletion and hypermethylation [22, 42, 43]. A lot of effort has focused on finding FBXW7 mutations in various types of human cancer, which have shown that the overall point mutation frequency is only 6% in human cancers [43]. In breast cancer especially, the somatic mutation rate of the FBXW7 gene was found to be less than 1%, based on The Catalogue of Somatic Mutations in Cancer Database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/). Our study identifies an alternative mechanism for FBXW7 inactivation in breast cancer through overexpression/amplification of FAM83D. This finding may be extended to a variety of other human cancers.
In summary, this study demonstrates that FAM83D promotes oncogenic properties upon overexpression, and physically interacts with FBXW7. Overexpression of FAM83D in tumor cells leads to downregulation of FBXW7, resulting in elevated levels of numerous oncoproteins downstream of FBXW7. Targeting the interaction between FAM83D and FBXW7 could serve as an effective cancer therapy.
MATERIALS AND METHODS
Cell lines and plasmids
The cell lines MCF10A and BT549 were maintained as previously described [32]. C20ORF129 (1758) cDNA was a gift from Dr. Erich A. Nigg. Full length FAM83D (1848bp) was generated by ligation of the 90bp fragment into 5’ of C20ORF129. FAM83D was cloned into a 3XFLAG-HA tagged CMV10 vector, which was subsequently cloned into a pBabe.puro retroviral vector (Cell Biolabs).
Knockdown of FAM83D by short hairpin RNA
The shRNA-expression vector was generated by cloning the annealed sense (GATCCCCatggacggatggcaaattaTTCAAGAG AtaatttgccatccgtccatTTTTTGGAAA) andantisense (AGCTTTTCCAAAAAatggacggatggcaaatta TCTCTTGA AtaatttgccatccgtccatGGG) oligos into the pSUPER.puro retroviral vector (Oligoengine). Viral particles were produced by transfection of the pSUPER vector into 293T ectopic Phoenix cells using Lipofectamine 2000 (Invitrogen). Stable shFAM83D BT549 clones were selected with puromycin (2ug/ml), and empty pSuper.Retro.puro vector was used as control.
RNA isolation and real-time RT-PCR
RNA was isolated using the TRIzol Reagent (Invitrogen). First strand cDNA was synthesized with the SuperScript III (Invitrogen) or iScript (Bio-Rad) cDNA synthesis kits. All primers for qRT-PCR are: FAM83D, forward: 5’-CGCGAGAGGTGATTGCAGTGGT-3’; reverse: 5’-CGCGAGAGGTGATTGCAGTGGT-3’. GAPDH: forward: 5’-AAGGTGAAGGTCGGAGTCAA-3’; reverse: 5’-AATGAAGGGGTCATTGATGG-3’. qRT-PCR was performed using the Power SYBR Green PCR Master Mix (ABI) and the CFX96 Real-time PCR detection system (Bio-Rad), according to manufacturers’ protocols.
Cell proliferation assay
Cells were seeded in 96-well plates in triplicate at densities of 1×103 per well. Cell proliferation was monitored at desired time points using 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) (Promega). In brief, the MTT assay was performed by adding 20 μl MTT (5mg/ml) for 4 hrs. Light absorbance of the solution was measured at 570 nm on a microplate reader.
Colony formation assay
Cells were seeded in triplicate at 500 cells/6-cm dishes in complete medium. After 3 weeks of growth, cells were fixed and stained with crystal violet (0.1%, w/v in 20nM 4-morpholinepropanesulfonic acid), and visible colonies were counted to determine cell numbers in each colony.
Scratch-healing assay
Monolayers of MCF10A and MCF10A-FAM83D cells grown in 6-well plates were scratched by a sterile 200 µl pipette tip, washed 3 times with DPBS then cultured for the indicated time. Photographs focusing on the same position were taken immediately and after indicated time with a Nikon camera.
Motility and invasion assay
2.5 x 105 (invasion) or 2.5 x 104 (motility) cells were resuspended in DMEM/F12 1:1 medium and seeded on BD Falcon Cell Culture Inserts with or without a thin layer of MATRIGEL Basement Membrane Matrix. The inserts were then placed on 6-well plates containing complete medium acting as chemo-attractant. After incubation for 16 hours, inserts were washed with PBS, fixed with 4% formalin (Sigma), permeabilized with methanol and stained with Giemsa staining solution (Invitrogen). The unmigrated cells on the surface of the membrane were removed using cotton swabs. Images were taken and analyzed with ImageJ to acquire cell numbers.
Apoptosis Assay
BT549 cells were transfected with a negative control or siFAM83D RNA oligos (Ambion) for 24, 48 and 72 hours. Caspase 3/7 activity was measured at each time point with the Caspase-Glo 3/7 Assay kit (Promega) according to the manufacturer’s instructions.
Antibodies and Western blotting
Antibodies against E-cadherin and Vimentin, are from Cell Signaling, and anti-flag and anti-HA antibodies are from Sigma. For Western blots, cells were washed with cold PBS and lysed in RIPA lysis buffer (1% Triton X-100, 0.1% SDS, 50mM Tris PH 7.5, 150mM NaCl, 0.5% sodium deoxycholate, 10mM NaF) supplemented with 2 mM PMSF, 2 mM Na3VO4, and protease inhibitors (Roche). Cell lysates were fractionated on Novex® Tris-Glycine gels (Invitrogen) and transferred to PDVF membranes (Millipore) before antibody detection.
Confocal immunofluorescence microscopy
Cells were seeded on glass coverslips in 4-well plates then fixed with 4% paraformaldehyde or methanol, then permeabilized for 5 min with PBS containing 0.1% Triton X-100. These cells were then blocked for 30min in 10% BSA and then incubated with primary antibodies overnight at 4oC. After three washes in PBS, the slides were incubated for 1 hour in the dark with FITC-conjugated secondary antibodies (Invitrogen, Grand Island, NY, USA). After three further washes, the slides were stained with DAPI for 5 min to visualize the nuclei, and examined using a Confocal Zeiss Axio Observer D1 microscope and images were analysis by ImageJ software.
Statistical analysis
Differences in FAM83D mRNA expression levels between normal and breast cancers was analyzed by Mann-Whitney U (two groups) or Kruskal-Wallis (three groups) tests using publicly available datasets downloaded from the Gene Expression Omnibus (GEO). Kaplan-Meier plots were constructed and a long rank test was used to evaluate differences in FAM83D expression levels for disease-free and distant metastasis-free survival curves. In addition, the relationship between FAM83D expression and survival was explored in microarray datasets by dividing the cases from each cohort into a group with high (top one-third), moderate (middle one-third), and low (bottom one-third) levels of expression. All analyses were performed using SPSS 11.5.0 for Windows. A two-tailed p-value of less than 0.05 was considered to indicate statistical significance. We determined the significance of differences in cell proliferation, migration, invasion and apoptosis using two-tailed Student’s t-test (MS EXCEL 2007 or Graphpad Prism 5). Real-time RT-PCR data analysis was performed using CFX96 Manager (Bio-Rad).
ACKNOWLEDGEMENTS
The authors thank Dr. Erich A Nigg for providing expression constructs of FAM83D, Dr. Bert Vogelstein for providing HCT116 and DLD-1 and their derivative FBXW7-/- cell lines, and Dr. Gary H Karpen for reading and editing manuscript. This work was supported by National Natural Science Foundation of China No. 81172528, 31271461, Doctoral Fund of Ministry of Education of China No. 20110131110035 and Natural Science Foundation of Shandong Province No. ZR2011HM034 (G. Wei); by the National Institutes of Health, National Cancer Institute grant R01 CA116481, and the Low Dose Scientific Focus Area, Office of Biological & Environmental Research, US Department of Energy (DE-AC02-05CH11231) (J.-H. Mao).
References
1. Tsafrir D, Bacolod M, Selvanayagam Z, Tsafrir I, Shia J, Zeng Z, Liu H, Krier C, Stengel RF, Barany F, Gerald WL, Paty PB, Domany E, Notterman DA. Relationship of gene expression and chromosomal abnormalities in colorectal cancer. Cancer Res. 2006; 66: 2129-37.
2. Scotto L, Narayan G, Nandula SV, Arias-Pulido H, Subramaniyam S, Schneider A, Kaufmann AM, Wright JD, Pothuri B, Mansukhani M, Murty VV. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: potential role in progression. Genes Chromosomes Cancer. 2008; 47: 755-65.
3. Ramakrishna M, Williams LH, Boyle SE, Bearfoot JL, Sridhar A, Speed TP, Gorringe KL, Campbell IG. Identification of candidate growth promoting genes in ovarian cancer through integrated copy number and expression analysis. PLoS One. 2010; 5: e9983.
4. Karhu R, Mahlamaki E, Kallioniemi A. Pancreatic adenocarcinoma - genetic portrait from chromosomes to microarrays. Genes Chromosomes Cancer. 2006; 45:721-30.
5. van Dekken H, Paris PL, Albertson DG, Alers JC, Andaya A, Kowbel D, van der Kwast TH, Pinkel D, Schroder FH, Vissers KJ, Wildhagen MF, Collins C. Evaluation of genetic patterns in different tumor areas of intermediate-grade prostatic adenocarcinomas by high-resolution genomic array analysis. Genes Chromosomes Cancer. 2004; 39: 249-56.
6. Wullich B, Riedinger S, Brinck U, Stoeckle M, Kamradt J, Ketter R, Jung V. Evidence for gains at 15q and 20q in brain metastases of prostate cancer. Cancer Genet. Cytogenet. 2004; 154: 119-23.
7. Postma C, Terwischa S, Hermsen MA, van der Sijp JR, Meijer GA. Gain of chromosome 20q is an indicator of poor prognosis in colorectal cancer. Cell Oncol. 2007; 29: 73-5.
8. Carvalho B, Postma C, Mongera S, Hopmans E, Diskin S, van de Wiel MA, van Criekinge W, Thas O, Matthäi A, Cuesta MA, Terhaar Sive Droste JS, Craanen M, Schröck E, Ylstra B, Meijer GA. Multiple putative oncogenes at the chromosome 20q amplicon contribute to colorectal adenoma to carcinoma progression. Gut. 2009; 58:79-89.
9. Hodgson JG, Chin K, Collins C, Gray JW. Genome Amplification of Chromosome 20 in Breast Cancer. Breast Cancer Res. Treat. 2003; 78: 337-345.
10. Hao Z, Zhang H, Cowell J. Ubiquitin-conjugating enzyme UBE2C: molecular biology role in tumorigenesis and potential as a biomarker. Tumour Biol. 2012; 33: 723-30.
11. Aguirre-Portolés C, Bird AW, Hyman A, Cañamero M, Perez de Castro I, Malumbres M. Tpx2 controls spindle integrity genome stability and tumor development. Cancer Res. 2012; 72: 1518-28.
12. Chang H, Wang J, Tian Y, Xu J, Gou X, Cheng J. The TPX2 gene is a promising diagnostic and therapeutic target for cervical cancer. Oncol. Rep. 2012; 27: 1353-9.
13. Nadler Y, Camp RL, Schwartz C, Rimm DL, Kluger HM, Kluger Y. Expression of Aurora A (but not Aurora B) is predictive of survival in breast cancer. Clin. Cancer Res. 2008; 14: 4455-62.
14. Lok W, Klein RQ, Saif MW. Aurora kinase inhibitors as anti-cancer therapy. Anticancer Drugs. 2010; 21: 339-50.
15. Littlepage LE, Adler AS, Kouros-Mehr H, Huang G, Chou J, Krig SR, Griffith OL, Korkola JE, Qu K, Lawson DA, Xue Q, Sternlicht MD, Dijkgraaf GJ, Yaswen P, Rugo HS, Sweeney CA, Collins CC, Gray JW, Chang HY, Werb Z. The transcription factor ZNF217 is a prognostic biomarker and therapeutic target during breast cancer progression Cancer Discov. 2012; 2: 638-51.
16. Vendrell JA, Thollet A, Nguyen NT, Ghayad SE, Vinot S, Bièche I, Grisard E, Josserand V, Coll JL, Roux P, Corbo L, Treilleux I, Rimokh R, Cohen PA. ZNF217 Is a Marker of Poor Prognosis in Breast Cancer That Drives Epithelial-Mesenchymal Transition and Invasion. Cancer Res. 2012; 72: 3593-606.
17. Sauer G, Körner R, Hanisch A, Ries A, Nigg EA, Sillje HH. Proteome analysis of the human mitotic spindle. Mol. Cell Proteomics. 2005; 4: 35-43.
18. Santamaria A, Nagel S, Sillje HH, Nigg EA. The spindle protein CHICA mediates localization of the chromokinesin Kid to the mitotic spindle. Curr. Biol. 2008; 18: 723-9
19. Chang Q, Chen J, Beezhold KJ, Castranova V, Shi X, Chen F. JNK1 activation predicts the prognostic outcome of the human hepatocellular carcinoma. Mol. Cancer. 2009; 8: 64.
20. Ramakrishna M, Williams LH, Boyle SE, Bearfoot JL, Sridhar A, Speed TP, Gorringe KL, Campbell IG. Identification of candidate growth promoting genes in ovarian cancer through integrated copy number and expression analysis. PLoS One. 2010; 5: e9983.
21. Inamura K, Shimoji T, Ninomiya H, Hiramatsu M, Okui M, Satoh Y, Okumura S, Nakagawa K, Noda T, Fukayama M, Ishikawa Y. A metastatic signature in entire lung adenocarcinomas irrespective of morphological heterogeneity. Hum. Pathol. 2007; 38: 702-9.
22. Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division growth and differentiation. Nat. Rev. Cancer. 2008; 8: 83-93.
23. Welcker M, Orian A, Grim JE, Eisenman RN, Clurman BE. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr. Biol. 2004; 14: 1852-7.
24. Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG, Jr. The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell. 2005; 8: 25-33.
25. Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001; 294: 173-7.
26. Liu N, Li H, Li S, Shen M, Xiao N, Chen Y, Wang Y, Wang W, Wang R, Wang Q, Sun J, Wang P. The Fbw7/human CDC4 tumor suppressor targets proproliferative factor KLF5 for ubiquitination and degradation through multiple phosphodegron motifs. J. Biol. Chem. 2010; 285: 18858-67.
27. Oberg C, Li J, Pauley A, Wolf E, Gurney M, Lendahl U. The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel-10 homolog. J. Biol. Chem. 2001; 276: 35847-53.
28. Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, Balmain A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008; 321: 1499-502.
29. Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, Lapuk A, Neve RM, Qian Z, Ryder T, Chen F, Feiler H, Tokuyasu T, Kingsley C, Dairkee S, Meng Z, Chew K, Pinkel D, Jain A, Ljung BM, Esserman L, Albertson DG, Waldman FM, Gray JW. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006; 10: 529-41.
30. Climent J, Dimitrow P, Fridlyand J, Palacios J, Siebert R, Albertson DG, Gray JW, Pinkel D, Lluch A, Martinez-Climent JA. Deletion of chromosome 11q predicts response to anthracycline-based chemotherapy in early breast cancer Cancer Res. 2007; 67: 818-26.
31. Fridlyand J, Snijders AM, Ylstra B, Li H, Olshen A, Segraves R, Dairkee S, Tokuyasu T, Ljung BM, Jain AN, McLennan J, Ziegler J, Chin K, Devries S, Feiler H, Gray JW, Waldman F, Pinkel D, Albertson DG. Breast tumor copy number aberration phenotypes and genomic instability. BMC Cancer. 2006; 6: 96.
32. Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo WL, Stilwell JL, Pinkel D, Albertson DG, Waldman FM, McCormick F, Dickson RB, Johnson MD, Lippman M, Ethier S, Gazdar A, Gray JW. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006; 10: 515-27.
33. Fulp W, Rubio R, Wang T, Agrawal D, McCarthy SM, Gruidl M, Bloom G, Anderson T, White J, Quackenbush J, Yeatman T. Proliferative genes dominate malignancy-risk gene signature in histologically-normal breast tissue. Breast Cancer Res. Treat. 2010; 119: 335-46.
34. Brown M, Miron A, Liao X, Iglehart JD, Livingston DM, Ganesan S. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006; 9:121-32.
35. Sgroi DC. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res. 2009; 11: R7.
36. Pawitan Y, Bjohle J, Amler L, Borg AL, Egyhazi S, Hall P, Han X, Holmberg L, Huang F, Klaar S, Liu ET, Miller L, Nordgren H, Ploner A, Sandelin K, Shaw PM, Smeds J, Skoog L, Wedren S, Bergh J. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts Breast Cancer Res. 2005; 7: R953-64.
37. Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, Pawitan Y, Hall P, Klaar S, Liu ET, Bergh J. An expression signature for p53 status in human breast cancer predicts mutation status transcriptional effects and patient survival. Proc. Natl. Acad. Sci. U.S.A. 2005; 102: 13550-5.
38. Loi S, Haibe-Kains B, Desmedt C, Lallemand F, Tutt AM, Gillet C, Ellis P, Harris A, Bergh J, Foekens JA, Klijn JG, Larsimont D, Buyse M, Bontempi G, Delorenzi M, Piccart MJ, Sotiriou C. Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J. Clin. Oncol. 2007; 25: 1239-46.
39. Correlation of microarray-based breast cancer molecular subtypes and clinical outcomes: implications for treatment optimization. BMC Cancer. 2011; 11:143.
40. Wang Y, Liu Y, Lu J, Zhang P, Wang Y, Xu Y, Wang Z, Mao JH, Wei G. Rapamycin inhibits FBXW7 loss-induced epithelial-mesenchymal transition and cancer stem cell-like characteristics in colorectal cancer cells. Biochem. Biophys. Res. Commun. 2013; 434: 352-356.
41. Kalluri R and Weinberg RA. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009; 119: 1420-8.
42. Akhoondi S, Lindstrom L, Widschwendter M, Corcoran M, Bergh J, Spruck C, Grander D and Sangfelt O. Inactivation of FBXW7/hCDC4-beta expression by promoter hypermethylation is associated with favorable prognosis in primary breast cancer. Breast Cancer Res. 2010; 12: R105.
43. Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, Mueller-Holzner E, Corcoran M, Dagnell M, Nejad SZ, Nayer BN, Zali MR, Hansson J, Egyhazi S, Petersson F, Sangfelt P, Nordgren H, Grander D, Reed SI, Widschwendter M, Sangfelt O and Spruck C: FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res 2007, 67: 9006-12.