
INTRODUCTION
Breast cancer continues to be the most prevalent and frequently diagnosed malignancy in women worldwide [1]. In the past two decades, advances in early diagnostic and treatment strategies have decreased overall breast cancer mortality by more than 30% [2, 3]. However, once metastases have developed, the prognosis is poor [4–7].
Epidemiological and pre-clinical studies indicate an inverse correlation between serum concentrations of vitamin D and the development and progression of breast cancer [8–10]. Vitamin D deficiency in patients with early stage breast cancer is also associated with a poor prognosis [11].
The active metabolite of vitamin D, 1,25-dihydroxy-vitamin D3 (1,25D3) is a steroid hormone with pleiotropic effects on multiple tissues including bone, the immune system and cancer cells [12, 13]. The effects of vitamin D are mediated through binding of 1,25D3 to the vitamin D receptor (VDR). Upon ligand binding, the VDR forms a heterodimer complex with the retinoic acid receptor (RXR), which facilitates nuclear translocation and subsequent binding of the complex to specific vitamin D response elements (VDRE) within the promoters of target genes [14–17]. Well established ligand-mediated nuclear actions of the VDR include inhibition of cancer cell proliferation, induction of apoptosis, and inhibition of metastasis by regulating cell migration and invasion [15, 18–21].
Of note, VDR expression also negatively correlate with tumor progression: relatively high VDR concentrations are found in non-malignant hyperplastic tissue while progression to malignant growth is often associated with a reduction or complete loss of VDR expression [18, 22, 23]. Consistent with these reports, invasive breast cancers have been found to have relatively low VDR expression compared to normal mammary epithelia [22, 24]. Finally, VDR expression in breast cancer tissues may be inversely associated with the prognosis and survival of patients with breast cancer [25]. Collectively, these data suggest that vitamin D action through VDR in breast epithelial cells, which may act to prevent a switch toward tumorigenicity.
Previous studies in rodents have demonstrated that vitamin D deficiency promotes the growth of human breast cancer cells in bone [26, 27]. Using a vitamin D deficiency mouse model, these studies have shown that the acceleration in cancer cell growth was in part due to the effects of vitamin D deficiency on the bone microenvironment, as hypovitaminosis D induced a significant increase in osteoclast-mediated bone resorption due to secondary hyperparathyroidism [26, 28]. To further determine the interaction between tumor cells and vitamin D, bone resorption was inhibited through treatment with osteoprotegrin (OPG) in both vitamin D deficient and control mice. Interestingly, tumor burden in OPG-treated mice remained increased in vitamin D deficient mice relative to controls. It is thus likely that under these circumstances the absence of vitamin D had a direct effect on breast cancer growth [26, 27], which in turn poses the question as to whether vitamin D exerts direct inhibitory effects on cancer cell growth.
To determine the molecular mechanism of these direct effects, we disrupted vitamin D signaling in cancer cells and studied tumor growth in bone. Based on the known anti-proliferative function of vitamin D signaling, we hypothesized that silencing VDR expression in breast cancer cells would promote breast cancer growth. We therefore knocked down VDR expression in the human breast cancer cell line MCF-7 and followed up with clonal selection to generate highly efficient knockdown clones. In contrast to our initial hypothesis, we discovered that VDR knockdown inhibited cancer cell proliferation in the absence of vitamin D, suggesting a novel function of the VDR in promoting breast cancer cell growth.
RESULTS
Generation of stable VDR knockdown clones
Parental MCF-7 cells were transduced with either the shVDR or shNT construct, then continuously maintained with complete media containing puromycin and allowed to grow exponentially before being used for single cell clonal selection. Out of 30 NT clones, NT#13 expressed VDR mRNA and protein levels similar to PA (Parental MCF-7) cells (Figure 1A, 1C) and was therefore selected for all subsequent experiments.
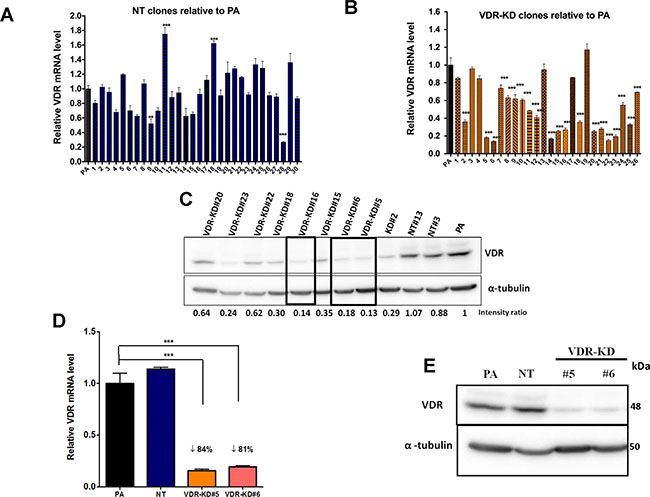
Figure 1: Stable knockdown of VDR in MCF-7 cells. MCF-7 cells were transduced with a lentivirus expressing either a non-target shRNA (NT) or shRNA against VDR (VDR-KD) and single cell clones were selected. (A–B) Level of expression of VDR mRNA was measured using quantitative RT-PCR in (A) MCF-7 NT clones and (B) MCF-7 VDR-KD clones and compared to parental MCF-7 cells (PA). (C) Level of VDR protein in cell lysates of PA, NT and VDR-KD clones was assessed using Western blotting. (D–E) MCF-7 PA cells and NT and VDR-KD clones were grown for 8 weeks in absence of antibiotic and VDR mRNA and protein levels were reassessed to ensure stable knockdown.
Results are expressed as the mean ± SEM (n = 3). ***p < 0.001 using one-way ANOVA with Tukey’s post-test.
Out of 27 VDR-KD clones screened, clones #5, 6 and 16 exhibited knockdown of both VDR mRNA and protein expression between 80–85% compared to PA cells and NT clones (Figure 1B, 1C). Clones were retested for stability of VDR knockdown after culture in the absence of puromycin for 8 weeks. After 8 weeks, out of 3 clones, VDR knockdown in clones #5 and #6 remained stable both at mRNA and protein levels and were used for further experiments (Figure 1D, 1E). The overall level of VDR gene knockdown among the different VDR-KD clones is ~50%, which may be due to variability within puromycin-resistant populations. The average of VDR mRNA levels of all VDR-KD clones was significantly reduced as compared to the average of VDR mRNA levels of all VDR NT clones (Mean ± SEM: 0.961 ± 0.0575 relative VDR mRNA in NT clones versus 0.515 ± 0.0553 relative VDR mRNA in VDR-KD clones, p < 0.001).
VDR knockdown abrogates vitamin D signaling in MCF-7 cells
Treatment with 10–8M 1,25D3 for 24 hours increased VDR mRNA and protein expression by NT cells, while the two MCF-7-VDR-KD clones showed only marginal responses to ligand exposure (Figure 2A, 2B). CYP24 is a direct VDR target gene [23, 29] and treatment with 1,25D3 induced a robust increase in CYP24 mRNA in NT cells (Figure 2C). In contrast, CYP24 mRNA induction was attenuated in VDR-KD#5 and VDR-KD#6 knockdown clones (Figure 2C), indicating effective disruption of VDR signaling in both clones.
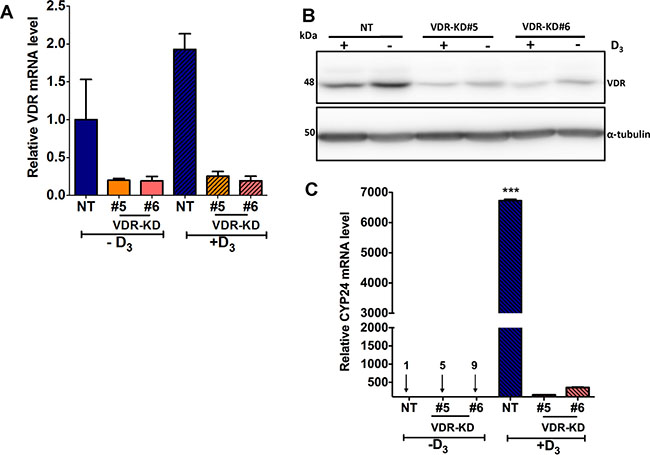
Figure 2: VDR knockdown abrogates vitamin D signaling in MCF-7 cells. (A) In response treatment with 1,25D3 for 24 hours, VDR mRNA was increased by 2-fold in NT cells as compared to vehicle treated cells. In contrast, VDR-KD clones showed marginal response to the 1,25D3 treatment. (B) After 48-hour treatment with 1,25D3, VDR protein levels were significantly increased in NT treated cells. The VDR-KD clones show marginally increased VDR protein levels following treatment. (C) After 24-hour treatment of NT cells, a significant induction of CYP24 mRNA was observed compared to vehicle treated cells. In contrast, CYP24 mRNA induction was attenuated in VDR-KD clones with 1,25D3 treatment.
Results are expressed as the mean ± SEM (n = 3). ***p < 0.001 NT(-D3) compared to NT (+D3) using one-way ANOVA with Tukey’s post test.
VDR knockdown reduces MCF-7 cell growth and induces apoptosis in ligand-free culture
To test the effect of 10–8M 1,25D3 on the in vitro growth of NT and VDR-KD cells, clonal and non-clonal lines was tested over 6 days, cells were cultured in charcoal-stripped media that lacks 1,25D3. The growth rate of MCF-VDR-KD cells prior to clonal selection, when the population was still heterogeneous, was first examined. Compared to vehicle, treatment with 1,25D3 significantly reduced the growth of NT cells but not that of VDR-KD cells, consistent with the disruption of VDR signaling. Surprisingly, however, the MCF7-VDR-KD non-clonal cells showed ligand-independent growth inhibitory and pro-apoptotic effects, when compared with MCF7-NT non-clonal cells (Figure 3A, 3B). These results were further confirmed using MCF-7-VDR-clonal lines. Similar to the non-clonal cell line, we found that in the absence of 1,25D3 the growth of both VDR-KD clones was significantly decreased compared to NT cells. Remarkably, growth of VDR-KD clones in absence of 1,25D3 was similar to that of NT cells in the presence of 1,25D3 (Figure 3C, 3D). Furthermore, treatment with 1,25D3 increased apoptosis in NT cells. In the absence of ligand, apoptosis in VDR-KD clones was significantly increased compared to NT cells. However, 1,25D3 treatment of VDR-KD clones did not increase apoptosis (Figure 3E). These data indicate that loss of the VDR decreases the growth of breast cancer cells and induces apoptosis independently of its ligand, 1,25D3.
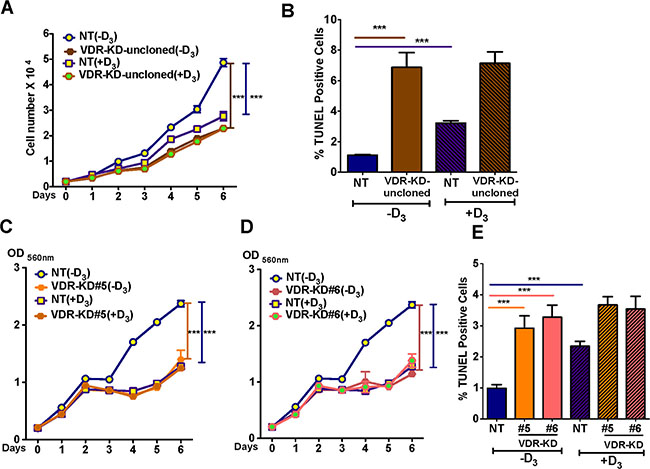
Figure 3: VDR knockdown reduces MCF-7 cell growth and induces apoptosis in a ligand-independent manner. (A) In ligand-free culture, MCF7-VDR-KD non-clonal cells showed reduced growth by 53% compared to NT cells. Treatment of NT cells with 1,25D3 reduced cell growth by 44% compared to untreated cells. (B) In ligand free culture, MCF7-VDR-KD uncloned cells showed 6.8-fold increased apoptosis compared to NT cells, as measured by TUNEL assay. In response to 1,25D3, NT uncloned cells showed 3-fold increased apoptosis as compared to untreated cells.(C) Similar to non-clonal cells, VDR-KD#5 showed 41% growth reduction compared to MCF7-NT cells. Treatment of MCF7-NT cells with 10–8 M 1,25D3 reduced cell growth by ~50% compared to untreated MCF7-NT cells. In contrast, the same treatment has no effect on growth of VDR-KD#5 cells. (D) Similarly, on day 6, VDR-KD#6 had a decreased growth by 51%, compared to NT in ligand-free culture. The magnitude of growth inhibition in VDR knockdown clones is comparable to the ligand mediated growth inhibition in control cell lines. Treatment of VDR-KD#6 cells with 1,25D3 does not reduce cell growth. (E) Both VDR-KD clones exhibited a 3-fold increased rate of apoptosis compared to NT cells in ligand-free culture. Upon 1,25D3 treatment, NT cell apoptosis increased 1.3-fold while VDR knockdown cells showed slightly (but insignificant) increased apoptosis compared to vehicle treated cells.
Results are expressed as the mean ± SEM. For growth assay (n = 6) ***p < 0.001 compared to NT (-D3) using 2-way ANOVA with Bonferroni post-test. For TUNEL using 1-way ANOVA with Tukey’s post-test.
VDR knockdown in MCF-7 cells reduces orthotopic tumor growth in vivo
To determine the effects of VDR knockdown in MCF-7 cells on orthotopic cell growth, NT and VDR-KD cells (clone #5) were implanted into the mammary fat pad of vitamin D-replete nude mice. As shown in Figure 4A, VDR knockdown significantly reduced tumor growth compared to MCF-7-NT cells. At endpoint (day 50 post implantation), tumor weight was 72% lower in tumors derived from MCF-7-VDR-KD cells compared to those grown from MCF-7-NT cells (Figure 4B).
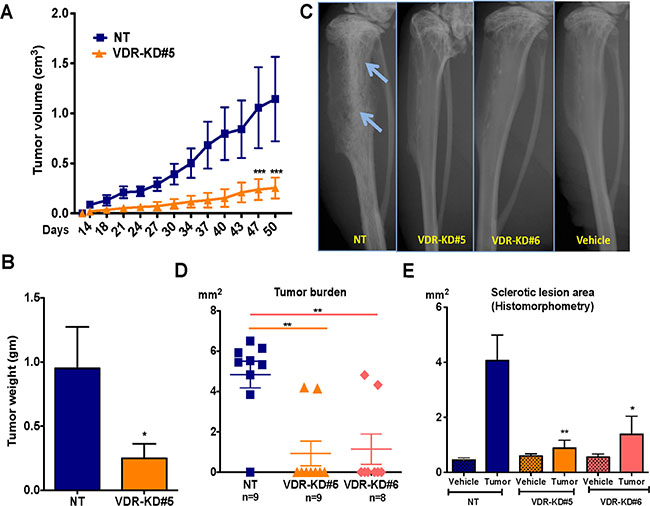
Figure 4: VDR knockdown in MCF-7 cells reduces orthotopic tumor growth and also reduces its capability to form tumor in the bone environments. (A) Orthotopic tumor growth: Tumors derived from VDR-KD#5 cells grew significantly slower than NT cell-derived tumors. (B) Orthotopic tumor weight: At the experiment endpoint (day 50), the tumor mass of mice that received the VDR-KD clone was reduced by 72% compared to MCF7-NT. (C) Representative radiographs: Representative images showing osteoblastic lesions as indicated by arrows in tibiae injected with MCF7-NT cells 21 weeks p.i. In contrast, no bone changes can be seen in bones inoculated with VDR-KD clonal cells. (D) Quantitative histomorphometry: Of the mice implanted with MCF7-NT, 88% developed tumors as confirmed by the presence of cancer cells in tibiae. In contrast, only 25% of mice implanted with VDR-KD cells developed tumors. (E) Bone histomorphometry: Sclerotic lesion area was significantly greater in MCF7-NT injected bones than in tibiae inoculated with VDR-KD cells.
Results are expressed as the mean ± SEM: Figure A and B (n = 5/group). ***p < 0.001 compared to MCF-N7 NT tumors, using 2-way ANOVA with Bonferroni post-test. Figure C to E (n = 8–9/group)., *p < 0.05 , **p < 0.01 compared to NT tumor using 1-way ANOVA with Tukey’s post-test.
VDR knockdown in MCF-7 cells impairs tumor formation in bone
To determine whether the bone microenvironment affects the growth of VDR-KD cells, NT and VDR-KD clones were inoculated into the proximal tibiae of vitamin D-replete nude mice. Osteosclerotic tumors derived from NT cells became radiographically visible 6 weeks post inoculation (p.i.), and continued to develop until study completion at 21 weeks p.i. In contrast, no obvious X-ray changes were identified in the two VDR-KD groups throughout the duration of the experiment (Figure 4C). Quantitative histomorphometry demonstrated that in the NT-inoculated group, 8 out of 9 mice formed tumors. In contrast, only 2 out of 9 mice inoculated with VDR-KD cells developed histologically identifiable tumors, while in the remaining 7 mice no tumor was present (Figure 4D). Tumor area was 4.7-fold lower in tibiae inoculated with VDR-KD#5 compared to NT cells, and similar differences were observed with VDR-KD#6 cells (Figure 4D). Trabecular bone volume (sclerotic lesion area assessed histomorphometrically) was increased in all groups compared to vehicle-inoculated tibiae. However, compared to NT controls, the amount of trabecular bone volume was significantly lower in tibiae inoculated with VDR-KD cells (Figure 4E).
MicroCT analysis revealed that trabecular bone volume of tibiae inoculated with NT cells was increased by 44-fold compared to vehicle-injected bones. In contrast, trabecular bone volume in tibiae inoculated with VDR-KD#5 and VDR-KD#6 cells increased by only 8 and 18-fold, respectively, compared to vehicle-injected controls, again significantly less than bones inoculated with NT cells (Figure 5A, 5B). Trabecular number (Tb.N) in the tibiae inoculated with NT was significantly higher than in contralateral tibiae as well as the VDR-KD inoculated tibiae (Figure 5C). Similarly, trabecular separation (Tb.Sp) in VDR-KD containing bones was 2-fold higher than in NT bearing bones (Figure 5D).
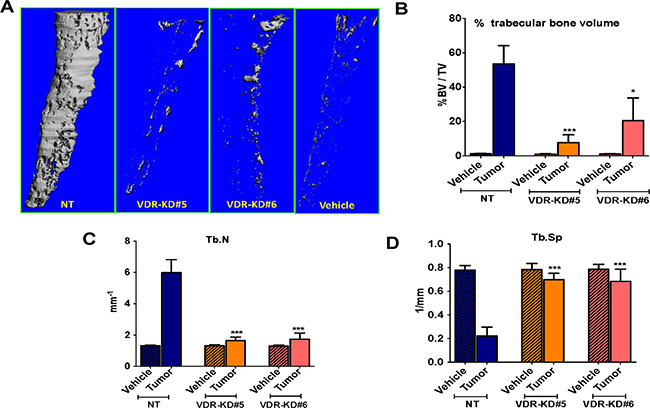
Figure 5: VDR knockdown in MCF-7 cells decreased osteosclerotic bone formation in bones of nude mice. Bone micro CT analysis was performed only for trabecular bone by excluding cortical bone (A) Bone microCT: Representative images showing tumor-mediated trabecular bone formation in tibiae injected with MCF7-NT cells 21 weeks p.i. In contrast, negligible amounts of trabecular bone formation seen in bones inoculated with VDR-KD clonal cells. (B–D) Quantitative analysis of micro-CT scans: Increased trabecular bone volume (BV/TV %) was observed in tibiae injected with NT cells compared to contralateral tibiae and VDR-KD inoculated tibiae. Trabecular number in the tibiae inoculated with NT cells was significantly higher compared to vehicle control. Tibiae injected with VDR knockdown cells showed significantly higher trabecular number compared to vehicle. Similarly, trabecular separation was decreased by 72% in tibiae with NT cells, compared to vehicle inoculated tibiae. Trabecular separation in VDR-KD inoculated tibia was reduced up to 13% compared to vehicle-inoculated tibiae.
Results are expressed as the mean ± SEM. *p < 0.05 ,***p < 0.001 compared to NT tumor using 1-way ANOVA with Tukey’s post-test.
Trabecular number in the tibiae inoculated with NT cells was significantly higher (+456%) compared to vehicle control. Tibiae injected with VDR knockdown cells showed an increase in trabecular number of up to 141% compared to vehicle (Figure 5C). Similarly, trabecular separation was decreased by 72% in tibiae with NT cells, compared to vehicle inoculated tibiae. Trabecular separation in VDR-KD inoculated tibia was reduced up to 13% compared to vehicle-inoculated tibiae (Figure 5D).
Collectively, these data indicate that VDR knockdown negatively affects the capability of MCF-7 cancer cells to form tumors and induce osteosclerosis in bone following direct intra-tibial inoculation in mice.
Stably expressed mutant VDR (mutVDR) accumulates in the cytoplasm
As shown above, knockdown of the VDR in MCF-7 breast cancer cells reduced growth in ligand-free conditions, suggesting that the VDR functions independently of 1,25D3 binding to promote cell growth in vitro. To further elucidate the function of the VDR in breast cancer cells and the underlying molecular events. Therefore, we next tested the hypothesis that the cytoplasmic VDR controls breast cancer cell growth.
To explore this hypothesis, we introduced a mutant VDR construct which, due to a mutation in the nuclear localization signal in the VDR gene, is unable to translocate to the nucleus and thus accumulates in the cytoplasm [30]. Importantly, the ligand-binding domain of the mutant VDR is not modified and thus is available to bind 1,25D3 [31]. As shown in Figure 6A, mRNA expression levels of the mutVDR (measured using specific primers that detect mutVDR expression only) were similar in mutVDR-NT and mutVDR-VDR-KD cells. To confirm that expression of the mutant VDR in VDR-KD cells did not affect the efficiency of knockdown of endogenous VDR, we determined the levels of endogenous VDR mRNA using specific primers. As shown in Figure 6B, VDR-KD cells expressing the mutant VDR continued to show a knockdown of the endogenous VDR, confirming that the efficiency of shRNA knockdown of VDR is not reduced with concurrent mutVDR expression in these cells. VDR Western blots demonstrated ~3-fold increase in VDR protein levels in the cytoplasmic fraction of mutVDR-VDR-KD cells compared to empty vector (EV)-NT cells (Figure 6C). Upon treatment with 1,25D3, VDR is normally detectable in the nuclear fraction, as is evident in EV-transfected cells. In contrast, mutVDR is much less responsive to 1,25D3–induced nuclear translocation (Figure 6D). Of note, complete abrogation of nuclear VDR translocation was not demonstrated with this mutation, which may have resulted in a marginal increase in nuclear VDR signals in mutVDR expressing VDR-KD cells.
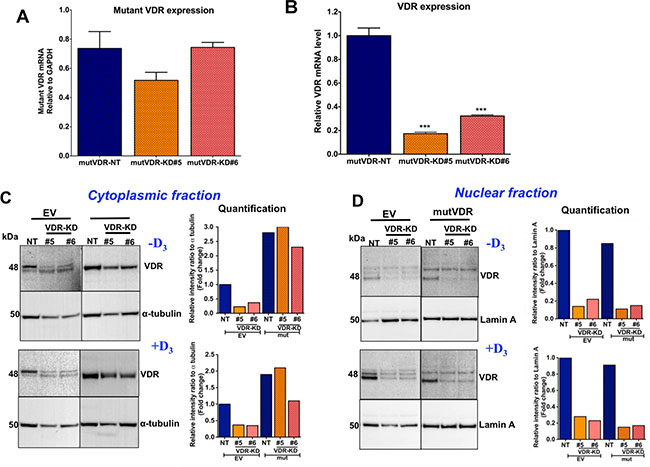
Figure 6: Stable expression of nuclear localization mutant VDR (mutVDR) in VDR knockdown clones. (A) After stable transfection of mutVDR into VDR knockdown cells the level of mutVDR mRNA in VDR knockdown clones was comparable to MCF7-NT cells using mutant VDR specific primers. (B) Expression of mutVDR in VDR-KD#5 and VDR-KD#6 does not change its shRNA knockdown effects. (C) Western blot of cytoplasmic fractions and its quantification. After stable transfection of the mutVDR and empty vector (EV) into VDR knockdown cells, Western blots demonstrated a ~3-fold increase in mutVDR protein in the cytoplasmic fraction of VDR-KD clones compared to EV-transfected NT cells in the absence of vitamin D. (D) Western blot of nuclear fractions and its quantification. Despite stable transfection of mutVDR and EV into VDR-KD cells, the mutVDR marginally translocated into the nucleus following treatment with 10–8M 1,25D3 for 48 hours. In contrast, the intact VDR considerably translocated to the nucleus in EV transfected control cells in response to treatment with 1,25D3.
Results are expressed as the mean ± SEM. ***p < 0.001 compared to mutVDR-NT using 1-way ANOVA with Tukey’s post-test.
Cytoplasmic VDR positively controls MCF-7 breast cancer cell growth in the absence of vitamin D
We next compared the growth of EV and mutVDR expressing cells in vitro in the presence and absence of 1,25D3. The growth of the EV-transfected cells (including EV-NT and EV-VDR-KD) was comparable to that of non-transfected cells. In the absence of ligand, the growth of mutVDR -transfected NT cells was similar to EV transfected NT cells (Figure 7A). As expected, the EV-VDR-KD clones demonstrated reduced growth in the absence of ligand compared to EV-NT cells. However, stable expression of the mutVDR in VDR-KD clones increased their growth rate to that of EV-NT cells (Figure 7A, 7B). These results indicate that the VDR is involved in the regulation of MCF-7 cell growth through its cytoplasmic accumulation. When mutVDR-VDR-KD cells were treated with 1,25D3, they were unresponsive to the treatment and there was no suppression of cell growth until day 5 (Figure 7C).
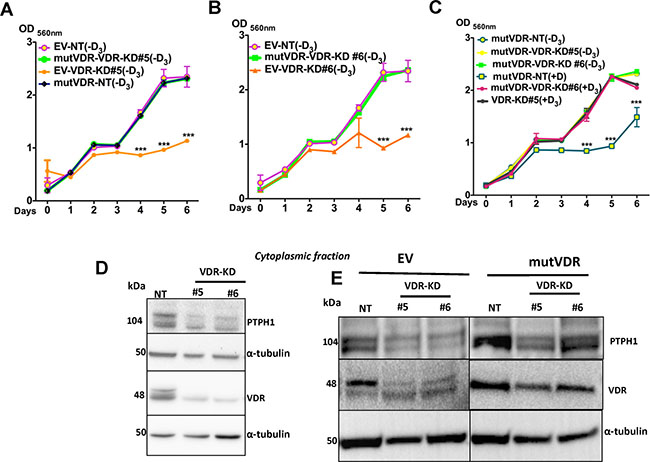
Figure 7: Expression of mutVDR into the VDR knockdown cells restores their growth. (A–B) Similar to VDR-KD cells, in ligand-free culture the EV-VDR-KD clones were slow in growth compared to EV-NT. Stable expression of the mutVDR in VDR-KD clones rescued the growth phenotype of VDR knockdown cells. mutVDR expression in both knockdown clones resulted in similar increases in growth rate. This was comparable to the growth rate of EV-NT cells. (C) When mutVDR -VDR-KD clones were treated with 1,25D3, they were unresponsive to treatment, thus cell growth remained the same until day 5. A slight reduction in growth was observed on day 6. (D) Western blots of the cytoplasmic fraction showed that compared to NT, PTPH1 protein levels were decreased by 50% in VDR-KD#5 and 40% in VDR-KD#6 (E) After mutVDR expression, PTPH1 protein was increased by 2 fold in cytoplasm of mutVDR -VDR-KD clones compared to their respective EV controls.
Results are expressed as the mean ± SEM (n = 6). ***p < 0.001 compared to EV-NT (−D3), mutVDR-VDR-KD#5 (-D3), mutVDR-VDR-KD#6 (-D3), mutVDR-NT(-D3), mutVDR-VDR-KD#5 (+D3), mutVDR-VDR-KD#6 (+D3) using 2-way ANOVA with Bonferroni post-test.
Protein tyrosine kinases and protein tyrosine phosphatases (PTPs) play a critical role in malignant transformation through their tightly regulated activity on tyrosine phosphorylation. Protein tyrosine phosphatase 1 (PTPH1) has been identified as a specific phosphatase for p38γ mitogen-activated protein kinase (MAPK) that shows oncogenic activity in cooperation with MAPK through direct binding. In human breast cancer cells, PTPH1 regulates cancer cell growth by its stimulatory effect on VDR via its binding to VDR in the cytoplasm [32, 33]. To confirm the ligand-independent cytoplasmic function of VDR, we measured PTPH1 protein levels in NT and VDR-KD cells. In ligand-free conditions, PTPH1 protein in cytoplasmic extracts was decreased by 50% in VDR-KD#5 and by 40% in VDR-KD#6 clones compared to NT cells (Figure 7D). When the mutVDR was expressed in VDR-KD cells (mutVDR-VDR-KD), PTPH1 protein levels were increased by 2-fold in mutVDR-VDR-KD clones relative to their respective EV controls (Figure 7E). These results support the hypothesis that cytoplasmic VDR promotes MCF-7 breast cancer cell growth possibly through its action on PTPH1.
DISCUSSION
By using a stable VDR knockdown approach in vitro and in vivo in mouse models, our study demonstrates that the cytoplasmic VDR plays an important role in breast cancer cell growth. This novel ligand-independent function of the VDR to promote cancer cell growth contrasts with its canonical ligand-dependent inhibitory nuclear actions on cell growth in the presence of vitamin D.
The MCF-7 breast cancer cell line expresses an abundance of endogenous VDR, which makes it difficult to achieve efficient VDR knockdown. Therefore, we performed single cell clonal selection to isolate two clones with marked reduction in VDR expression (~ −85%) using two controls, PA and NT. The knockdown of VDR efficiently blocked VDR-mediated signaling as shown by reduced expression of the VDR primary response gene CYP24 in the presence of vitamin D.
Consistent with previous reports [23, 26, 34], we showed that vitamin D treatment inhibited MCF-7 cell growth. Based on previous studies that vitamin D deficiency promotes breast cancer growth in bone [26, 27, 35] as well as the known anti-proliferative effects of vitamin D on breast cancer cells, we originally hypothesized that knockdown of the VDR in breast cancer cells would promote tumor cell growth. As expected, knockdown of the VDR in MCF-7 cells rendered the cells insensitive to vitamin D. However, in contrast to our hypothesis, we were surprised to find that VDR knockdown alone, in the absence of vitamin D, impaired cell growth. These results suggest that the VDR may have the hitherto unknown ability to stimulate cell growth independent of ligand binding. Experiments measuring the growth of two VDR-KD clones and VDR-KD uncloned cells using two different assays (MTT and direct cell count) provided the same results, indicating that the growth inhibitory phenotype of VDR-knockdown cells is not due to off-target effects or an artifact in the MCF-7-KD cells.
Our in vivo studies were performed in vitamin D-replete mice. Of note, the VDR-KD cells were consistently non-responsive to vitamin D treatment in the in vitro experiments, which provides evidence that the growth suppression seen in VDR-KD cells in vivo is independent of vitamin D. The bone microenvironment provides a fertile soil for breast cancer cells, and bone is a preferred target organ for breast cancer metastasis [7, 36, 37]. MCF-7 cells have previously been reported to form mixed osteolytic and osteoblastic bone lesions in which both osteoclasts and osteoblasts act to support tumor growth [36, 38, 39]. Our in vivo experiments indicate that ablation of the VDR in MCF-7 cells strongly reduces tumor formation within the bone environment, since about 87% of the mice failed to develop histologically visible tumors or tumor-related changes in the bone. In contrast, visible osteoblastic tumors were seen in 8 out of 9 tibiae inoculated with NT cells. The tumors seen in the two mice that did develop visible lesions following injection of VDR-KD clones were comparable in size and structure to those formed in mice injected with NT. These tibiae also showed a radiographic phenotype equivalent to that seen in the MCF7-NT group. One reason for the apparent impairment in tumor development with VDR-KD cells could be that the time required for inoculated cells to commence rapid proliferation and form tumours in VDR-KD cells is longer than that for the control cells. Alternatively, it could represent a reduced ability of VDR-KD cells to survive inoculation and introduction to a bone environment.
As the ligand-independent function of the VDR has not been widely studied, little information is available on the ligand-independent properties of the VDR. In skin cells, the unliganded VDR is required for keratinocyte stem cell function through interaction with the Wnt-β-catenin signaling pathway [40, 41]. In breast cancer, a single in vitro study has identified that PTPH1 positively regulates breast cancer growth by its stimulatory effect on VDR protein expression [32]. VDR localization and its specific effects are an emerging field of research and the findings we reported in this study contribute to our understanding of the phenotypic effects of cytoplasmic VDR on cancer cells. Furthermore, it has been reported that in the absence of its ligand, the VDR is distributed between the cytoplasm and the nucleus [30, 42]. VDR localization and its dependence on ligand binding are not absolute, though, as shown by us and others [24, 26, 27, 29], ligand binding is associated with enhanced shuttling of the VDR from the cytoplasm to the nucleus. However, recent evidence suggests that shuttling of the unliganded VDR to the nucleus produces basal transcriptional activity via binding to co-repressors or co-activators [43–47]. Global chromatin immunoprecipitation (ChIP) analyses have also indicated that a significant number of DNA-binding sites are occupied by the VDR in the absence of its cognate ligand [48]. However, the role of cytoplasmic VDR in the absence of ligand is not well studied. Our VDR knockdown results suggest a different role than expected and have led us to propose that the cytoplasmic VDR could be a factor contributing to the regulation of breast cancer cell proliferation. To investigate this further, we successfully created conditions that induced accumulation of VDR in the cell cytoplasm using stable expression of a mutant form of VDR.
Stable expression of mutVDR increased the growth of VDR knockdown cells to that of unmodified NT cells. This experiment demonstrated that in absence of its ligand, the cytoplasmic VDR promotes cell growth, a function that is in stark contrast to the inhibitory effects of the liganded VDR following nuclear translocation. Using Western immunoblotting, we have detected the presence of PTPH1 protein in cytosolic extracts of NT cells and its absence in VDR-KD cells. When the VDR was artificially accumulated in the cytoplasm of MCF7-KD cells using the mutant VDR, PTPH1 also increased corresponding to increased cytoplasmic VDR expression. These results support the concept that the cytoplasmic VDR controls MCF-7 cell growth. Altered PTPH1 levels with respect to VDR localization may be the responsible factor mediating the actions of unliganded cytoplasmic VDR but the detailed molecular interactions between their cytoplasmic association and cellular response remain to be elucidated.
Vitamin D deficiency is common in women with breast cancer at advanced disease stages [8–10]. However, the relationship between vitamin D status and clinical outcomes remain controversial. Numerous studies have linked low serum concentrations of 25-hydroxyvitamin D with increased cancer incidence and more aggressive tumor behavior, including increased bone metastasis and decreased survival [8, 10, 49]. However, most of these reports are epidemiological or clinical association studies, which cannot establish cause and effect, nor do they provide plausible mechanistic concepts. It is therefore not surprising that other reports are inconsistent with the relationship suggested by clinical and epidemiological studies [50, 51]. As the VDR is the central modulator of the autocrine/paracrine response to vitamin D, some of the inconsistencies in the above quoted clinical studies regarding the correlation between vitamin D status and breast cancer might be due to differences in VDR status [18, 52]. Our mostly observational study indicates that apart from vitamin D levels, VDR status and its compartmental distribution may contribute to regulating breast cancer cell growth. The use of a single model cell line is a limitation of this study and it is currently unknown if cytoplasmic VDR accumulation causes similar effects in other cancer cell lines. Detailed investigation into mechanisms behind ligand-independent cytoplasmic activity of VDR under conditions of vitamin D deficiency would have clinical applicability in deciding the treatment strategies in patients with breast cancer. Measuring nuclear and cytoplasmic VDR expression in human tumors at different stages of progression and correlating these results with the patient’s vitamin D status would help to clarify whether cytoplasmic accumulation of VDR might be relevant in regulating tumor growth in patients. A prospective clinical trial would be required to determine such a relationship, and this could help clarify the clinical discrepancies reported with regard to vitamin D and tumor outcomes [50].
In conclusion, this study demonstrates that cytoplasmic VDR can control MCF-7 breast cancer cell growth independent of vitamin D. Further investigations into the underlying mechanisms may deepen our understanding of the role of the VDR in cancer cell behavior and thus may open new therapeutic approaches for patients with metastatic breast cancer.
MATERIALS AND METHODS
Cell line
The human breast cancer cell line MCF-7 was selected for these studies as it expresses the VDR at relatively high levels and is responsive to 1,25D3 [24]. MCF-7 cells were obtained from Garvan’s Cancer Group which is now known as Kinghorn Cancer Centre. We have authenticated these cells as human by probing RNA with human specific probes for housekeeping genes and confirmed their expected phenotype of high estrogen dependence and high sensitivity to growth inhibition by 1,25D3 treatment, and by their induction of mixed osteogenic/osteoblastic lesions when implanted in tibiae. MCF-7 cells were maintained in RPMI medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin solution and 200 μg/mL insulin (Sigma, MO, USA) in a 37°C humidified atmosphere with 5% CO2. All the experiments used cells of ≤ 5 passages. Cells were routinely tested for the absence of mycoplasma contamination using mycoplasma GoTaq M5005 detection kit (Promega Corporation, WI, USA).
Stable shRNA knockdown and preparation of stable clones
Parental MCF-7 cells (PA) were transduced with a lentivirus encoding shRNA against the VDR (shVDR, TRCN000019506, MISSION shRNA, Sigma) (VDR-KD). Cells transduced with a non-target shRNA control vector (shNT, SHC002V, Sigma) were used as controls (NT). Two VDR-shRNA constructs were tested and the one with best knockdown efficiency was used for VDR knockdown and subsequent clonal selection. VDR-KD and NT cells were selected from heterogeneous pools using 2 μg/mL puromycin (Sigma). Clonal selection for both NT and VDR-KD was carried out using limiting dilution in the presence of puromycin in a 96-well plate format. Using this method, 30 NT and 27 VDR-KD clones were prepared, maintained and screened at mRNA and protein levels to identify clones with high VDR knockdown efficiency. From these, one NT and two VDR-KD (#5 and #6) clones were selected for further in vitro and in vivo experiments. The selection of two VDR knockdown clones reduced the possibility of clonal as well as off-target effects. Throughout this study both the knockdown clones showed a similar phenotype. Initially, we used both PA and NT as controls, however as they each showed a similar phenotype we continued all experiments using NT as a control since it had been generated in parallel with the VDR-KD clones.
Nuclear localization signal (NLS) mutant VDR (mutVDR) overexpression
The NT and VDR-KD MCF-7 cells were transfected with a mutated form of the VDR (mutVDR), which due to a mutation in the nuclear localization (NLS) signal is unable to translocate to the nucleus (kindly provided by Prof Chen, Medical College of Wisconsin, USA). The mutVDR was originally generated by Prufer et al. through point mutations of the K53Q, R54G and K55E basic amino acids in the NLS-I of VDR and cloned into a V5-tagged pcDNA3.1 vector [30]. A pcDNA3.1 empty vector (EV) was used as a control. MCF-7 cells (NT and VDR-KD clones) were plated in 6-well plates at 3 × 105 cells/well and transfected using LipofectamineTM (10 μg DNA per well) [53]. From the next day forward, cells were selected using the G418 antibiotic (1 mg/mL) for 3–5 days. After selection, mutVDR -expression was confirmed using real time-PCR and western blotting. For the purpose of this study, the cells resulting from this transfection will be referred to as mutVDR -NT, mutVDR -VDR-KD, EV-NT and EV-VDR-KD.
Quantitative RT-PCR
For gene expression studies, total RNA was isolated using GenElute RNA miniprep kits (Sigma) or Nucleospin RNA II kits (Macherey-Nagel GmbH & Co. KG) from the cells treated with 10–8M 1,25D3 (Biomole, ENZO life sciences, NY, USA) or its vehicle (ethanol) for the times indicated under serum-free conditions (0.1% BSA containing RPMI media supplemented with 1% penicillin streptomycin solution and 200μg/mL insulin). Total RNA (500 ng/sample) was reverse-transcribed using Superscript III (Life Technologies, CA, USA) after oligo(dT) primers (Promega Corp, WI, USA). Real-time PCR was performed using Hot start-IT SYBR green Q-PCR master mix (Affymetrix, Inc. USB products, CA, USA) with Bio-Rad CFX96 Thermal Cycler (Bio-Rad, CA, USA), or alternately, using iQSYBR-Green SuperMix (Bio-Rad) with Bio-Rad thermal cycler (Bio-Rad). Primers were optimized for RT-PCR target gene expression, which was normalized against the housekeeping gene, GAPDH. mRNA levels for gene expression of human GADPH, VDR, CYP24A1, and mutant VDR were assessed by real-time RT-PCR using species-specific primers. Primer sequences were: hGAPDH (Forward 5′-TATGACAACGAATTTGGCTACAG-3′; Reverse 5′-GATGGTACATGACAAGGTGC-3′); hVDR (Forward 5′-ACCTGGACAACAAGAGCGA-3′; Reverse 5′-CTCCTTCCTTCTCCTTCTGATG-3′); hCYP24A1 (Forward 5′-GCATCTTCCATTT GGCGT-3′; Reverse 5′-AATACCACCATCTGAGGCGT-3′); hmutantVDR (Forward 5′-CCACCTGCTCTATGCCAAG-3′; Reverse 5′-GAAAGGACAGTGGGAGTG-3′).
Cell fractionation and Western blot analysis
NT and VDR-KD cells or their mutVDR or EV transfected counterparts were treated with 10–8M 1,25D3 or vehicle (0.1% ethanol) in RPMI medium supplemented with 2% charcoal stripped, heat-inactivated FBS and 200 μg/mL insulin. In our hands, untreated FBS contains 1,25D3 at a concentration of 360pM when measured by specific RIA (Diasorin, USA). In contrast, 1,25D3 concentrations were undetectable in media supplemented with 2% charcoal stripped heat-inactivated FBS. Moreover, when MCF-7 cells were cultured in serum-free media or in media supplemented with 2% charcoal stripped heat-inactivated FBS, induction of CYP24 expression was not observed. We therefore considered culture media supplemented with 2% charcoal stripped heat-inactivated serum, 1% penicillin streptomycin solution and 200 μg/mL insulin to be free of 1,25D3 and refer to it as ligand-free media.
Media was changed every 24 hours. After 48 hours, whole cell lysates were prepared using RIPA buffer supplemented with protease inhibitor cocktail (Roche Pharmaceutical Co., Germany). Total protein concentration was determined using a Bradford assay (BioRad, CA, USA). Samples were loaded on 8% polyacrylamide gels and electrophoresis was performed using a Mini Trans-Blot cell (BioRad). Proteins were transferred onto a Hybond™-P membrane (GE Healthcare, WI, USA) using a Mini-PROTEAN Cell transfer system (Bio-Rad). Alternatively, cell fractions were prepared using NE-PER Nuclear and Cytoplasmic Extraction reagent kits (Thermo Scientific, IL, USA) followed by protein estimation using a BCA protein assay (Thermo Scientific, IL, USA). Samples were loaded on Bolt, 4–12% mini gels, separated and transferred using the Bolt electrophoresis system and reagents (Invitrogen, CA, USA).
Membranes were blocked in TBS-T-5% milk for 1 h, incubated overnight with the primary antibody and for 1 h with the secondary antibody. Antibody detection was performed using Immobilon™ Western Chemiluminescent HRP Substrate (Millipore, MA, USA) according to the manufacturer’s directions, and signal was detected either on Image Quant LAS4000 (GE healthcare) or BioRad Image gel system (BioRad). Primary antibodies included anti-VDR (Clone 9A7γ.E10.E4, Neo Markers, CA, USA), anti-PTPH1 (Santa Cruz Biotechnology, CA, USA), anti-α-tubulin (Sigma), anti-Lamin-A (AbCam, MA, USA). Secondary antibodies included anti-Rat (R&D system, MN, USA), anti-goat (Santa Cruz Biotechnology) Anti-mouse IgG, and anti-rabbit IgG (Sigma). A Western blot of VDR from cell extracts showed several bands, consistent with previously published studies using an anti-VDR antibody clone similar to that employed in our study [54–56].
Cell growth assay
Proliferation of NT and VDR-KD cells, or their mutVDR or EV transfected counterparts, was assessed either by MTT assay or using direct cell counting. Cells were seeded in 2% charcoal stripped FBS (see above) containing RPMI media and were treated with either 10–8M 1,25D3 or vehicle (0.1% ethanol) and media were replaced every 24hrs. For the MTT assay, MTT reagent (5mg/mL) (Calbiochem, CA, USA) was added to each well. After incubation for 4hrs at 37°C, cells were lysed using a PBS solution with SDS (10%) and HCl (0.01N), and the plate was incubated at 37°C for an additional 16 hrs. Absorbance was measured at 570 nm using an OPTIMA (BMG lab tech, VI, USA) plate reader. Alternatively, treated cells were counted daily by trypan blue exclusion after trypsinizing until they reached 100% confluence on day four. Experiments were repeated 3 times in independent settings to ensure validity of the results.
Cell apoptosis assay
NT and VDR-KD cells were cultured in medium containing 2% charcoal stripped FBS and treated with either 0.1% ethanol or 10−8M 1,25D3 for 6 days. Culture media was replaced every 24 hrs. Apoptosis was measured by TUNEL assay using the in situ cell death detection kit, POD (Roche Pharmaceutical Co., Germany). TUNEL-positive and total cells were counted in 3–5 random fields/well at 400× magnification.
Animal experiments
Four-week-old female athymic nude mice with body weight of 18–20 grams were purchased from Harlan (Indianapolis, IN, USA). Animal experiments were performed at Indiana University School of Medicine, IN, USA following approval by Indiana University Institutional Animal Care and Use Committee.
Mammary fat pad tumor model
Twenty 4-week-old female nude mice were randomized into 2 groups (n = 5/group). Both groups of mice were implanted with sustained release pellets containing 0.72 mg of 17ß estradiol (E2) (Innovative Research of America, FL, USA) subcutaneously on the lateral side of the neck, followed by subcutaneous implantation of NT and VDR-KD#5 cells [54]. Cells were suspended in cold Matrigel/PBS (1:1) at a concentration of 2 × 107 cells/mL. 100 μL of the suspension (2 × 106 cells) was implanted into the fourth mammary fat pad using a 27-gauge needle [26]. Starting from day 14, tumor dimensions were measured every three days using a precision calliper and tumor volume was calculated using the formula (length × width2)/2. Tumor weight was measured at the termination of the study (day 50).
Intratibial xenograft model
Fifty-two 4-week-old female nude mice were divided into three groups (n = 13/group): NT, VDR-KD#5, VDR-KD#6. Under general anaesthesia with ketamine/xylazine 75/10 mg/kg i.p., 2 × 105 cells in 20 μL of cell suspension (NT and VDR-KD clonal lines) were slowly injected through the knee joint into the tibial plateau of the left tibia using a Hamilton syringe [53, 57]. Contralateral tibiae were injected similarly with vehicle alone (PBS). As MCF-7 cells produce predominantly osteoblastic lesions, tumor growth was monitored every three weeks by digital radiography using a Kubtec Xpert80 (Kubtec digital X-ray, Milford, CT, USA).
Micro-computed tomography (Micro-CT)
MicroCT analyses were performed using a VivaCT 40 scanner (Scanco Medical, Bassersdorf, Switzerland). Mice were positioned on the microCT bed under continuous isoflurane anesthesia and oxygen. After defining the region of interest, the proximal tibia was scanned at high resolution with 10 μm voxel size and 200 ms integration time. For the trabecular bone analysis, trabecular contours were drawn beginning 300 μm below the growth plate and extending for 7.6 mm -9.5 mm. The same number of slices were analysed in both legs. Parameters measured included: trabecular bone volume per total volume (BV/TV, %), trabecular number (Tb.N, mm−1) and trabecular separation (Tb.Sp, mm). All values were compared with the contralateral vehicle injected tibia.
Bone histology and histomorphometry
Tibiae collected from 6–8 mice/group were used for paraffin embedding and 5–6 mice/group for plastic embedding. Tibiae from mice inoculated with tumor/sham intratibially were collected after euthanasia and fixed in 10% neutral buffered formalin for 48 h, followed by decalcification in 10% EDTA for 3 weeks. After embedding in paraffin, longitudinal mid-sagittal sections of 3.5 μm in thickness were cut and stained with haematoxylin & eosin (MUTVDR&E). Plastic embedded specimens were cut using an automated microtome (Microm HM 360, Thermo Scientific). Longitudinal mid-sagittal sections of 4.5μm in thickness were placed on slides coated with Haupt’s solution and incubated at 37ºC overnight. Sections were then stained with Von Kossa MacNeal’s Tetrachrome to distinguish newly formed osteoid from mineralized bone. Both paraffin and plastic embedded sections were visualized, captured and analysed at 12.5× to 400× magnification using an OsteoMeasure Image Analysis software v.13.2 System (Osteometrics, Atlanta, GA, USA) to determine total tissue area, tumor area and new woven bone formation in sclerotic lesions.
Statistical analysis
Data were analysed using GraphPad Prism version 5 (GraphPad, La Jolla, CA, USA). For simple comparison of two means, Student’s t test was performed. One-way ANOVA followed by Tukey’s post-test was used when comparing three groups or more. A 2-way ANOVA followed by Bonferroni post-test was used to compare groups affected by 2 variables. In vitro experiments were performed three times. Results shown are from a representative experiment. Data are presented as mean ± SEM. A p-value < 0.05 was considered significant.
ACKNOWLEDGMENTS
We thank Ms. Xiang H. Peng for help in histology and Ms. Maria Niewolna for her assistance in tissue culture. We also thank Dr. Jenna N. Regan for her help in manuscript review and further thanks to Ms. Colette Fong-Yee and Ms. Shu-Oi Chow for their technical help. We also thank Dr. Chen, Med. College of Wisconsin, USA, for kindly providing the mutVDR construct.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
GRANT SUPPORT
This work has been supported in part by the following grants: University of Sydney Cancer Research Fund (M.J.S), Cure Cancer Foundation of Australia (Y.Z), Cancer Council New South Wales (M.J.S, Y.Z, H.Z, C.R.D), Cancer Institute NSW CDF Fellowship (Y.Z), National Health and Medical Research Council, Australia (NHMRC) (Y.Z, Early Career Fellowship 596870), University of Sydney Postgraduate Award (T.T).
The work undertaken at Indiana University has been supported by grants to T.A.G. (U01CA143057 from the NCI tumor Microenvironment Network; R01CA69158), the Susan G. Komen Foundation, the Indiana Economic Development Grant, the Jerry and Peggy Throgmartin Endowment of the IU Simon Cancer Center, IU Simon Cancer Center Breast Cancer Program and a generous donation from the Withycombe family.
REFERENCES
1. Bray F, Ren JS, Masuyer E, Ferlay J. Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int J Cancer. 2013; 132:1133–1145.
2. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013; 63:11–30.
3. Yip CH, Smith RA, Anderson BO, Miller AB, Thomas DB, Ang ES, Caffarella RS, Corbex M, Kreps GL, McTiernan A. Guideline implementation for breast healthcare in low- and middle-income countries: early detection resource allocation. Cancer. 2008; 113:2244–2256.
4. Coleman RE, Rubens RD. The clinical course of bone metastases from breast cancer. Br J Cancer. 1987; 55:61–66.
5. Smith HS, Barkin RL. Painful Boney Metastases. Am J Ther. 2014; 21:106–30.
6. Guise TA, Brufsky A, Coleman RE. Understanding and optimizing bone health in breast cancer. Curr Med Res Opin. 2010; 26:3–20.
7. Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002; 2:584–593.
8. Bilinski K, Boyages J. Association between 25-hydroxyvitamin D concentration and breast cancer risk in an Australian population: an observational case-control study. Breast Cancer Res Treat. 2013; 137:599–607.
9. Mohr SB, Gorham ED, Alcaraz JE, Kane CJ, Macera CA, Parsons JK, Wingard DL, Garland CF. Serum 25-hydroxyvitamin D and prevention of breast cancer: pooled analysis. Anticancer Res. 2011; 31:2939–2948.
10. Peppone LJ, Rickles AS, Janelsins MC, Insalaco MR, Skinner KA. The association between breast cancer prognostic indicators and serum 25-OH vitamin D levels. Ann Surg Oncol. 2012; 19:2590–2599.
11. Goodwin PJ, Ennis M, Pritchard KI, Koo J, Hood N. Prognostic effects of 25-hydroxyvitamin D levels in early breast cancer. J Clin Oncol. 2009; 27:3757–3763.
12. Lin R, White JH. The pleiotropic actions of vitamin D. Bioessays. 2004; 26:21–28.
13. Verstuyf A, Carmeliet G, Bouillon R, Mathieu C. Vitamin D: a pleiotropic hormone. Kidney Int. 2010; 78:140–145.
14. Carlberg C, Campbell MJ. Vitamin D receptor signaling mechanisms: integrated actions of a well-defined transcription factor. Steroids. 2013; 78:127–136.
15. Deeb KK, Trump DL, Johnson CS. Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer. 2007; 7:684–700.
16. Carlberg C, Seuter S. A genomic perspective on vitamin D signaling. Anticancer Res. 2009; 29:3485–3493.
17. Christakos S, Dhawan P, Benn B, Porta A, Hediger M, Oh GT, Jeung EB, Zhong Y, Ajibade D, Dhawan K, Joshi S. Vitamin D: molecular mechanism of action. Ann N Y Acad Sci. 2007; 1116:340–348.
18. Townsend K, Banwell CM, Guy M, Colston KW, Mansi JL, Stewart PM, Campbell MJ, Hewison M. Autocrine metabolism of vitamin D in normal and malignant breast tissue. Clin Cancer Res. 2005; 11:3579–3586.
19. Wang Q, Lee D, Sysounthone V, Chandraratna RAS, Christakos S, Korah R, Wieder R. 1,25-dihydroxyvitamin D3 and retonic acid analogues induce differentiation in breast cancer cells with function- and cell-specific additive effects. Breast Cancer Res Treat. 2001; 67:157–168.
20. Krishnan AV, Feldman D. Mechanisms of the anti-cancer and anti-inflammatory actions of vitamin D. Annu Rev Pharmacol Toxicol. 2011; 51:311–336.
21. Jensen SS, Madsen MW, Lukas J, Binderup L, Bartek J. Inhibitory effects of 1alpha,25-dihydroxyvitamin D(3) on the G(1)-S phase-controlling machinery. Mol Endocrinol. 2001; 15:1370–1380.
22. Lopes N, Sousa B, Martins D, Gomes M, Vieira D, Veronese LA, Milanezi F, Paredes J, Costa JL, Schmitt F. Alterations in Vitamin D signalling and metabolic pathways in breast cancer progression: a study of VDR, CYP27B1 and CYP24A1 expression in benign and malignant breast lesions. BMC Cancer. 2010; 10:483.
23. Anderson MG, Nakane M, Ruan X, Kroeger PE, Wu-Wong JR. Expression of VDR and CYP24A1 mRNA in human tumors. Cancer Chemother Pharmacol. 2006; 57:234–240.
24. Buras RR, Schumaker LM, Davoodi F, Brenner RV, Shabahang M, Nauta RJ, Evans SR. Vitamin D receptors in breast cancer cells. Breast Cancer Res Treat. 1994; 31:191–202.
25. Ditsch N, Toth B, Mayr D, Lenhard M, Gallwas J, Weissenbacher T, Dannecker C, Friese K, Jeschke U. The association between vitamin D receptor expression and prolonged overall survival in breast cancer. J Histochem Cytochem. 2012; 60:121–129.
26. Ooi LL, Zhou H, Kalak R, Zheng Y, Conigrave AD, Seibel MJ, Dunstan CR. Vitamin D deficiency promotes human breast cancer growth in a murine model of bone metastasis. Cancer research. 2010; 70:1835–1844.
27. Ooi LL, Zheng Y, Zhou H, Trivedi T, Conigrave AD, Seibel MJ, Dunstan CR. Vitamin D deficiency promotes growth of MCF-7 human breast cancer in a rodent model of osteosclerotic bone metastasis. Bone. 2010; 47:795–803.
28. Zheng Y, Zhou H, Ooi LL, Snir AD, Dunstan CR, Seibel MJ. Vitamin D deficiency promotes prostate cancer growth in bone. Prostate. 2011; 71:1012–1021.
29. Vaisanen S, Dunlop TW, Sinkkonen L, Frank C, Carlberg C. Spatio-temporal activation of chromatin on the human CYP24 gene promoter in the presence of 1alpha,25-Dihydroxyvitamin D3. J Mol Biol. 2005; 350:65–77.
30. Prufer K, Racz A, Lin GC, Barsony J. Dimerization with retinoid X receptors promotes nuclear localization and subnuclear targeting of vitamin D receptors. The Journal of biological chemistry. 2000; 275:41114–41123.
31. Hsieh JC, Shimizu Y, Minoshima S, Shimizu N, Haussler CA, Jurutka PW, Haussler MR. Novel nuclear localization signal between the two DNA-binding zinc fingers in the human vitamin D receptor. J Cell Biochem. 1998; 70:94–109.
32. Zhi HY, Hou SW, Li RS, Basir Z, Xiang Q, Szabo A, Chen G. PTPH1 cooperates with vitamin D receptor to stimulate breast cancer growth through their mutual stabilization. Oncogene. 2011; 30:1706–1715.
33. Hou SW, Zhi HY, Pohl N, Loesch M, Qi XM, Li RS, Basir Z, Chen G. PTPH1 dephosphorylates and cooperates with p38gamma MAPK to increase ras oncogenesis through PDZ-mediated interaction. Cancer research. 2010; 70:2901–2910.
34. Zinser GM, McEleney K, Welsh J. Characterization of mammary tumor cell lines from wild type and vitamin D3 receptor knockout mice. Mol Cell Endocrinol. 2003; 200:67–80.
35. Alimirah F, Vaishnav A, McCormick M, Echchgadda I, Chatterjee B, Mehta RG, Peng X. Functionality of unliganded VDR in breast cancer cells: repressive action on CYP24 basal transcription. Molecular and cellular biochemistry. 2010; 342:143–150.
36. Guise TA, Mohammad KS, Clines G, Stebbins EG, Wong DH, Higgins LS, Vessella R, Corey E, Padalecki S, Suva L, Chirgwin JM. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin Cancer Res. 2006; 12:6213s-6216s.
37. Waning DL, Mohammad KS, Reiken S, Xie W, Andersson DC, John S, Chiechi A, Wright LE, Umanskaya A, Niewolna M, Trivedi T, Charkhzarrin S, Khatiwada P, et al. Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nature medicine. 2015; 21:1262–1271.
38. Zheng Y, Zhou H, Fong-Yee C, Modzelewski JR, Seibel MJ, Dunstan CR. Bone resorption increases tumour growth in a mouse model of osteosclerotic breast cancer metastasis. Clin Exp Metastasis. 2008; 25:559–567.
39. Yoneda T, Michigami T, Yi B, Williams PJ, Niewolna M, Hiraga T. Actions of bisphosphonate on bone metastasis in animal models of breast carcinoma. Cancer. 2000; 88:2979–2988.
40. Cianferotti L, Cox M, Skorija K, Demay MB. Vitamin D receptor is essential for normal keratinocyte stem cell function. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104:9428–9433.
41. Beildeck ME, Gelmann EP, Byers SW. Cross-regulation of signaling pathways: An example of nuclear hormone receptors and the canonical Wnt pathway. Exp Cell Res. 2010; 316:1763–1772.
42. Huet T, Laverny G, Ciesielski F, Molnar F, Ramamoorthy TG, Belorusova AY, Antony P, Potier N, Metzger D, Moras D, Rochel N. A vitamin D receptor selectively activated by gemini analogs reveals ligand dependent and independent effects. Cell reports. 2015; 10:516–526.
43. Sanchez-Martinez R, Zambrano A, Castillo AI, Aranda A. Vitamin D-dependent recruitment of corepressors to vitamin D/retinoid X receptor heterodimers. Mol Cell Biol. 2008; 28:3817–3829.
44. Tagami T, Lutz WH, Kumar R, Jameson JL. The interaction of the vitamin D receptor with nuclear receptor corepressors and coactivators. Biochem Biophys Res Commun. 1998; 253:358–363.
45. Dwivedi PP, Muscat GE, Bailey PJ, Omdahl JL, May BK. Repression of basal transcription by vitamin D receptor: evidence for interaction of unliganded vitamin D receptor with two receptor interaction domains in RIP13delta1. J Mol Endocrinol. 1998; 20:327–335.
46. Gardiner EM, Esteban LM, Fong C, Allison SJ, Flanagan JL, Kouzmenko AP, Eisman JA. Vitamin D receptor B1 and exon 1d: functional and evolutionary analysis. J Steroid Biochem Mol Biol. 2004; 89–90:233–238.
47. Prufer K, Barsony J. Retinoid X receptor dominates the nuclear import and export of the unliganded vitamin D receptor. Mol Endocrinol. 2002; 16:1738–1751.
48. Pike JW, Meyer MB, Benkusky NA, Lee SM, St John H, Carlson A, Onal M, Shamsuzzaman S. Genomic Determinants of Vitamin D-Regulated Gene Expression. Vitam Horm. 2016; 100:21–44.
49. Garland CF, Gorham ED, Mohr SB, Garland FC. Vitamin D for cancer prevention: global perspective. Ann Epidemiol. 2009; 19:468–483.
50. Chlebowski RT. Vitamin d and breast cancer incidence and outcome. Anti-cancer agents in medicinal chemistry. 2013; 13:98–106.
51. Rohan TE, Negassa A, Chlebowski RT, Ceria-Ulep CD, Cochrane BB, Lane DS, Ginsberg M, Wassertheil-Smoller S, Page DL. A randomized controlled trial of calcium plus vitamin D supplementation and risk of benign proliferative breast disease. Breast Cancer Res Treat. 2009; 116:339–350.
52. Brunner RL, Wactawski-Wende J, Caan BJ, Cochrane BB, Chlebowski RT, Gass ML, Jacobs ET, LaCroix AZ, Lane D, Larson J, Margolis KL, Millen AE, Sarto GE, et al. The effect of calcium plus vitamin D on risk for invasive cancer: results of the Women’s Health Initiative (WHI) calcium plus vitamin D randomized clinical trial. Nutr Cancer. 2011; 63:827–841.
53. Zheng Y, Chow SO, Boernert K, Basel D, Mikuscheva A, Kim S, Fong-Yee C, Trivedi T, Buttgereit F, Sutherland RL, Dunstan CR, Zhou H, Seibel MJ. Direct crosstalk between cancer and osteoblast lineage cells fuels metastatic growth in bone via auto-amplification of IL-6 and RANKL signaling pathways. Journal of bone and mineral research. 2014; 29:1938–1949.
54. Dall G, Vieusseux J, Unsworth A, Anderson R, Britt K. Low Dose, Low Cost Estradiol Pellets Can Support MCF-7 Tumour Growth in Nude Mice without Bladder Symptoms. J Cancer. 2015; 6:1331–1336.
55. Lange TS, Zou Y, Singh RK, Kim KK, Kristjansdottir K, Sholler GL, Brard L. Chemotherapeutic effect of calcidiol derivative B3CD in a neuroblastoma xenograft model. Chem Biol Drug Des. 2010; 76:164–173.
56. Narvaez CJ, Byrne BM, Romu S, Valrance M, Welsh J. Induction of apoptosis by 1,25-dihydroxyvitamin D3 in MCF-7 Vitamin D3-resistant variant can be sensitized by TPA. J Steroid Biochem Mol Biol. 2003; 84:199–209.
57. Zheng Y, Zhou H, Modzelewski JR, Kalak R, Blair JM, Seibel MJ, Dunstan CR. Accelerated bone resorption, due to dietary calcium deficiency, promotes breast cancer tumor growth in bone. Cancer research. 2007; 67:9542–9548.