
INTRODUCTION
Pediatric Burkitt lymphoma (BL) is the most common histological subtype of non-Hodgkin lymphoma (NHL) in children and adolescents [1, 2]. We and others have demonstrated that the prognosis of pediatric BL has remarkably improved over the past 40 years following the introduction of short intensive multi-agent chemotherapy [1–4]. However, this successful approach is limited by significant chemotherapy-induced acute toxicity, suggesting the need to identify less toxic and more targeted therapy [1, 2]. We investigated the addition of the anti-CD20 monoclonal antibody, rituximab, to the FAB (French American British) chemotherapy regimen in children and adolescents with intermediate-risk and advanced BL [5, 6]. Immunotherapy, by targeting CD20 by rituximab, has been demonstrated to alter a variety of signal transduction pathways, including NF-κB, MAPK/ERK, JAK/STAT, PI3K/AKT, and B-cell receptor pathways. These alterations lead to cell death and/or sensitization of cells to the effects of cytotoxic chemotherapeutic agents [7–9]. Furthermore, children and adolescents with BL who relapse or progress after initial chemoimmunotherapy have a dismal prognosis, suggesting the development of chemoimmunotherapy resistant disease [1, 2]. Novel strategies will be required for the reduction of both acute morbidities and prolonged hospital stays secondary to intensive multi-agent chemotherapy in newly diagnosed patients with BL and/or circumvention of chemoimmunotherapy resistance in patients with relapsed/refractory disease.
We have previously identified secondary chromosomal aberrations in 70% of pediatric BL patients with a C-MYC gene rearrangement [10]. Specifically, we identified a significantly inferior event free and overall survival (EFS and OS) in children and adolescents with a specific loss of the 13q14.3 locus [10, 11]. In multivariate analysis controlling for stage, lactate dehydrogenase (LDH) levels, country of treatment, and group classification, children and adolescents with BL who had a 13q deletion had significantly poorer EFS compared to patients treated with the same French American British (FAB) chemotherapy regimen [10].
We compared the molecular signature and gene expression profile of pediatric BL patients treated within the Children’s Oncology Group, National Cancer Institute, and Berlin-Frankfurt-Munster (BFM) - pediatric NHL Group [12] and found consistency in the pediatric BL molecular signature [12–15]. Interestingly, Dave et al. suggested that DLEU1 gene located at chromosome 13q14.3 region [16, 17] is significantly amplified in adult BL vs. diffuse large B-cell lymphoma (DLBCL), and is one of the BL molecular classifying genes and a target of the oncogene, C-MYC [14]. Deletion of DLEU1, therefore, may in part form the molecular basis for the significantly inferior EFS in children with BL and a concomitant 13q14.3 deletion when treated with FAB chemotherapy [10].
DLEU1 has been found frequently deleted and a potential tumor suppressor gene in hematopoietic tumors including chronic lymphocytic leukemia (CLL) and mantle cell lymphoma [16, 17]. An open reading frame corresponding to a 78 amino acids sequence has been identified in DLEU1 gene by human transcriptome, functional genomics and proteomic analysis [18, 19]. DLEU1 protein has been predicted to interact with several cancer-related proteins, including c-Myc, Tubulin beta-2C chain (TUBB2C), E3 ubiquitin-protein ligase (UBR1), cellular tumor antigen p53, and Ras association (RalGDS/AF-6) domain family member 1 (RASSF1) [18]. Interestingly, TUBB2C and RASSF1 are frequently silenced in human cancers and enhance apoptosis and tumor suppression [20, 21]. UBR1 affects the cell cycle via PI3K-AKT signaling and loss of UBR1 accelerates B-cell lymphomagenesis [22]. We have observed that the expression levels of RASSF1, TUBB2C and UBR1 were significantly higher in BL compared to DLBCL cell lines [23]. These data, taken together, suggest that DLEU1 may function as a tumor suppressor in c-Myc activated BL by repressing cell cycle progression and enhancing programmed cell death via protein-protein interaction. In the current study, we set out to investigate the hypothesis that the deletion of DLEU1 in BL may affect the expression of DLEU1 network genes and alter signal transduction pathways leading to the inhibition of programmed cell death and in part be responsible for the mechanism of resistance to chemoimmunotherapy in patients with BL with a 13q14.3 deletion.
RESULTS
Generation of TALEN mediated DLEU1 knockdown BL cell line
Three pairs of TALENs (TALEN1, TALEN2, and TALEN3) targeting the endogenous DLEU1 gene were constructed based on modified restriction enzyme and ligation (REAL) assembly methods for DLEU1 gene modification (Figure 1A). To excise the entire DLEU1 locus, TALEN1 and TALEN3 (T13), and TALEN2 and TALEN3 (T23) were transfected into Raji cells. Single DLEU1 knockdown Raji cell clones were screened for the desired 23 Kb deletion which was confirmed by PCR and sequencing analysis (Figure 1B). To ensure the purity of a single clone, one of the positive single clones (T13-2) was re-plated and its daughter cells, four clones T13-2-2, T13-2-4, T13-2-14 and T13-2-16 were re-screened as above. Quantitative RT-PCR showed significant reduction in expression of DLEU1 compared to WT, with reductions of 75% (p < 0.01), 80% (p < 0.05), 83% (p < 0.01), and 77% (p < 0.01) in clones T13-2-2, T13-2-4, T13-2-14, and T13-2-16, respectively (Figure 1C). Since clone T13-2-14, hereafter referred to as “DLEU1 knockdown (DLEU1-KD)”, showed the highest reduction of DLEU1 mRNA, we used this clone for all further experimentation in this study. The DLEU1-KD clones had no significant reduction in DLEU2 mRNA (data not shown).
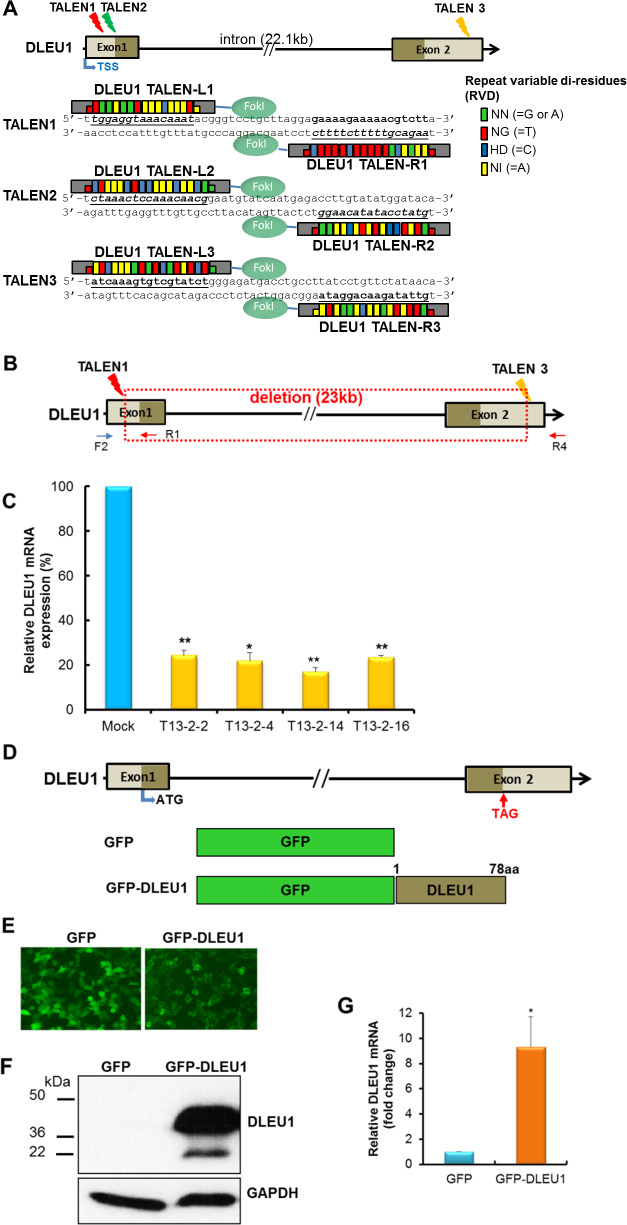
Figure 1: TALENs-induced DLEU1 knockdown and DLEU1 stably overexpressing Raji cell line. (A) A diagram showing the targeting of the DLEU1 locus by three different TALENs and TALE repeat arrays are shown with the repeat-variable di-residue (RVD) underlined and represented by small colored rectangles boxes. Two monomeric TALENs (L and R) are required to bind the DLEU1 target site to cleave DNA by fused FokI. TSS, transcription start site. (B) Single clones with TALEN1 and 3-induced DLEU1 locus disruption (22,843bp deletion, boxed with red dot line) were isolated. (C) Comparison of expression of DLEU1 mRNA between WT control (WT) and DLEU1 knockdown single clones T13-2-2, T13-2-4, T13-2-14 and T13-2-16 by qRT-PCR. Data are presented as mean ± SD (n = 3, paired t test) and p-value are displayed on plot. (D) Diagram of DLEU1 gene and ORF (78 a.a). Empty GFP vector (GFP) and GFP fused DLEU1 (GFP-DLEU1) constructs are shown. ATG, start codon; TAG, stop codon. (E) GFP and GFP-DLEU1 fusion plasmids were stably transfected into Raji cells, and then western blotting (F) and qRT-PCR (G) were performed. Data are presented mean ± SD of triplicates (paired t test). *p < 0.005.
Establishment of DLEU1 stably overexpressing BL cell line
DLEU1 full-length cDNA was cloned into pEGFP-N3 expression vector and GFP-DLEU1 plasmid was transfected into HEK293 cells to confirm expression of the fusion protein under the fluorescent microscope (Figure 1D and 1E). GFP-DLEU1 construct was then stably transfected into Raji cells. The expression of DLEU1 at mRNA level was detected by RT-PCR (Figure 1F), and the predicted size of the fusion protein (approximately 36 kDa) was confirmed by western blotting analysis (Figure 1G) whereas endogenous DLEU1 protein was not detectable in GFP control.
DLEU1 expression levels have significant effects on BL cell proliferation and programmed cell death
To examine whether DLEU1 gene affects cell proliferation and programmed cell death in BL, Raji cells with or without DLEU1 knocked out (DLEU1-KD or WT), were plated into 48-well plates and evaluated every 24 hours for cell proliferation by MTS assay and programmed cell death by Caspase 3/7 assay. DLEU1-KD cells showed a significant increase in cell proliferation (20% at 24 hrs, p < 0.05; 25% at 48 hrs, p < 0.05) (Figure 2A), and a significant reduction in caspase 3/7 activity (25% at 24 hrs, p < 0.01; 33% at 48 hrs, p < 0.05) (Figure 2B), when compared to those of WT cells. Conversely, when we overexpressed GFP or GFP-DLEU1 in Raji cells, GFP-DLEU1 cells showed a significant decrease in cell proliferation (15% reduction, p < 0.03; 19% reduction, p < 0.03; and 15% reduction, p < 0.01 at 24, 48 and 72 hours, respectively) (Figure 2C) and a significant increase in programmed cell death (10.2% increase, p < 0.02; 9.7% increase, p < 0.05 at 24 and 48 hours, respectively) (Figure 2D) compared to GFP control cells. We further analyzed the expression of anti- and pro-apoptotic genes in these cells, and found that DLEU1 knockdown resulted in a significant increase in expression of the anti-apoptotic genes (Bcl-2, Bcl-xL and Mcl-1 ), and decrease in that of the pro-apoptotic gene (Bax) at mRNA level (Figure 2E). Consistently, GFP-DLEU1 expression induced a significant decrease in Mcl-1 and increase in Bax expression (Figure 2F). We observed similar changes in the expression of these genes at protein levels (Figure 2G and 2H).
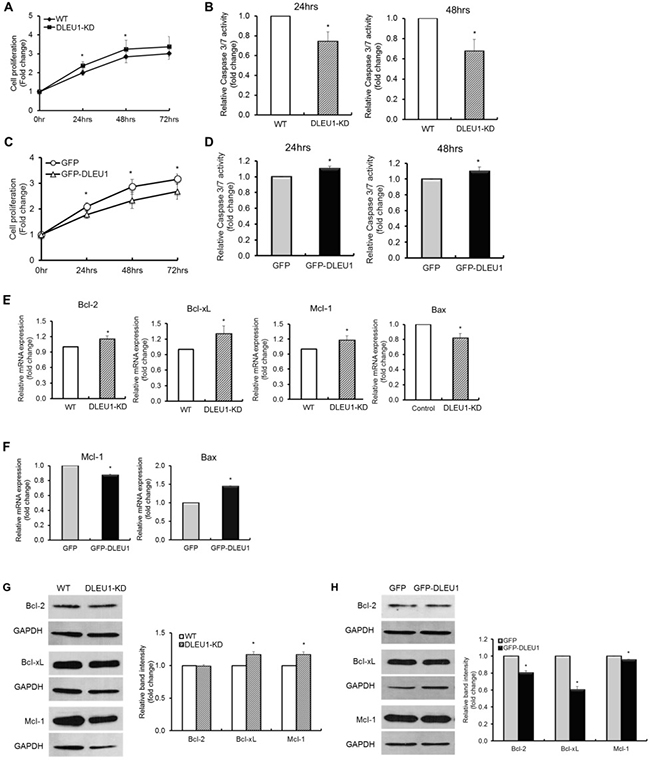
Figure 2: DLEU1 downregulates cell proliferation and upregulates caspase-dependent apoptosis. (A, C) Comparison of DLEU1-KD vs WT (A) and GFP-DLEU1 vs GFP (C) cell proliferation. (B, D) Caspase 3/7 activity in DLEU1-KD vs WT cells (B) and GFP-DLEU1 vs GFP cells (D). (E–H) mRNA (E, F) and protein (G, H) expression of anti-apoptotic and pro-apoptotic genes in the Bcl-2 family in DLEU1-KD vs WT (E, G) and GFP-DLEU1 vs GFP (F, H) cells. Data are represented as the mean ± SD of triplicates (paired t test). *p < 0.05; **p < 0.01.
DLEU1 significantly affects the expression of network genes and signaling pathways in BL
To monitor the expression of DLEU1 network genes, TUBB2C and UBR1, in DLEU1-KD cells compared to WT, quantitative reverse-transcriptase PCR (qRT-PCR) was performed. There was significantly reduced expression of TUBB2C and UBR1 mRNA (60%, p < 0.05; 25%, p < 0.01, respectively) (Figure 3A) in DLEU1-KD compared to WT cells. Furthermore, we compared protein activation related to Akt and NF-κB signaling pathways in DLEU1-KD and WT cells. DLEU1 knockdown resulted in significant increase in phosphorylation level of IκBα (2.6 fold, p < 0.01) and Akt (1.5 fold, p < 0.05) (Figure 3B and 3C). Conversely, GFP-DLEU1 expression induced significant inhibition of phosphorylation of IκBα (30% reduction, p < 0.05) and Akt (46% reduction, p < 0.05) (Figure 3D and 3E).
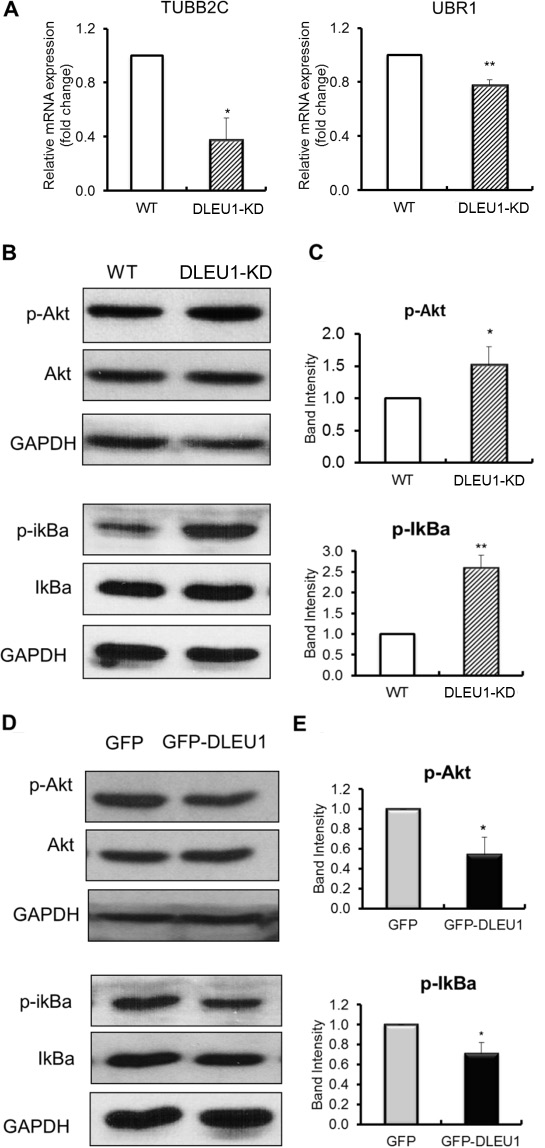
Figure 3: DLEU1 regulates the expression of network genes and signaling pathways. (A) mRNA expression of DLEU1 network genes in DLEU1-KD vs WT cells. (B, C) Significant increase in p-Akt and p-IκBα levels in DLEU1-KD vs WT cells. (D, E) Significant decrease in p-Akt and p-IκBα levels in GFP-DLEU1 vs GFP cells. Data are represented as the mean ± SD of triplicates (paired t test). *p < 0.05; **p < 0.01.
DLEU1 knockdown results in significant inhibition of programmed cell death, and increase in cell proliferation, in rituximab and/or cyclophosphamide treated cells
We next examined the changes in cell proliferation and caspase 3/7 dependent apoptosis in DLEU1-KD cells. These cells were treated with rituximab (RTX) (10 ug/ml) and cyclophosphamide (CTX) (10 mM), alone or in combination, for 48 hours. A significant reduction was noted in caspase 3/7-dependent apoptosis in DLEU1-KD cells treated with RTX alone (15% reduction, p < 0.05), CTX alone (24% reduction, p < 0.05) and in combination (35% reduction, p < 0.05) compared to WT cells (Figure 4A). Conversely, a significant increase in cell proliferation was observed in DLEU1-KD cells treated with RTX alone (10% increase, p < 0.05), CTX alone (9.4% increase, p < 0.05) and in combination (7.7% increase, p < 0.05) (Figure 4B). Moreover, RTX treated DLEU1-KD cells showed a significant increase in expression of anti-apoptotic genes, Bcl-2 and Bcl-xL (>1.3-fold, p < 0.05 and >2.0-fold, p < 0.05, respectively), and a significant decrease in expression of the pro-apoptotic gene, Bax (~25%, p < 0.05) (Figure 4C). These results were further confirmed at protein level by western blotting analyses. (Figure 4D).
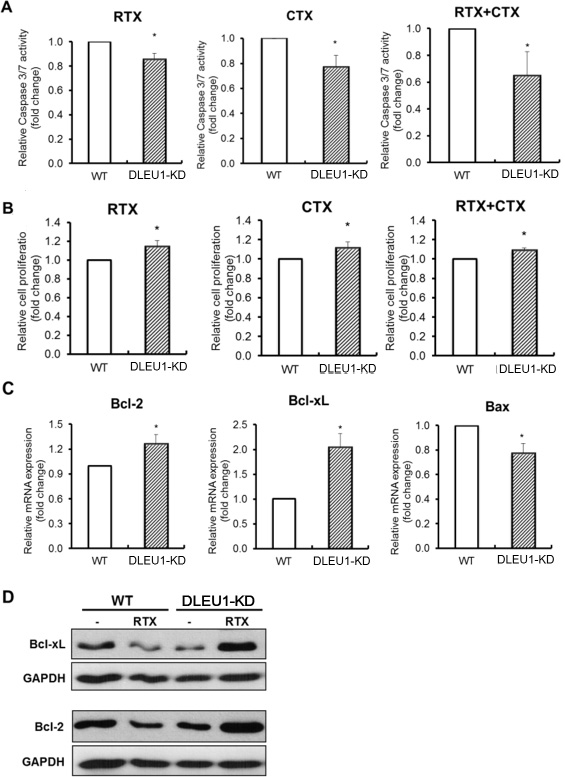
Figure 4: Caspase-dependent apoptosis and cell proliferation in rituximab and cyclophosphamide alone or in combination-treated DLEU1-KD vs WT cells. (A) Caspase 3/7 activity of rituximab (RTX, 10 ug/ml), cyclophosphamide (CTX, 10 mM) and RTX plus CTX combination (RTX/CTX) treated DLEU1-KD vs WT cells. (B) Cell proliferation of RTX, CTX and RTX/CTX treated DLEU1-KD vs WT cells. (C, D) The mRNA (C) and protein (D) expression of anti-apoptotic genes in RTX treated DLEU1-KD vs WT cells. Data are represented as the mean ± SD of triplicates (paired t test). *p < 0.05.
Global gene expression profiling in DLEU1 knockdown cells
To understand and elucidate the function and mechanism of DLEU1 gene in BL, we took a genomic approach employing Affymetrix GeneChip technology in DLEU1-KD vs. WT cells. A total of 2,501 differentially expressed genes (DEGs) from 21,381 human genes were identified (>2-fold, p < 0.05, 11.7%) between TALENs-induced DLEU1-KD cells compared to WT cells (Figure 5A). From among the 2,501 filtered genes, 1,995 genes (1,722 known genes plus 273 unknown genes, 79.8%) were up-regulated and 506 genes (415 known genes plus 91 unknown genes, 20.2%) were down-regulated (Figure 5B). When DEG-associated gene ontology (GO) terms were assessed, 126, 26 and 29 GO terms were associated with up-regulated genes whereas 78, 15 and 27 GO terms were associated with down-regulated genes (p < 0.05) among biological process, cellular component, and molecular function categories, respectively (Figure 5C and Supplementary Table 1). Functional clustering results of up-regulated genes suggest that genes related to anti-inflammatory response and anti-apoptotic response were activated including STAT1, IRAK1, ATK1, IκBα, CCNG2 and Bcl-2. Functional clustering results of down-regulated genes suggest that chromatin remodeling associated processes were inhibited. Notably, hematopoietic processes, especially B cell activation or possibly lymphoid lineage, were inhibited due to down-regulation of cell adhesion. A selected subset of DLEU1 target genes with biological function were validated by real time qRT-PCR. The results indicated a pattern consistent with gene expression profiling data (Figure 5D).
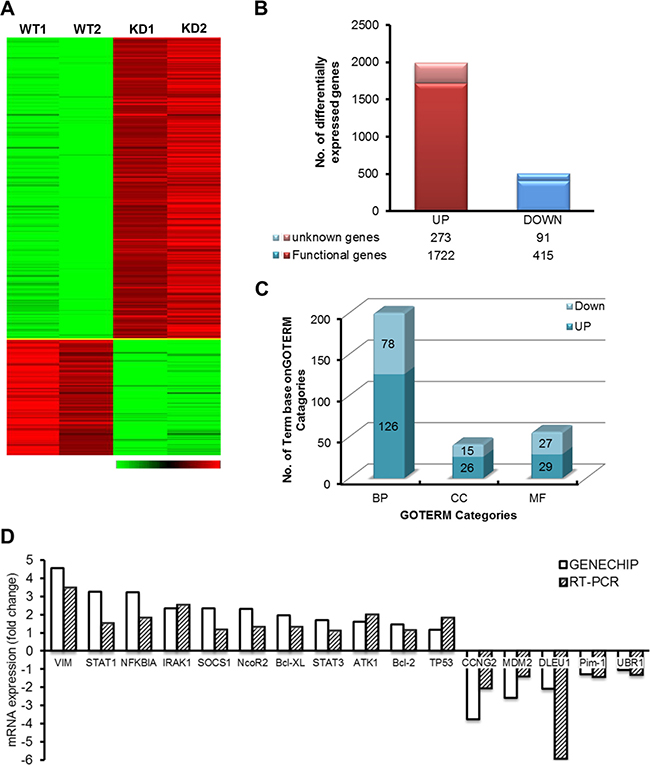
Figure 5: Gene expression patterns in DLEU1-KD vs WT. (A) Hierarchical clustering comparing the gene expression patterns between DLEU1-KD and WT cells. Green represents decreased expression and red increased expression, according to the scale. (B) Number of up- (left column) and down- (right column) regulated genes by DLEU1. (C) The numbers of genes in each functional category are demonstrated. (D) Verification of DLEU1 regulated genes identified by Affymetrix Genechip profiling by qRT-PCR. GAPDH was used as an endogenous control for normalization.
Effect of DLEU1 on sensitivity of xenograft tumors to RTX and/or CTX in vivo
We further investigated whether DLEU1 expression level has any effect on the sensitivity to RTX and/or CTX treatment of xenografted NSG mice. DLEU1-KD or WT Raji cells engineered to express luciferase were injected into NSG mice. Xenografted mice were then treated with RTX (30 mg/kg), CTX (25 mg/kg), or RTX together with CTX, weekly for 4 weeks. Mice receiving PBS served as a vehicle control. We found that tumors formed by DLEU1-KD cells grew faster than those by WT cells under the treatment of RTX, CTX, or combined (Supplementary Figure 2), indicating that the DLEU1 knockdown cells were more resistant to the treatment of these drugs. Consistently, we found that DLEU1-KD cells injected mice had significantly shortened survival time compared to those injected with WT cells when treated with RTX (42 days vs. 52 days, p < 0.005), or RTX combined with CTX (48 days vs 55.5 days, p < 0.05) (Figure 6A–6D). In the GFP-DLEU1 overexpressed xenograft model, we found that GFP-DLEU1 mice had significantly extended survival compared to GFP mice following treatment of RTX (57.5 days vs 51 days, p < 0.05, n = 8 per group), CTX (50.5 days vs 37.5 days, p < 0.0001, n = 12 per group), and RTX/CTX-combination treatment (60.5 days vs 53 days, p < 0.05, n = 12 per group) (Figure 6E–6H and Supplementary Figure 3).
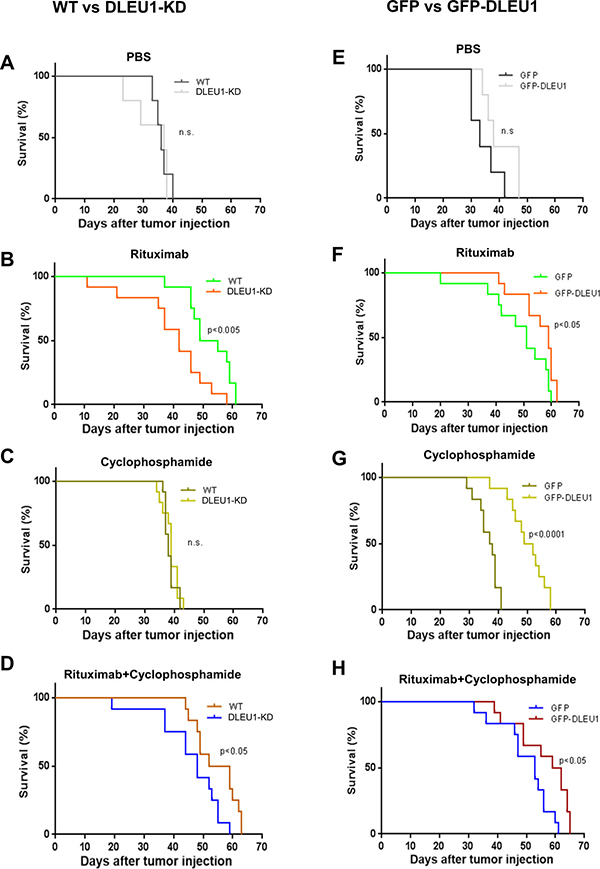
Figure 6: Survival of DLEU1-KD and GFP-DLEU1 mice treated with rituximab, cyclophosphamide and rituximab/cyclophosphamide combination. (A–D) Pair-wise comparison of survival curves of WT and DLEU1-KD mice (A–D) following the treatment of RTX, CTX, or RTX/CTX. *p < 0.05; **p < 0.005. (E–H) Pair-wise comparison of control (GFP) and DLEU1 overexpressed (GFP-DLEU1) xenograft mice (E–H) following the treatment of RTX, CTX, or RTX/CTX. ns, not significant; *p < 0.05; **p < 0.005; ***p < 0.0001.
DISCUSSION
Children and adolescents with 13q14.3 deletion have a significant poorer EFS compared to pediatric BL patients without this abnormality [10, 11]. Furthermore, this region has been determined to be the most frequently genetically altered locus in B-CLL [24, 25] and a major control locus of B-cell proliferation in CLL [24]. In the current preclinical study, we investigated if DLEU1 expression alters 1) BL programmed cell death, cell proliferation and the expression of DLEU1 network genes in vitro and 2) the survival of DLEU1 knockdown and overexpressing BL xenografted mice following chemoimmunotherapy.
We generated TALENs-mediated DLEU1 KD (83% reduction, p < 0.01) as well as GFP-DLEU1 stable expressing Raji BL cell lines. We investigated the effect of DLEU1 expression on cell proliferation and programmed cell death, and analyzed the expression of network genes and signaling pathways in BL following treatment with RTX and/or CTX using qRT-PCR and western blotting analyses. Gene expression profiling was performed using Affymetrix microarrays in DLEU1 KD BL cells compared to control to identify cellular processes regulated by DLEU1. We found that DLEU1 in part regulates BL apoptosis, cell proliferation and associated signaling pathways. Gene expression data additionally support the hypothesis that DLEU1 plays in part a role in BL cell signaling by demonstrating that anti-inflammatory and anti-apoptotic response genes, STAT1, IRAK1, ATK1, IκBα, CCNG2 and Bcl-2 were activated whereas hematopoietic-associated and chromatin remodeling-associated genes were downregulated in DLEU1-KD BL in comparison to wild type BL cells. These observations are consistent with the recent report of tumor suppressive function within the genetic region of 13q14.3 which is associated with suppressing tumor cell viability in cancers [24].
Furthermore, we observed a significant decrease in both chemotherapeutic and immunotherapeutic responses of DLEU1 KD cells as compared to WT with CTX and RTX, respectively, suggests that a DLEU1 loss may in part be a potential mechanism for therapeutic resistance. We then established in vivo xenograft NSG mouse models with DLEU1 knockdown and overexpressing Raji cells and investigated the sensitivity of these xenograft tumors to the treatment of RTX and/or CTX. More importantly, the deletion of DLEU1 results in significant enhancement of tumor progression and shortened survival in RTX and CTX treated DLEU1 KD cells xenografted mice compared to WT cell injected mice. In contrast, GFP-DLEU1 BL xenografted mice treated with RTX and/or CTX exhibited a significant survival advantage over control BL xenografted mice. These data in part suggest that DLEU1 level is associated with chemoimmunotherapy resistance.
The use of a TALENs method provided an opportunity to investigate the loss of function of a specific gene [26–29]. While this study sheds some light on at least one mechanism of chemoimmunotherapy resistance in BL, it will be important to investigate the role of other gene products found in the 13q14.3 region such as microRNA clusters, miR-15a/miR-16-1 [24, 30], and long non-coding RNA (lncRNA) gene, DLEU2 [24], and RFP2 and/or KCNRG [31]. DLEU1 may perhaps work in tandem with miR-15a/miR-16-1, DLEU2 and/or RFP2 to create an additive or synergistic tumor suppressive or oncogenic effect. We plan to investigate in future studies the individual and combined roles of miR-15a, miR-16-1, DLEU2 and/or RFP2 by molecular excision via TALENs in BL lymphomagenesis and their importance in regulating BL programmed cell death.
Interestingly, DLEU1 has been considered as a lncRNA gene [16, 17, 24]. However, DLEU1 was defined as putative functional protein coding gene that encodes a 78 amino acids based on Ensemle database [32] and the universal protein resource (UniProt) [33, 34]. It has recently been implicated that predicted open reading frame in lncRNA can be translated to functional peptides [35, 36]. A human protein-protein interaction study showed an interaction between DLEU1 and TUBB2C, TUBB2C and Bruton’s tyrosine kinase (BTK) [37, 38], suggesting that DLEU1 may interact with BTK. BTK is a regulator of normal B-cell development and is activated upon B-cell receptor (BCR) stimulation [39]. In addition, BTK has recently become an important target in hematological malignancies, with the development of various targeted therapies specifically targeted to inhibit BCR signaling [40, 41]. Specifically, a BTK-targeting molecule, ibrutinib has been approved by FDA for treatment of CLL and MCL patients who have failed prior therapy and is currently under pre-clinical investigation for treatment of BL [42]. We aim to further investigate the potential connection between DLEU1 and the BCR signaling pathways, and the role of this relationship in BL.
In summary, our results suggest that the deletion of DLEU1 at 13q14.3 in BL may in part result in chemoimmunotherapy resistance in BL. Furthermore, there is also the implication for pretreatment screening for the presence of the 13q14.3 deletion in children and adolescent BL and the potential investigation of alternative therapeutic strategies. In future studies, we will address which therapies may perhaps re-sensitize BL cells lacking the 13q14.3 region to improve therapy induced programmed cell death.
MATERIALS AND METHODS
Ethics statement
Investigation has been conducted in accordance with the ethical standards and according to the Declaration of Helsinki and according to national and international guidelines and has been approved by the Institutional Animal Care and Use Committee at New York Medical College.
Engineering of transcription activator-like effector nucleases (TALENs) targeting DLEU1
Potential TALENs target sites of DLEU1 were identified using web-based ZiFiT Targeter software (http://zifit.partners.org/). The BLAST analysis and Repeat Masker Web server (www.repeatmasker.org) were utilized for uniqueness of targeted sequences. The designed DLEU1 TALEN pairs were constructed based on Restriction Enzyme And Ligation (REAL) assembly methods using the Joung Lab REAL Assembly TALENS kit from Addgene [43]. TALENs were designed to target the DLEU1 transcription start and stop site on chromosome 13q14.3. To examine the validity of constructed TALENs pairs in mammalian cells, plasmids expressing each TALENs pair were transfected into Human embryonic kidney (HEK)293 cells and genomic DNA was extracted 24 hours post-transfection. Disrupted target sequences caused by non-homologous end joining (NHEJ) were confirmed by surveyor mutation detection assay (Supplementary Figure 1) and sequencing analysis (data not shown).
Cell culture and transfection
HEK293 and Raji human BL cells were obtained from ATCC (Manassas, VA) and maintained as previously described [44, 45]. These cell lines were authenticated by ATCC, and were provided us within 6 months before the use of cells, and cells were passaged for fewer than 8 weeks of resuscitation in our laboratory. Plasmid constructs were transfected into HEK293 and Raji cells using Lipofectamine® reagent (Invitrogen) and Amaxa Nucleofector™ Kit V (Lonza), respectively according to the manufacturer instructions.
Surveyor nuclease assay
Genomic DNA was extracted from cells 24–48 hours post-transfection using a genomic DNA extraction kit (Promega). The measurement of NHEJ - mediated mutation was performed using the Surveyor endonuclease assay kit (Transgenomic, Inc.) according to the manufacturer’s instructions. The PCR primers used are as follows: F2, 5′-TCTTGCTTTCCCGACATTT TTACG-3′, F4, 5′-CTAGAAGAGCCAACCAACAG-3′, and R1, 5′-AGTTGTTCCGAGGCTTAAGTGC GA-3′.
Generation of TALENs mediated DLEU1 knockdown cell line
Cells were transfected with TALEN expression plasmids and genomic DNA was extracted at 48 hours post-transfection followed by Surveyor nuclease assay as above. Raji cells with DLEU1 gene modification identified by NHEJ were seeded into 96 well plates to isolate a single clone. Individual colonies were picked, expanded and re-genotyped as described above.
Establishment of BL cell line stably overexpressing DLEU1
The DLEU1 overexpression construct (GFP-DLEU1) was generated by cloning the full length DLEU1 cDNA into pEGFP-N3 vector. GFP-DLEU1 was then transfected into 293T or Raji cells. Cells were selected under G418 (500 ug/ml) for stable clones and expression of GFP-DLEU1 was confirmed by qRT-PCR and Western blotting analyses.
Quantitative reverse-transcriptase PCR (qRT-PCR)
Total RNA was prepared using TRIzol reagent (Invitrogen) according to the manufacturer’s instruction and 1 ug of RNA was used for cDNA synthesis using qScript™ cDNA Synthesis Kit (Quantas). qRT-PCR was performed on the CFX96 Real-time system (Bio-rad) using SsoFast™ EvaGreen® Supermix (Bio-rad). Primers used in qRT-PCR are provided in Supplementary Table 2. Relative quantification (ddCt) of mRNA expression of genes was determined by normalizing to the housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Western blotting analysis
Western blot analysis was performed as we have previously described [44]. Band intensities on SDS-PAGE gel were measured using ImageJ software program. Antibodies specific for GAPDH, phospho-Akt and Akt, phospho-IκBα and IκBα and GFP were purchased from Cell Signaling Technology and an antibody specific for DLEU1 was provided by ProteinTech. Antibodies specific for anti-apoptotic genes, Bcl-2, Bcl-xL, and Mcl-1 and pro-apoptotic proteins, Bad and Bax were purchased from Cell Signaling Technology.
Cell proliferation assay
Prior to cell proliferation measurements, cells were treated without or with various doses of RTX (0 – 100 ug/ml), CTX (0 – 10 mM) alone and in combination. Cells (1 × 104) were plated into 48 well plates and then counted every 24 hrs as previously described [44]. Cell growth was determined using the non-radioactive CellTiter 96® Aqueous One solution cell proliferation assay (MTS) (Promega) and measured by a Multiabel Counter (Perkin Elmer) at OD490.
Caspase 3/7 assay
Caspase 3/7 activity was directly measured at 48 and 72 hours after treatment using Caspase-Glo 3/7 Activity kit (Promega) according to the manufacturer’s protocol. Relative light intensity was measured in each well using Clarity Luminescence Microplate Reader (BioTek).
Antibodies and reagents
A chimeric human and mouse anti-CD20 type I mAb, Rituximab (RTX), was purchased from Genentech Inc. and CTX was purchased from Sigma Aldrich and dissolved as a stock solution in PBS.
Gene expression profiling
Gene expression profiling experiments were performed using Affymetrix Human Genome 133 Plus 2.0 arrays at the Rockefeller University Genomics Facility. Two hundred nanograms of total RNA were biotin-labeled using Ambion MessageAmp™ Premier RNA Amplification Kit (Life Technologies). The fragmented aRNA was hybridized to the array as described in the Affymetrix Technical Analysis Manual (Affymetrix). Gene Chips were stained with streptavidin-phycoerythrin, and then scanned with the Affymetrix GeneChip Scanner 3000 7G. Data has been deposited in the NCBI’s Gene Expression Omnibus data base (accession number GSE65674).
Microarray data processing and analysis
Signal intensity was processed using MAS5 algorithm. The Probe sets that were not tagged as present in any samples according to the presence-absence-call and control probe sets were discarded. Among 21,437 surviving probe sets, we identified probe sets whose signal ratio between test group average and control group average exceeded two-fold as DEG [46] and retained for further analyses. We used DAVID (version 6.7) to investigate the biological processes and pathways implicated by DEG [47]. Specifically, we employed the functional annotation clustering utility that clusters functionally related terms linked to DEG. We interrogated over-expressed and under-expressed probe sets separately.
In vivo xenograft models
All experimental animals were γ- irradiated (2.5 Gy) 1 day before cell transplantation. DLEU1-KD and mock control (WT), or GFP-DLEU1 and mock control (GFP) were transfected with a firefly luciferase expression plasmid (ffluc-zeo, kindly provided by Laurence Cooper MD, PhD) into Raji cells followed by zeocin selection for stable clones. These selected cells (1 × 106) were then subcutaneously injected into four- to six-week-old female NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice from Jackson laboratory. After verification of tumor burden by bioluminescence imaging using the Xenogen IVIS-200 (Caliper Life Sciences), mice were treated with either PBS control (n = 5 per group), or RTX (30 mg/kg, n = 12 per group), or CTX (25 mg/kg, n = 12 per group) or RTX together with CTX (n = 12 per group) by intraperitoneal (i.p) injection at 7 day intervals as we have previously described [48]. Tumor progression was monitored at day 7 and once every week by bioluminescence imaging. All mice were housed and maintained under specific pathogen-free conditions under protocols (51-2-0913H and 59-2-1113H) approved by the Institutional Animal Care and Use Committee at New York Medical College.
Statistical analysis
Significant differences between DLEU1-KD vs WT or GFP-DLEU1 vs GFP groups (RT-PCR, cell proliferation assay, Caspase 3/7 assay and band intensity by Western blot) were determined by using one-tailed paired Student’s t-test in Microsoft Excel and p-values less than 0.05 were considered significant. Error bars are described in the Figure legends as ± SD. Survival rates were analyzed by the Kaplan-Meier method and differences evaluated by log-rank test using the Prism Version 6.0 software.
Abbreviations
BCR: B-cell receptor; BFM: Berlin-Frankfurt-Munster; BL: Burkitt lymphoma; BTK: Bruton’s tyrosine kinase; CLL: chronic lymphocytic leukemia; CTX: cyclophosphamide; DEG: differentially expressed genes; DLBCL: diffuse large B-cell lymphoma; DLEU1: Deleted in Lymphocytic Leukemia 1; DLEU1-KD: DLEU1 knockdown; EFS: event free survival; FAB: French American British; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; HEK: Human embryonic kidney; lncRNA: long non-coding RNA; NHEJ: non-homologous end joining; NHL: non-Hodgkin lymphoma; OS: Overall survival; qRT-PCR: Quantitative reverse-transcriptase PCR; REAL: restriction enzyme and ligation; RTX: rituximab; TALEN: transcription activator-like effector nuclease; TUBB2C: Tubulin beta-2C chain; UBR1: ubiquitin-protein ligase; WT: wild-type.
Authors’ contributions
SL organized, designed the study and performed experiments, acquired and analyzed the data and wrote the manuscript. WL and TS analyzed the data, and wrote and revised the manuscript. CY performed experiments, analyzed the data and wrote the manuscript. TC analyzed the data and wrote the manuscript. TO wrote the manuscript. RRM, SLP, EM, JA, LH, and CV, revised and approved the manuscript. MSC conceived and designed the study, analyzed the data and wrote the manuscript.
ACKNOWLEDGMENTS
The authors would like to dedicate this manuscript in memory of Warren Sanger, PhD, Professor of Pediatrics, and Director of the Human Genetics Laboratory at the University of Nebraska Medical Center who contributed significantly to the original identification of the significant importance of 13q14.3 deletion in pediatric Burkitt lymphoma and who recently passed away after a long productive career in pediatric NHL cytogenetics. The authors also would like to thank the Genomics resource center, Rockefeller University for assistance in the microarray analysis.
CONFLICTS OF INTEREST
All authors declare no financial conflicts of interest.
FUNDING
Funding was provided by grants from the Pediatric Cancer Research Foundation, Hyundai Hope on Wheels, and St. Baldrick’s Foundation (Mitchell S Cairo, MD).
REFERENCES
1. Cairo MS, Gerrard M, Sposto R, Auperin A, Pinkerton CR, Michon J, Weston C, Perkins SL, Raphael M, McCarthy K, Patte C. Results of a randomized international study of high-risk central nervous system B non-Hodgkin lymphoma and B acute lymphoblastic leukemia in children and adolescents. Blood. 2007; 109:2736–2743.
2. Cairo MS, Sposto R, Gerrard M, Auperin A, Goldman SC, Harrison L, Pinkerton R, Raphael M, McCarthy K, Perkins SL, Patte C. Advanced stage, increased lactate dehydrogenase, and primary site, but not adolescent age (>/= 15 years), are associated with an increased risk of treatment failure in children and adolescents with mature B-cell non-Hodgkin’s lymphoma: results of the FAB LMB 96 study. J Clin Oncol. 2012; 30:387–393.
3. Gerrard M, Cairo MS, Weston C, Auperin A, Pinkerton R, Lambilliote A, Sposto R, McCarthy K, Lacombe MJ, Perkins SL, Patte C. Excellent survival following two courses of COPAD chemotherapy in children and adolescents with resected localized B-cell non-Hodgkin’s lymphoma: results of the FAB/LMB 96 international study. Br J Haematol. 2008; 141:840–847.
4. Patte C, Auperin A, Gerrard M, Michon J, Pinkerton R, Sposto R, Weston C, Raphael M, Perkins SL, McCarthy K, Cairo MS. Results of the randomized international FAB/LMB96 trial for intermediate risk B-cell non-Hodgkin lymphoma in children and adolescents: it is possible to reduce treatment for the early responding patients. Blood. 2007; 109:2773–2780.
5. Goldman S, Smith L, Anderson JR, Perkins S, Harrison L, Geyer MB, Gross TG, Weinstein H, Bergeron S, Shiramizu B, Sanger W, Barth M, Zhi J, et al. Rituximab and FAB/LMB 96 chemotherapy in children with Stage III/IV B-cell non-Hodgkin lymphoma: a Children’s Oncology Group report. Leukemia. 2013; 27:1174–1177.
6. Goldman S, Smith L, Galardy P, Perkins SL, Frazer JK, Sanger W, Anderson JR, Gross TG, Weinstein H, Harrison L, Shiramizu B, Barth M, Cairo MS. Rituximab with chemotherapy in children and adolescents with central nervous system and/or bone marrow-positive Burkitt lymphoma/leukaemia: a Children’s Oncology Group Report. Br J Haematol. 2014; 167:394–401.
7. Suzuki E, Umezawa K, Bonavida B. Rituximab inhibits the constitutively activated PI3K-Akt pathway in B-NHL cell lines: involvement in chemosensitization to drug-induced apoptosis. Oncogene. 2007; 26:6184–6193.
8. Bonavida B. Rituximab-induced inhibition of antiapoptotic cell survival pathways: implications in chemo/immunoresistance, rituximab unresponsiveness, prognostic and novel therapeutic interventions. Oncogene. 2007; 26:3629–3636.
9. Kheirallah S, Caron P, Gross E, Quillet-Mary A, Bertrand-Michel J, Fournie JJ, Laurent G, Bezombes C. Rituximab inhibits B-cell receptor signaling. Blood. 2010; 115:985–994.
10. Poirel HA, Cairo MS, Heerema NA, Swansbury J, Auperin A, Launay E, Sanger WG, Talley P, Perkins SL, Raphael M, McCarthy K, Sposto R, Gerrard M, et al. Specific cytogenetic abnormalities are associated with a significantly inferior outcome in children and adolescents with mature B-cell non-Hodgkin’s lymphoma: results of the FAB/LMB 96 international study. Leukemia. 2009; 23:323–331.
11. Nelson M, Perkins SL, Dave BJ, Coccia PF, Bridge JA, Lyden ER, Heerema NA, Lones MA, Harrison L, Cairo MS, Sanger WG. An increased frequency of 13q deletions detected by fluorescence in situ hybridization and its impact on survival in children and adolescents with Burkitt lymphoma: results from the Children’s Oncology Group study CCG-5961. Br J Haematol. 2010; 148:600–610.
12. Klapper W, Szczepanowski M, Burkhardt B, Berger H, Rosolowski M, Bentink S, Schwaenen C, Wessendorf S, Spang R, Moller P, Hansmann ML, Bernd HW, Ott G, et al. Molecular profiling of pediatric mature B-cell lymphoma treated in population-based prospective clinical trials. Blood. 2008; 112:1374–1381.
13. Cairo MS, Day N, Goldman S, Sanger WG, Harrison L, Lim MS, Miles R, Perkins LP. Genomic Pathways and Potential Therapeutic Targets in Pediatric Burkitt Lymphoma (PBL): A Children’s Oncology Group Report Blood (ASH abstract). 2011; 118:1587.
14. Dave SS, Fu K, Wright GW, Lam LT, Kluin P, Boerma EJ, Greiner TC, Weisenburger DD, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Delabie J, et al. Molecular diagnosis of Burkitt’s lymphoma. N Engl J Med. 2006; 354:2431–2442.
15. Hummel M, Bentink S, Berger H, Klapper W, Wessendorf S, Barth TF, Bernd HW, Cogliatti SB, Dierlamm J, Feller AC, Hansmann ML, Haralambieva E, Harder L, et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006; 354:2419–2430.
16. Liu Y, Corcoran M, Rasool O, Ivanova G, Ibbotson R, Grander D, Iyengar A, Baranova A, Kashuba V, Merup M, Wu X, Gardiner A, Mullenbach R, et al. Cloning of two candidate tumor suppressor genes within a 10 kb region on chromosome 13q14, frequently deleted in chronic lymphocytic leukemia. Oncogene. 1997; 15:2463–2473.
17. Stilgenbauer S, Nickolenko J, Wilhelm J, Wolf S, Weitz S, Dohner K, Boehm T, Dohner H, Lichter P. Expressed sequences as candidates for a novel tumor suppressor gene at band 13q14 in B-cell chronic lymphocytic leukemia and mantle cell lymphoma. Oncogene. 1998; 16:1891–1897.
18. Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, Timm J, Mintzlaff S, Abraham C, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005; 122:957–968.
19. Dimmer EC, Huntley RP, Alam-Faruque Y, Sawford T, O’Donovan C, Martin MJ, Bely B, Browne P, Mun Chan W, Eberhardt R, Gardner M, Laiho K, Legge D, et al. The UniProt-GO Annotation database in 2011. Nucleic Acids Res. 2012; 40:D565–570.
20. Liu L, Amy V, Liu G, McKeehan WL. Novel complex integrating mitochondria and the microtubular cytoskeleton with chromosome remodeling and tumor suppressor RASSF1 deduced by in silico homology analysis, interaction cloning in yeast, and colocalization in cultured cells. In Vitro Cell Dev Biol Anim. 2002; 38:582–594.
21. Agathanggelou A, Cooper WN, Latif F. Role of the Ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res. 2005; 65:3497–3508.
22. Chen E, Kwon YT, Lim MS, Dube ID, Hough MR. Loss of Ubr1 promotes aneuploidy and accelerates B-cell lymphomagenesis in TLX1/HOX11-transgenic mice. Oncogene. 2006; 25:5752–5763.
23. Day N, Ayello J, Waxman I, Shereck E, McGuinn C, van de Ven C, Lim M, Cairo MS. Differential gene signatures in burkitt (BL) vs diffuse large b-cell lymphoma (DLBCL): disruption of the DLEU1 signal transduction pathways in BL vs DLBCL. Cancer Research. 2008; abstract:2993.
24. Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, Ambesi-Impiombato A, Califano A, Migliazza A, Bhagat G, Dalla-Favera R. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010; 17:28–40.
25. Migliazza A, Bosch F, Komatsu H, Cayanis E, Martinotti S, Toniato E, Guccione E, Qu X, Chien M, Murty VV, Gaidano G, Inghirami G, Zhang P, et al. Nucleotide sequence, transcription map, and mutation analysis of the 13q14 chromosomal region deleted in B-cell chronic lymphocytic leukemia. Blood. 2001; 97:2098–2104.
26. Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science. 2011; 333:1843–1846.
27. Carlson DF, Fahrenkrug SC, Hackett PB. Targeting DNA With Fingers and TALENs. Mol Ther Nucleic Acids. 2012; 1:e3.
28. Maeder ML, Linder SJ, Reyon D, Angstman JF, Fu Y, Sander JD, Joung JK. Robust, synergistic regulation of human gene expression using TALE activators. Nat Methods. 2013; 10:243–245.
29. Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol. 2012; 30:460–465.
30. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, Negrini M, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002; 99:15524–15529.
31. Ivanov DV, Tyazhelova TV, Lemonnier L, Kononenko N, Pestova AA, Nikitin EA, Prevarskaya N, Skryma R, Panchin YV, Yankovsky NK, Baranova AV. A new human gene KCNRG encoding potassium channel regulating protein is a cancer suppressor gene candidate located in 13q14.3. FEBS Lett. 2003; 539:156–160.
32. Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, Gil L, Giron CG, Gordon L, et al. Ensembl 2015. Nucleic Acids Res. 2015; 43:D662–669.
33. Bateman A and UniPort-consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2015; 43:D204–212.
34. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347:1260419.
35. Niazi F, Valadkhan S. Computational analysis of functional long noncoding RNAs reveals lack of peptide-coding capacity and parallels with 3′ UTRs. RNA. 2012; 18:825–843.
36. Bazzini AA, Johnstone TG, Christiano R, Mackowiak SD, Obermayer B, Fleming ES, Vejnar CE, Lee MT, Rajewsky N, Walther TC, Giraldez AJ. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 2014; 33:981–993.
37. de Weers M, Brouns GS, Hinshelwood S, Kinnon C, Schuurman RK, Hendriks RW, Borst J. B-cell antigen receptor stimulation activates the human Bruton’s tyrosine kinase, which is deficient in X-linked agammaglobulinemia. J Biol Chem. 1994; 269:23857–23860.
38. Qiu Y, Kung HJ. Signaling network of the Btk family kinases. Oncogene. 2000; 19:5651–5661.
39. Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013; 12:229–243.
40. Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013; 369:32–42.
41. Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, Barrientos JC, Chmielowska E, Radford J, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013; 369:507–516.
42. Lee S, Yin C, O’Connell T, Barth M, Ayello J, Harrison L, Van de Ven C, Miles R, Galardy P, Goldman SC, Lim MS, Hermiston M, McAllister-Lucas L, et al. Ibrutinib significantly improves survival in a human Burkitt lymphoma (BL) xenograft NSG mouse model: Ibrutinib may be a potential adjuvant agent in the treatment of BL. Cancer Research. 2015; abstract:2608.
43. Sander JD, Maeder ML, Joung JK. Engineering designer nucleases with customized cleavage specificities. Curr Protoc Mol Biol. 2011; Chapter 12:Unit12 13.
44. Lee S, Roy F, Galmarini CM, Accardi R, Michelon J, Viller A, Cros E, Dumontet C, Sylla BS. Frameshift mutation in the Dok1 gene in chronic lymphocytic leukemia. Oncogene. 2004; 23:2287–2297.
45. Lee S, Huang H, Niu Y, Tommasino M, Lenoir G, Sylla BS. Dok1 expression and mutation in Burkitt’s lymphoma cell lines. Cancer Lett. 2007; 245:44–50.
46. Dennis G, Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003; 4:3.
47. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44–57.
48. Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, Czuczman M, Cairo MS. Targeting CD20+ Aggressive B-cell Non-Hodgkin Lymphoma by Anti-CD20 CAR mRNA-Modified Expanded Natural Killer Cells In Vitro and in NSG Mice. Cancer Immunol Res. 2015; 3:333–344.