
INTRODUCTION
With the increasing of elderly population in the world, more and more individuals are suffering from senile dementia. It has been estimated that 115.4 million people globally will develop this cognitive disorder by 2050 [1]. Understanding the pathological mechanism of dementia could help us find new biomarker and yield novel therapeutic strategy. Previous studies have found that age-related loss of sex-steroid hormone can increase the risk of dementia, and women after menopause have greater risk to suffer from Alzheimer’s disease (AD) as compared to age-matched men [2, 3], suggesting that estrogen may play a key role in the pathological development of dementia. Even though estrogen therapy has been applied to improve the cognitive function of postmenopausal women, the effects of this hormone replacement therapy (HRT) on dementia remain controversial in clinical trials [4, 5, 6, 7]. Instead of benefiting the cerebral functions, HRT was found detrimental to women aged 65 or older [5, 6], while no apparent negative effect was found in women aged from 50 to 59 [8]. This disparity of therapeutic effects may due to the difficulty for HRT to diminish the established pathological changes in patients more than 65 years old [9, 10, 11]. Overall, these phenomena suggest that initiating estrogen therapy at the early years of postmenopause (before ages 50 to 60 years) would in all probability be better than that at the late stage of postmenopause (at ages of more than 65 to 79 years). This also indicates that replenishing the insufficient estrogen level at the beginning of postmenopause may be the optimum timing to prevent cognitive impairment of women in the following years, which contributes to a new concept about HRT for the prevention of dementia. However, in terms of this early-initiated HRT, changes in the brain of postmenopausal women are largely unknown. Exploring the pathophysiological alterations of brain at the early stage of postmenopause and identifying the effects of estrogen (E2) in this process will greatly improve our understandings of HRT for the prevention of cognitive impairment in women.
Fortunately, many studies have been implemented to investigate the action of E2 in brain. Based on studies in postmenopausal female rodents, E2 was found to regulate the permeability of blood-brain barrier (BBB) [12] and improve the spatial reference memory evaluated by both morris water maze (MWM) [13, 14] and radial arm maze [15, 16, 17]. Besides that, E2 was also found to regulate synapse plasticity by increasing the expression of hippocampal SNAP25 and synaptophysin [18, 19, 20], and stimulate neurogenesis in dentate gyrus by activating estrogen receptors [21, 22, 23]. Moreover, the expression of neurotrophin like BDNF [24, 25] and the activity of NOS [26] could also be promoted by E2. E2 has been found to modulate glia-mediated neuronal inflammation pathway in aged female mice as well [27]. Experiments in aged monkeys revealed that E2 administration could restore the cognitive function of these non-human primates [28]. In addition, E2 was confirmed to improve the synaptic function [29] and exert the antioxidative effects [30, 31] in ovariectomized rats. It has the ability to inhibit the activity of apoptosis signaling [32] and increase the expression of anti-apoptosis gene Bcl2 [33]. Furthermore, in hippocampal neurons in vitro, it has been documented that E2 can induce spinogenesis [34] and ameliorate excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity [35]. These studies fundamentally evidenced that E2 played a pivotal role in dementia. However, the morphological changes of the brain regarding E2 deficiency are extremely limited, especially in the initiation of menopause. In this study, by transmission electron microscopy (TEM), we observed the ultrastructure of the hippocampus in mice at the early stage of postmenopause with and without E2 supplementation. The aim was to provide basic morphological evidences for early-initiated HRT.
RESULTS
Estrogen could prevent mitochondrial damage in the hippocampal DG region of early postmenopausal mice
Firstly, serum estrogen levels in 6 M group, 18 M group and 18 M + E2 group were tested. As illustrated in Figure 1, serum estrogen levels in 18 M group (10.21±2.43 pg/ml) are less than those in 6 M group (47.22±2.39 pg/ml), and the E2 levels in 18 M + E2 group (28.97±4.16 pg/ml) indicate that E2 replacement effectively improves the decreased estrogen level in 18 M group.
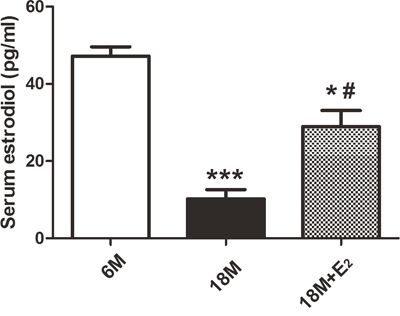
Figure 1: Estradiol concentration in mice serum. The concentration of serum estradiol (pg/ml) was decreased in 18 M group in comparison with 6 M group (n = 8) (* p < 0.05, *** p < 0.001). Estradiol administration in 18 M + E2 group partly reversed the low level of estradiol in 18 M mice (n = 8) (# p < 0.05, unpaired t-test). Data are presented as mean ± SEM.
Next, we used Transmission Electron Microscope (TEM) to observe the morphological changes of hippocampus. Compared with 6 M group (Figure 2A and 2D), the form of neuron and neuronal chromatin remained unchanged in both hippocampal CA1 (Figure 2B) and DG (Figure 2E) regions of 18 M group, and supplementation of E2 for 2 months did not affect the status as shown in 18 M + E2 group (Figure 2C and 2F). Importantly, the neuronal polarity was also preserved in CA1 subfield (Figure 2A-2C). Organelles including mitochondria, Golgi body and rough endoplasmic reticulum were all well developed in hippocampal CA1 region of experimental groups (Figure 2A1-2C1). In DG region, though Golgi body and rough endoplasmic reticulum presented no difference among 6 M, 18 M and 18 M + E2 groups, we found marked swelling of mitochondria coupled with disappeared cristae in 18 M group (Figure 2E1), which could be prevented by E2 administration (Figure 2F1).
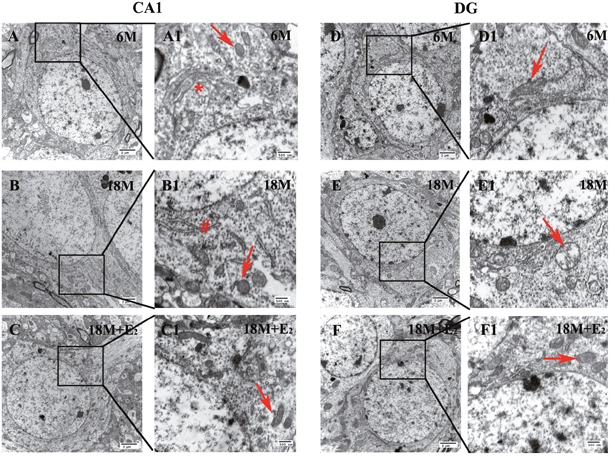
Figure 2: The morphology and ultrastructure of neurons in both CA1 and DG regions. A-F. Pictures were taken by TEM at 10000 × magnification, neurons presented integrated nuclear membrane and normal chromatin distribution in both CA1 and DG regions of 6 M (A, D), 18 M (B, E) and 18 M + E2 (C, F) group. A1-F1. Certain fields of A-F were further observed at 30000 × magnification. A1. Organelles in the CA1 neuron of 6 M group were well-organized, which is represented by Golgi body (signed by asterisk). B1. In the CA1 neuron of 18 M group, healthy endoplasmic reticulum (signed by well number) and mitochondrion (signed by arrow) were detected. C1. In the CA1 neuron of 18 M + E2 group, intact mitochondria with high electron density could be seen. D1. Organelles in the DG neuron of 6 M group were prominent. E1. The mitochondria in the DG neuron of 18 M group were swelling with disappeared cristae. F1. All the organelles were well developed in the DG neuron of 18 M + E2 group. Scale bar = 2 μm in A-F and 500 nm in A1-F1.
Estrogen ameliorated lipofuscin deposition in hippocampal DG neurons of early postmenopausal mice
Lipofuscin is composed of a pile of spherical structures with varying degrees of electron density, it is often deposited in perinuclear region of cytoplasm [40]. In comparison with the limited amount of lipofuscins in both CA1 and DG regions of 6 M group (Figure 3A and 3D), we found marked accumulation of lipofuscins in relative regions of 18 M group, as indicated by arrows in Figure 3B and 3E. Furthermore, after elderly mice were treated with E2, lipofuscin accumulation was difficult to find in the DG region of 18 M + E2 group (Figure 3F) while not CA1 region (Figure 3C). Then the number of deposited lipofuscins in each group was calculated, and the results were shown in Figure 3G and 3H. The number of lipofuscins in each neuron of CA1 region was increased from 1.25±0.91 in 6 M group to 2.88±1.83 in 18 M group (P < 0.01). And in DG region, the number of lipofuscins in each neuron was increased from 1.07±1.05 in 6 M group to 3.17±1.79 in 18 M group (P < 0.001). Importantly, with the administration of E2, the accumulated lipofuscin number per neuron in DG region was significantly decreased to 1.28±1.01 (P < 0.001), but the number was 2.03±1.24 in CA1 region, which was not statistically different compared with that in 18 M group.
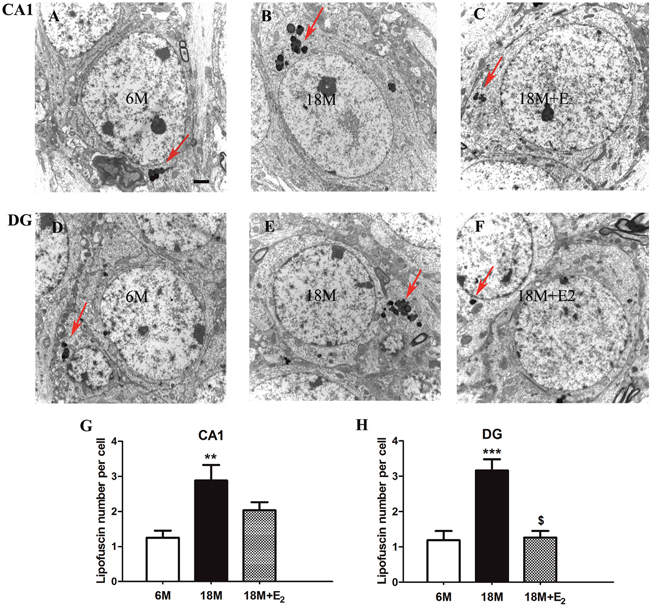
Figure 3: Lipofuscin deposition in the hippocampal neuron of mice. A. Single lipofuscin presented in the CA1 subfield of 6 M group. B. A pile of lipofuscins were accumulated in the perinuclear region of CA1 neurons in 18 M group. C. The amount of lipofuscins in the CA1 neuron of 18 M + E2 group was limited. D. Only small amount of lipofuscins could be seen in the DG subfield of 6 M group. E. Great amount of lipofuscins was found in the DG neuron of 18 M group. F. It is hard to trace lipofuscin in the DG neuron of 18 M + E2 group. G. The number of deposited lipofuscins was remarkably increased in the CA1 region of 18 M group. H. Estrogen replacement in 18 M + E2 group can mitigate the deposition of lipofuscin in the DG region of 18 M group. Lipofuscin is composed by a group of spherical structures with varying degrees of electron density which is pointed by rigid arrow in each image. Scale bar = 2 μm. ** p < 0.01, *** p < 0.001 compared to 6 M group; $ p < 0.05 compared to 18 M group. Data are presented as mean ± SEM.
Estrogen had no effect on the damaged microtubules of early postmenopausal mice
Microtubules play a key role in transporting neurotransmitters to the synapse, and they act as downstream effectors of neurotransmitter signaling in the target neuron [41]. Therefore, the morphology of hippocampal microtubule was detected in this study. As shown in Figure 4A and 4A1, neuronal microtubules were filled in axons with relative continuous and fiber-like texture in 6 M group. However, discontinuous and sparse microtubule arrays were observed in axons of 18 M group (Figure 4B and 4B1). To our surprise, the supplementation of E2 to postmenopausal mice failed to prevent the damage of microtubules (Figure 4C and 4C1). The statistical data in Figure 4D further manifested that the number of microtubules was significantly decreased in 18 M group compared with 6 M group, and E2 administration in 18 M + E2 group did not quantitatively reverse the defective microtubules of elderly mice.
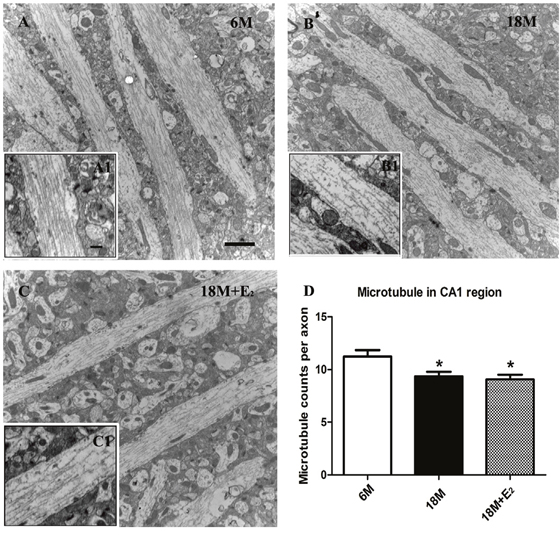
Figure 4: Estrogen was unable to ameliorate the destabilized microtubule in postmenopause mice. A. Regular and integrated microtubules were detected in CA1 region of 6 M group; A1. Well-organized fiber-like microtubules were visible in the enlarged region. B. The neurons of 18 M mice displayed inconsecutive microtubules; B1. Great intervals between microtubules in axon of neuron could be seen. C. Microtubules in 18 M + E2 mice also presented a less-organized morphology; C1. The continuity and amount of microtubules declined in neurons. D. Compared with 6 M group, the mean number of axonal microtubules in 18 M group was greatly decreased as well as in 18 M + E2 group. (* p < 0.05 compared to 6 M group). Data are expressed as mean ± SEM. Scale bar = 2 μm in A-C and 500 nm in A1-C1.
Estrogen had no effect on the unchanged synapses of early postmenopausal mice
Previous studies have reported that estrogen can regulate synapse plasticity [18, 19], so we calculated the number of synapses in both CA1 and DG regions. Intriguingly, there was no difference in these two subfields among three groups, indicating that menopause could not lead to the loss of synapses (Figure 5). In mice of 6 M group, many round vesicles containing neurotransmitters were filled in the presynaptic region. Likewise, these phenomena were found in both 18 M and 18 M + E2 groups. Moreover, no difference in the amount of vesicles was observed among these three groups (Figure 6A-6C). To further observe the synapse, enlarged images were obtained. We found that the typical morphologic features of synapse including presynaptic dense band, relative thin postsynaptic membrane and synaptic cleft were all similar among these three groups (Figure 6A1-6C1).
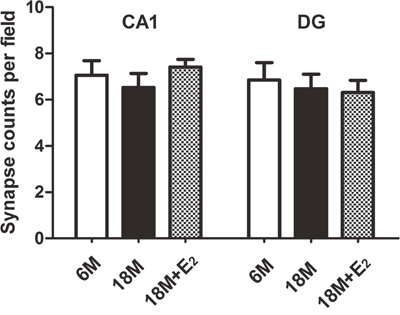
Figure 5: Synapse density in the hippocampus of mice. In hippocampal CA1 and DG subfields, the synapse number per field has no statistical difference among 6 M, 18 M and 18 M + E2 groups. (n = 22, one-way ANOVA). Data are presented as mean ± SEM.
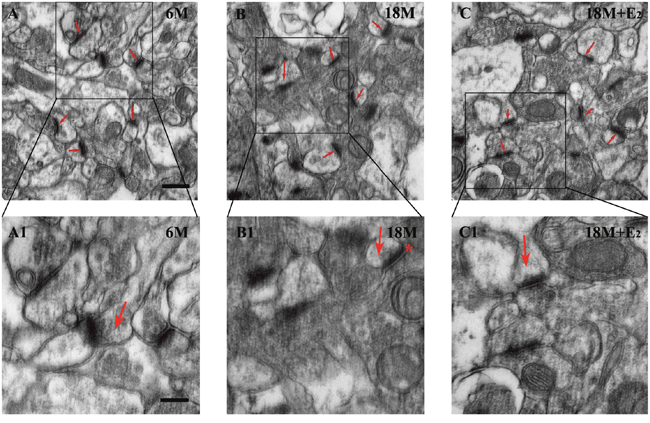
Figure 6: Estrogen had no effect on the morphology of synapse in postmenopause mice. A. Typical synapse including presynaptic and postsynaptic regions as well as synaptic cleft was observed in hippocampal CA1 neuron of 6 M mice; A1. Presynaptic vesicles were detected in the regional magnified version. B. In 18 M group, synapses were also prominent in morphology; B1. The synaptic cleft was clearly recognized and marked by an asterisk. C. The morphological appearance of synapse in 18 M + E2 mice was comparative with the other two groups; C1. Active zone thickness, synaptic cleft width and the amount of presynaptic vesicles were similar to the other groups. Active zone of synapse is arrowed in A-C. Arrows in A1-C1 point to the presynaptic region. Scale bar = 500 nm in A-C and 250 nm in A1-C1.
DISCUSSION
In the present study, we illustrated a comprehensive picture of morphological alterations in the hippocampus of early postmenopausal mice, and evaluated the role of E2 in these mice by TEM technique. We concluded that the main features of structure remodeling in the hippocampus of early postmenopausal mice were mitochondrial damage, lipofuscin deposition and microtubule degradation. Furthermore, we identified that E2 supplementation could prevent the onset of the mitochondrial damage and lipofuscin deposition rather than microtubule degradation caused by menopause. These findings suggest that hippocampal structure remodeling can be initiated at the early stage of postmenopause, but not all of the alterations can be reversed by E2 supplementation. Thus, the application of HRT in early postmenopausal women (before ages 50 to 60 years) cannot be too optimistic based on our data. To some extent, we also provide the morphological evidences for the failure of HRT in clinical trials of late postmenopausal women (at ages of more than 65 to 79 years).
Mitochondrial dysfunction is a common feature of both aging and neurodegenerative diseases [42, 43, 44]. Improvement of mitochondrial function is considered as a strategy to slow aging and aging-related neurodegenerative disorders [45, 46]. In this study, we found that abnormal mitochondrial structure was elicited in DG but not CA1 region of hippocampus at the early stage of postmenopause, and E2 supplementation could prevent the changes in DG region, indicating that E2 may be useful to preserve the structure of mitochondria in this early phase. The protective effect of E2 is probably exerted through its action in oxidative stress as previously reported [47, 48, 49]. The results also indicate that neurons in DG region are easier to be affected by E2 deficiency than neurons in CA1 region.
Interestingly, except for the abnormal mitochondria, the morphologies of organelles including Golgi apparatus, endoplasmic reticulum as well as cell membrane remained unchanged in the hippocampal neuron of 18 M group, suggesting that mitochondrial damage is prior to the injury of other organelles at the early stage of postmenopause. This is in line with a previous report [50]. Clinical data reported that, in women aged 65 and older, HRT negatively influenced verbal memory and brain volume [5], and HRT was ineffective for mild to moderate AD in women aged from 56 to 91 [51] as it missed the best time to reverse the established pathological changes. Here, we first demonstrated that developed mitochondrial structural remodeling may be the partial fundamental evidence of HRT resistance for the cognitive impairment treatment at both early and late stages of postmenopause.
As a marker of aging, lipofuscin has been reported to deposit in different areas of brain in models like 24 M female hamsters and rats [52, 53, 54, 55]. Lipofuscin is the intralysosomal electron-dense remnants filled by oxidized proteins and lipid clusters that cannot be digested by lysosome [56, 57]. The existence of lipofuscin is considered as the response to oxidative stress [58]. Mitochondrion is the key organelle responding to oxidative stress, the dysfunction of it will lead to the production of a large amount of reactive oxygen species [59, 60]. Hence, the deposition of lipofuscin is considered as the outcome of mitochondrial damage [61]. In this study, the number of lipofuscins was significantly increased in both CA1 and DG regions of hippocampus in 18 M mice, and E2 supply could effectively prevent the lipofuscin deposition in DG region. This result is consistent with changes of mitochondria, suggesting that the deposited lipofuscin might be associated with the damage of mitochondria. Of interest, it has been documented that E2 administration could reduce lipofuscin deposition in neurons of 12 M, 18 M and 24 M female rats [53, 62], but it failed to benefit the impaired cognitive ability of OVX rats aged 17-18 M [63]. These phenomena imply that the functional benefit of E2 is not simply associated with the decreased lipofuscin deposition.
Moreover, we observed the morphology of both neuronal microtubule and synapse of the hippocampus, due to their involvement in neurotransmission. We found that, though decreased number of microtubules was observed in the hippocampal neurons of 18 M mice, which is consistent with data from aging people [64, 65], the change was failed to be prevented by E2 supplementation. This result suggests that microtubule damage in mice at the early stage of postmenopause is not originated from E2 deficiency. During the process of estrogen supplementation in this study, the mice also went through the process of aging. Hence, some pathophysiological changes in aging may be involved in the damage of microtubules, including the increase of ROS and the decrease of neurotrophic factors [66, 67]. However, to figure out the mechanism of microtubule damage, further exploration is needed. Additionally, some literatures have reported that the expression of SNAP25 and synaptophysin, two components of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE), decreased in hippocampus of 18-24 M rats [20] and 27-28 M mice [13]. Since SNARE is an engine that mediates the fusion of vesicular and presynaptic membranes [68], the dysfunction of it may result in the accumulation of vesicles in presynapse. In the cortex of aging monkeys with low level of estrogen, accumulated vesicles in axon terminals were also reported [69]. While, in our work, no synaptic vesicle accumulation was detected in the hippocampus of both solvent-injected and E2-administrated 18 M mice.
The synapse density along with the active zone thickness and synaptic cleft width were also similar among groups in this study, which is in line with evidences from mouse [70], rat [71], monkey [72] and human being [73]. Nevertheless, the reduced number of synapse in aging rats like 30 M female Fisher 344 rats has also been published [74, 75, 76]. This discrepancy may due to the different ages of the animals. For example, the disturbance of neurotransmitter release in brain was detected only when the rats were over 18 M [77, 78]. In the present study, mice at the age of 18 M were detected, in which the degeneration of synapse may not exhibit yet. Therefore, the unchanged synapse density in our experimental mice is reasonable, based on the hints from a previous study which mentioned that aging rats with good memory tend to have the same synapse density in the DG region of hippocuampus like young rats [79].
Taken together, although E2 supplementation could prevent menopause-elicited mitochondrial damage and lipofuscin deposition, it failed to block the defective microtubules. These findings provide an experimental evidence of the unapparent effect of HRT in the early phase of postmenopause women and give a possible explanation for HRT failure at the late stage of postmenopause.
MATERIALS AND METHODS
Animals
Female C57BL/6 mice used in the experiments were separated into three groups: mice at the age of 6 months (6 M) without any injection were used as control 6 M group (n = 8), while mice at the age of 18 months (18 M) without and with the supplementation of E2 were designed as 18 M group (n = 8) and 18 M + E2 group (n = 8), respectively. The animals were raised at 23 ± 1°C and maintained on 12 h dark-light artificial cycle (lights on at 07:00 A.M.) with food and water available ad libium. All animal procedures were approved by the ethic committees of Harbin Medical University and the Institute of Laboratory Animal Science of China (A5655-01). Moreover, the protocols complied with the guidance for the Care and Use of Laboratory Animals of the US National Institutes of Health (NIH Publication No. 85–23, revised 1996).
Estrogen (E2) administration
Since the reproductive function of mice declines with time as the estrous cyclicity becomes acylic from 11 M to 16 M [80, 81], E2 administration was started in mice at the age of 16 M and lasted for two months, aiming to mimic a replenishment for the decreased E2 in early postmenopausal phase. Therefore, in the 18 M + E2 group, E2 (17β-estradiol, Sigma, E-2758, St Louis, MO, USA) dissolved in peanut oil was subcutaneously injected into mice with a dosage of 3.5 μg/kg every three days for two months. The injection was also performed in 18 M group, but only solvent was injected.
Mouse estradiol ELISA
At the age of 18 M, mice were anesthetized (pentobarbital sodium, 60 mg/kg, i.p.) before the blood was extracted from the left ventricle and maintained at room temperature for 3 h, the serum was collected after centrifugation (3000 rpm, 5 min), and stored at -80°C. Similarly, serum was obtained from 6 M mice. The concentration of estradiol in serum was measured using the mouse/rat estradiol ELISA kit (Calbiotech, ES180S-100, Spring Valley, CA) following the manufacturer’s instructions.
Transmission electron microscopy
Based on stereotaxic coordinates of hippocampus, coronal slices of mouse brain were prepared and soaked immediately in 2.5% glutaraldehyde. After 6-8 h at 4°C, they were cut into 1mm thick coronal slices and the hippocampi part was separated. Next, samples were rinsed with PBS (0.1 M) before post-fixed by osmium tetroxide for 1-2 h. The dehydration procedure was carried out using ethanol and acetone in concentration gradients. Subsequently, we used epoxy resin for embedding prior to the slicing of ultra-thin sections. Then, double staining by uranium acetate and lead citrate was performed. And finally, images were taken by transmission electron microscope (JEM-1220, JEOL Ltd., Tokyo, Japan).
Statistical analysis
The numbers of lipofuscin, synapse and microtubule were calculated in a blinded manner. And the amount of microtubules per axon was obtained by averaging three transverse sections. Data are expressed as mean ± SEM. GraphPad Prism 5.0 was used for statistical analysis. For the comparison among groups, one-way ANOVA was applied and P < 0.05 was considered as statistical significance.
ACKNOWLEDGMENTS
This work was supported by the Funds for Natural Science Foundation of China (81271207, 81070882).
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.
Author contributions
JA and YY conceived and designed the experiments. YY, LC, XC, QW, MD, JM, LZ and XJ performed the experiments and analyzed data. YY, LC and XC wrote and edited this manuscript. All authors read and approved the final manuscript.
REFERENCES
1. Khanna R, Wilson SM, Brittain JM, Weimer J, Sultana R, Butterfield A, Hensley K. Opening Pandora's jar: a primer on the putative roles of CRMP2 in a panoply of neurodegenerative, sensory and motor neuron, and central disorders. Future neurology. 2012; 7:749-771.
2. Birge SJ. The role of estrogen in the treatment and prevention of dementia: introduction. The American journal of medicine. 1997; 103:1S-2S.
3. Vina J, Lloret A. Why women have more Alzheimer's disease than men: gender and mitochondrial toxicity of amyloid-beta peptide. Journal of Alzheimer's disease. 2010; 20:S527-533.
4. Resnick SM, Coker LH, Maki PM, Rapp SR, Espeland MA, Shumaker SA. The Women's Health Initiative Study of Cognitive Aging (WHISCA): a randomized clinical trial of the effects of hormone therapy on age-associated cognitive decline. Clin Trials. 2004; 1:440-450.
5. Coker LH, Espeland MA, Rapp SR, Legault C, Resnick SM, Hogan P, Gaussoin S, Dailey M, Shumaker SA. Postmenopausal hormone therapy and cognitive outcomes: the Women's Health Initiative Memory Study (WHIMS). The Journal of steroid biochemistry and molecular biology. 2010; 118:304-310.
6. Shumaker SA, Legault C, Kuller L, Rapp SR, Thal L, Lane DS, Fillit H, Stefanick ML, Hendrix SL, Lewis CE, Masaki K, Coker LH. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women's Health Initiative Memory Study. JAMA. 2004; 291:2947-2958.
7. Brown S. WHI memory study fails to find oestrogen benefit in dementia. The journal of the British Menopause Society. 2004; 10:93.
8. Marjoribanks J, Farquhar C, Roberts H, Lethaby A. Long term hormone therapy for perimenopausal and postmenopausal women. The Cochrane database of systematic reviews. 2012; 7:CD004143.
9. Rocca WA, Grossardt BR, Shuster LT. Oophorectomy, menopause, estrogen treatment, and cognitive aging: clinical evidence for a window of opportunity. Brain research. 2011; 1379:188-198.
10. Espeland MA, Rapp SR, Shumaker SA, Brunner R, Manson JE, Sherwin BB, Hsia J, Margolis KL, Hogan PE, Wallace R, Dailey M, Freeman R, Hays J. Conjugated equine estrogens and global cognitive function in postmenopausal women: Women's Health Initiative Memory Study. JAMA. 2004; 291:2959-2968.
11. Henderson VW. Alzheimer's disease: Review of hormone therapy trials and implications for treatment and prevention after menopause. The Journal of steroid biochemistry and molecular biology. 2013.
12. Bake S, Sohrabji F. 17beta-estradiol differentially regulates blood-brain barrier permeability in young and aging female rats. Endocrinology. 2004; 145:5471-5475.
13. Frick KM, Fernandez SM, Bulinski SC. Estrogen replacement improves spatial reference memory and increases hippocampal synaptophysin in aged female mice. Neuroscience. 2002; 115:547-558.
14. Markham JA, Pych JC, Juraska JM. Ovarian hormone replacement to aged ovariectomized female rats benefits acquisition of the morris water maze. Hormones and behavior. 2002; 42:284-293.
15. Luine VN, Richards ST, Wu VY, Beck KD. Estradiol enhances learning and memory in a spatial memory task and effects levels of monoaminergic neurotransmitters. Hormones and behavior. 1998; 34:149-162.
16. Daniel JM, Fader AJ, Spencer AL, Dohanich GP. Estrogen enhances performance of female rats during acquisition of a radial arm maze. Hormones and behavior. 1997; 32:217-225.
17. Bohacek J, Daniel JM. Increased daily handling of ovariectomized rats enhances performance on a radial-maze task and obscures effects of estradiol replacement. Hormones and behavior. 2007; 52:237-243.
18. Foy M, Baudry M, Thompson R. Estrogen and hippocampal synaptic plasticity. Neuron glia biology. 2004; 1:327-338.
19. Woolley CS. Estrogen-mediated structural and functional synaptic plasticity in the female rat hippocampus. Hormones and behavior. 1998; 34:140-148.
20. Canas PM, Duarte JM, Rodrigues RJ, Kofalvi A, Cunha RA. Modification upon aging of the density of presynaptic modulation systems in the hippocampus. Neurobiology of aging. 2009; 30:1877-1884.
21. Tanapat P, Hastings NB, Reeves AJ, Gould E. Estrogen stimulates a transient increase in the number of new neurons in the dentate gyrus of the adult female rat. The Journal of neuroscience. 1999; 19:5792-5801.
22. Mazzucco CA, Lieblich SE, Bingham BI, Williamson MA, Viau V, Galea LA. Both estrogen receptor alpha and estrogen receptor beta agonists enhance cell proliferation in the dentate gyrus of adult female rats. Neuroscience. 2006; 141:1793-1800.
23. Qureshi HY, Han D, MacDonald R, Paudel HK. Overexpression of 14-3-3z promotes tau phosphorylation at Ser262 and accelerates proteosomal degradation of synaptophysin in rat primary hippocampal neurons. PloS one. 2013; 8:e84615.
24. Bimonte-Nelson HA, Nelson ME, Granholm AC. Progesterone counteracts estrogen-induced increases in neurotrophins in the aged female rat brain. Neuroreport. 2004; 15:2659-2663.
25. Singh M, Meyer EM, Simpkins JW. The effect of ovariectomy and estradiol replacement on brain-derived neurotrophic factor messenger ribonucleic acid expression in cortical and hippocampal brain regions of female Sprague-Dawley rats. Endocrinology. 1995; 136:2320-2324.
26. Grohe C, Kann S, Fink L, Djoufack PC, Paehr M, van Eickels M, Vetter H, Meyer R, Fink KB. 17 Beta-estradiol regulates nNOS and eNOS activity in the hippocampus. Neuroreport. 2004; 15:89-93.
27. Lei DL, Long JM, Hengemihle J, O'Neill J, Manaye KF, Ingram DK, Mouton PR. Effects of estrogen and raloxifene on neuroglia number and morphology in the hippocampus of aged female mice. Neuroscience. 2003; 121:659-666.
28. Erez H, Shemesh OA, Spira ME. Rescue of tau-induced synaptic transmission pathology by paclitaxel. Frontiers in cellular neuroscience. 2014; 8:34.
29. Ip JP, Fu AK, Ip NY. CRMP2: Functional Roles in Neural Development and Therapeutic Potential in Neurological Diseases. The Neuroscientist. 2014.
30. Xu W, Tse YC, Dobie FA, Baudry M, Craig AM, Wong TP, Wang YT. Simultaneous monitoring of presynaptic transmitter release and postsynaptic receptor trafficking reveals an enhancement of presynaptic activity in metabotropic glutamate receptor-mediated long-term depression. The Journal of neuroscience. 2013; 33:5867-5877.
31. Gandini MA, Henriquez DR, Grimaldo L, Sandoval A, Altier C, Zamponi GW, Felix R, Gonzalez-Billault C. Ca2.2 channel cell surface expression is regulated by the light chain 1 (LC1) of the microtubule-associated protein B (MAP1B) via UBE2L3-mediated ubiquitination and degradation. Pflugers Archiv. 2014.
32. Zhao M, Sun L, Yu XJ, Miao Y, Liu JJ, Wang H, Ren J, Zang WJ. Acetylcholine mediates AMPK-dependent autophagic cytoprotection in H9c2 cells during hypoxia/reoxygenation injury. Cellular physiology and biochemistry. 2013; 32:601-613.
33. Park JH, Lee JE, Shin IC, Koh HC. Autophagy regulates chlorpyrifos-induced apoptosis in SH-SY5Y cells. Toxicology and applied pharmacology. 2013; 268:55-67.
34. Nieto-Rostro M, Sandhu G, Bauer CS, Jiruska P, Jefferys JG, Dolphin AC. Altered expression of the voltage-gated calcium channel subunit alphadelta-1: A comparison between two experimental models of epilepsy and a sensory nerve ligation model of neuropathic pain. Neuroscience. 2014.
35. Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. Journal of neurochemistry. 1996; 66:1836-1844.
36. Velazquez-Zamora DA, Gonzalez-Tapia D, Gonzalez-Ramirez MM, Flores-Soto ME, Vazquez-Valls E, Cervantes M, Gonzalez-Burgos I. Plastic changes in dendritic spines of hippocampal CA1 pyramidal neurons from ovariectomized rats after estradiol treatment. Brain research. 2012; 1470:1-10.
37. Mehra RD, Sharma K, Nyakas C, Vij U. Estrogen receptor alpha and beta immunoreactive neurons in normal adult and aged female rat hippocampus: a qualitative and quantitative study. Brain research. 2005; 1056:22-35.
38. Adams MM, Fink SE, Shah RA, Janssen WG, Hayashi S, Milner TA, McEwen BS, Morrison JH. Estrogen and aging affect the subcellular distribution of estrogen receptor-alpha in the hippocampus of female rats. The Journal of neuroscience. 2002; 22:3608-3614.
39. Torres-Reveron A, Khalid S, Williams TJ, Waters EM, Jacome L, Luine VN, Drake CT, McEwen BS, Milner TA. Hippocampal dynorphin immunoreactivity increases in response to gonadal steroids and is positioned for direct modulation by ovarian steroid receptors. Neuroscience. 2009; 159:204-216.
40. Chapman MA. Interactions between cell adhesion and the synaptic vesicle cycle in Parkinson's disease. Medical hypotheses. 2014; 83:203-207.
41. Gardiner J, Overall R, Marc J. The microtubule cytoskeleton acts as a key downstream effector of neurotransmitter signaling. Synapse. 2011; 65:249-256.
42. Beal MF. Mitochondria take center stage in aging and neurodegeneration. Annals of neurology. 2005; 58:495-505.
43. Sastre J, Pallardo FV, Vina J. Mitochondrial oxidative stress plays a key role in aging and apoptosis. IUBMB life. 2000; 49:427-435.
44. Albers DS, Beal MF. (2000). Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. Advances in Dementia Research: Springer), pp. 133-154.
45. Gruber J, Fong S, Chen CB, Yoong S, Pastorin G, Schaffer S, Cheah I, Halliwell B. Mitochondria-targeted antioxidants and metabolic modulators as pharmacological interventions to slow ageing. Biotechnology advances. 2013; 31:563-592.
46. Reddy PH, Reddy TP. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Current Alzheimer research. 2011; 8:393-409.
47. Behl C, Widmann M, Trapp T, Holsboer F. 17-beta estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochemical and biophysical research communications. 1995; 216:473-482.
48. Behl C, Skutella T, Lezoualc'h F, Post A, Widmann M, Newton CJ, Holsboer F. Neuroprotection against oxidative stress by estrogens: structure-activity relationship. Molecular pharmacology. 1997; 51:535-541.
49. Stirone C, Duckles SP, Krause DN, Procaccio V. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Molecular pharmacology. 2005; 68:959-965.
50. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006; 443:787-795.
51. Mulnard RA, Cotman CW, Kawas C, van Dyck CH, Sano M, Doody R, Koss E, Pfeiffer E, Jin S, Gamst A, Grundman M, Thomas R, Thal LJ. Estrogen replacement therapy for treatment of mild to moderate Alzheimer disease: a randomized controlled trial. Alzheimer's Disease Cooperative Study. JAMA. 2000; 283:1007-1015.
52. Gerrits PO, Kortekaas R, de Weerd H, Veening JG and van der Want JJ. Regional differences in age-related lipofuscin accumulation in the female hamster brainstem. Neurobiology of aging. 2012; 33:625 e621-629.
53. Kumar P, Taha A, Kale RK, Cowsik SM, Baquer NZ. Physiological and biochemical effects of 17beta estradiol in aging female rat brain. Experimental gerontology. 2011; 46:597-605.
54. Bala K, Tripathy BC, Sharma D. Neuroprotective and anti-ageing effects of curcumin in aged rat brain regions. Biogerontology. 2006; 7:81-89.
55. Kaur J, Singh S, Sharma D, Singh R. Neurostimulatory and antioxidative effects of L-deprenyl in aged rat brain regions. Biogerontology. 2003; 4:105-111.
56. Jung T, Bader N, Grune T. Lipofuscin: formation, distribution, and metabolic consequences. Annals of the New York Academy of Sciences. 2007; 1119:97-111.
57. Jolly RD, Palmer DN, Dalefield RR. The analytical approach to the nature of lipofuscin (age pigment). Archives of gerontology and geriatrics. 2002; 34:205-217.
58. Kawauchi T. Cdk5 regulates multiple cellular events in neural development, function and disease. Development, growth & differentiation. 2014; 56:335-348.
59. Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proceedings of the National Academy of Sciences of the United States of America. 1994; 91:10771-10778.
60. Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007; 12:913-922.
61. Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. European journal of biochemistry/FEBS. 2002; 269:1996-2002.
62. Moorthy K, Yadav UC, Siddiqui MR, Mantha AK, Basir SF, Sharma D, Cowsik SM, Baquer NZ. Effect of hormone replacement therapy in normalizing age related neuronal markers in different age groups of naturally menopausal rats. Biogerontology. 2005; 6:345-356.
63. Foster TC, Sharrow KM, Kumar A, Masse J. Interaction of age and chronic estradiol replacement on memory and markers of brain aging. Neurobiology of aging. 2003; 24:839-852.
64. Cash AD, Aliev G, Siedlak SL, Nunomura A, Fujioka H, Zhu X, Raina AK, Vinters HV, Tabaton M, Johnson AB, Paula-Barbosa M, Avila J, Jones PK, Castellani RJ, Smith MA, Perry G. Microtubule reduction in Alzheimer's disease and aging is independent of tau filament formation. The American journal of pathology. 2003; 162:1623-1627.
65. Matus A, Green GD. Age-related increase in a cathepsin D like protease that degrades brain microtubule-associated proteins. Biochemistry. 1987; 26:8083-8086.
66. Budni J, Bellettini-Santos T, Mina F, Garcez ML, Zugno AI. The involvement of BDNF, NGF and GDNF in aging and Alzheimer's disease. Aging and disease. 2015; 6:331-341.
67. Patel R, Sesti F. Oxidation of ion channels in the aging nervous system. Brain research. 2016; 1639:174-185.
68. Jahn R, Scheller RH. SNAREs--engines for membrane fusion. Nature reviews Molecular cell biology. 2006; 7:631-643.
69. Soghomonian JJ, Sethares C, Peters A. Effects of age on axon terminals forming axosomatic and axodendritic inhibitory synapses in prefrontal cortex. Neuroscience. 2010; 168:74-81.
70. Xu X, Zhang Z. Effects of estradiol benzoate on learning-memory behavior and synaptic structure in ovariectomized mice. Life sciences. 2006; 79:1553-1560.
71. Geinisman Y, Ganeshina O, Yoshida R, Berry RW, Disterhoft JF, Gallagher M. Aging, spatial learning, and total synapse number in the rat CA1 stratum radiatum. Neurobiology of aging. 2004; 25:407-416.
72. Hara Y, Yuk F, Puri R, Janssen WG, Rapp PR, Morrison JH. Presynaptic mitochondrial morphology in monkey prefrontal cortex correlates with working memory and is improved with estrogen treatment. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111:486-491.
73. Scheff SW, Price DA, Sparks DL. Quantitative assessment of possible age-related change in synaptic numbers in the human frontal cortex. Neurobiology of aging. 2001; 22:355-365.
74. Genisman Y, Bondareff W. Decrease in the number of synapses in the senescent brain: a quantitative electron microscopic analysis of the dentate gyrus molecular layer in the rat. Mechanisms of ageing and development. 1976; 5:11-23.
75. Bertoni-Freddari C, Giuli C, Pieri C, Paci D. Quantitative investigation of the morphological plasticity of synaptic junctions in rat dentate gyrus during aging. Brain research. 1986; 366:187-192.
76. Bertoni-Freddari C, Fattoretti P, Paoloni R, Caselli U, Galeazzi L, Meier-Ruge W. Synaptic structural dynamics and aging. Gerontology. 1996; 42:170-180.
77. Wu CF, Bertorelli R, Sacconi M, Pepeu G, Consolo S. Decrease of brain acetylcholine release in aging freely-moving rats detected by microdialysis. Neurobiology of aging. 1988; 9:357-361.
78. Rastogi M, Ojha RP, Prabu PC, Devi BP, Agrawal A, Dubey GP. Prevention of age-associated neurodegeneration and promotion of healthy brain ageing in female Wistar rats by long term use of bacosides. Biogerontology. 2012; 13:183-195.
79. Geinisman Y, de Toledo-Morrell L, Morrell F. Loss of perforated synapses in the dentate gyrus: morphological substrate of memory deficit in aged rats. Proceedings of the National Academy of Sciences of the United States of America. 1986; 83:3027-3031.
80. Nelson JF, Felicio LS, Randall PK, Sims C, Finch CE. A longitudinal study of estrous cyclicity in aging C57BL/6J mice: I. Cycle frequency, length and vaginal cytology. Biology of reproduction. 1982; 27:327-339.
81. Felicio LS, Nelson JF, Finch CE. Longitudinal studies of estrous cyclicity in aging C57BL/6J mice: II. Cessation of cyclicity and the duration of persistent vaginal cornification. Biology of reproduction. 1984; 31:446-453.