
Introduction
Epithelial tissues cover the surface of the body and line the internal cavities [1]. The structural integrity of these tissues requires extensive cell-cell adhesion and interactions mediated by the adhesive junctional complexes consisting of the adherens junctions and desmosomes [2-5]. Adherens junctions are a ubiquitous type of intercellular junction and are present in both epithelial and non-epithelial cells [3, 6], whereas desmosomes are adhesive junctions that confer tensile strength and resilience on cells and are present not only in epithelial cells but also in non-epithelial cells that endure mechanical stress, such as cardiac muscle and meninges [7. 8]. Both adherens junctions and desmosomes are cadherin based. Cadherins are single-pass transmembrane glycoproteins that form homotypic interactions with cadherin proteins on neighboring cells and interact intracellularly with proteins of the catenin family [4, 5]. At the adherens junction in epithelia, the C-terminal domain of E-cadherin interacts, in a mutually exclusive manner, with β-catenin or γ-catenin (plakoglobin), which then interacts with α-catenin, an actin-binding protein. A fourth catenin protein, p120-catenin, interacts with the juxtamembrane domain of E-cadherin and stabilizes the cadherin dimers at the membrane (Figure 1; [9,10]). At the desmosome, the cytoplasmic domain of the desmosomal cadherins (desmocollins and desmogleins) interacts with plakophilin and plakoglobin, which in turn are associated with desmoplakin, an intermediate filament binding protein that connects the complex to the cytoskeleton (Figure 1; [7, 11]).
Although originally identified as structural proteins with a “glue-like” function, cadherin-based cell adhesion complexes (adherens junctions and desmosomes) have subsequently been shown to have significant interactions with elements of the signal transduction pathways that regulate growth and morphogenesis [12-17]. More specifically, cadherin-independent β-catenin and plakoglobin have been shown to have signaling functions through their interactions with an array of functionally diverse proteins, including receptor tyrosine kinases and phosphatases, tumor suppressors and transcription factors [18-20]. Consequently, these catenin proteins play important roles in regulating tumor development and metastasis progression. β-catenin has a well-documented oncogenic potential as a component of the Wnt signaling pathway, whereas plakoglobin generally acts as a tumor/metastasis suppressor. While some studies have reported a tumor promoting activity of plakoglobin in a cell/malignancy specific context as discussed in the following sections, this review is primarily focused on the potential mechanisms by which plakoglobin may inhibit tumorigenesis and metastasis.
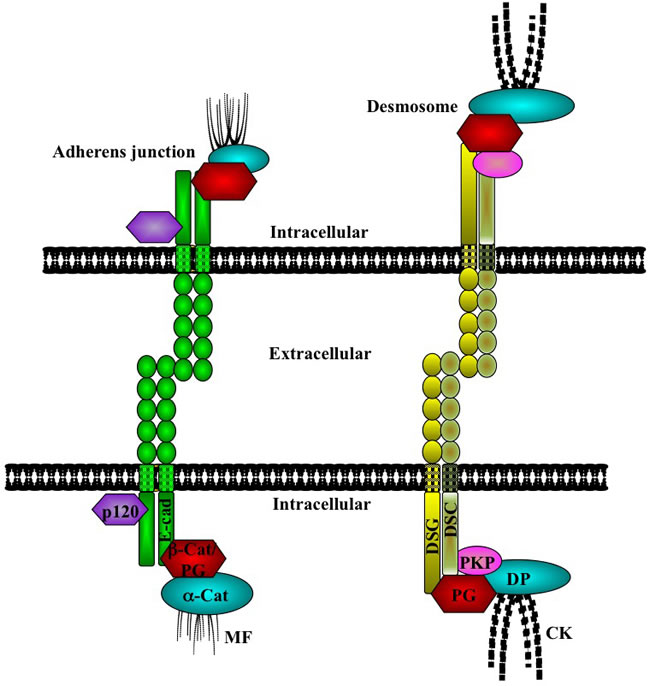
Figure 1: Cell adhesion complexes in epithelial cells. Cell-cell adhesion is maintained in epithelial tissues by the adherens junction and desmosomes. At the adherens junctions, E-cadherin (E-cad) forms extracellular interactions with E-cadherin molecules on neighboring cells. Intracellularly, E-cadherin interacts with either β-catenin (β-cat) or plakoglobin (PG), which then interact with α-catenin (a-cat), an actin binding protein. A fourth catenin, p120-catenin, also interacts with E-cadherin and regulates its stability at the membrane. The E-cadherin/catenin complex is stabilized at the membrane by interaction with the actin microfilaments (MF). At the desmosome, the desmosomal cadherins [desmoglein (DSG) and desmocollin (DSC)] interact with plakoglobin (PG) and plakophilin (PKP), which interact with desmoplakin (DP), which in turn associates with the cytokeratin intermediate filaments (CK). The basic, core protein composition of the desmosomes is represented here: the exact protein constituents of the desmosomes and their interactions vary among different types of cells and tissues. (See the text for references).
Plakoglobin: initial identification and early characterization
Plakoglobin was originally identified as an 83 kDa protein component of the desmosomal plaque [21). Following its initial identification, experiments using monoclonal antibodies, cDNA cloning and a combination of biochemical, morphological and molecular approaches demonstrated that this 83 kDa protein was present in both desmosomes and adherens junctions and thus, was given the name plakoglobin [21, 22).
Although identified as a junction protein, the role that plakoglobin played in the junctional complexes was unclear, and the partners it interacted with remained unidentified. Several years later, coimmunoprecipitation experiments showed that plakoglobin interacted with both desmoglein (thereby confirming plakoglobin as a constituent of the desmosomes; [23] and E-cadherin [24, 25]. However, since plakoglobin was found to be only loosely associated with E-cadherin, it was suggested that the main adhesive complexes primarily consisted of E-cadherin, β-catenin and α-catenin, although the existence of a separate E-cadherin-plakoglobin-α-catenin complex could not be ruled out [25].
Subsequently, it was demonstrated that plakoglobin interacted with both desmoglein and E-cadherin in both the soluble and cytoskeleton-associated pools of cellular proteins. Furthermore, a distinct, cadherin-independent pool of plakoglobin was observed, which suggested that plakoglobin may have a cellular function independent of cell adhesion [26]. Finally, phosphorylation experiments revealed that whereas the insoluble (cadherin-associated) pool of plakoglobin was serine phosphorylated, the soluble pool was serine, threonine and tyrosine phosphorylated, suggesting that these different pools of plakoglobin are differentially regulated and may potentially perform varying functions [26].
Plakoglobin and cell-cell adhesion
The most documented role of plakoglobin within the cell is in cell-cell adhesion. The identification of plakoglobin as a component of both the adherens junction and desmosomes suggested that it plays an important role in regulating cell-cell adhesion. However, since it was observed that the adherens junction could exist as a complex containing E-cadherin, β-catenin and α-catenin, independent of plakoglobin [25], the requirement for plakoglobin at the adherens junctions remained in question. Regardless, the essential role of plakoglobin in the regulation of cell-cell adhesion soon became apparent.
The function of plakoglobin in regulating cell-cell adhesion was clearly demonstrated when it was shown that the re-expression of E- or P-cadherin in cadherin-null murine spindle cell carcinomas with very low levels of plakoglobin was not sufficient to modify the morphology or tumorigenicity of the cells [27]. In these cells, although the exogenously expressed cadherins localized to the cell membrane and interacted with both α- and β-catenin, they did not interact with plakoglobin and desmosomes were not formed. From this work, the authors concluded that the presence of plakoglobin in the E-cadherin complex may be necessary for proper cell-to-cell adhesion.
The role of plakoglobin in regulating junction formation was also demonstrated when it was shown that A431 epithelial cells treated with dexamethasone (which decreased E-cadherin and plakoglobin levels) were unable to form adherens junctions and desmosomes and exhibited a fibroblastic morphology. Following the expression of E-cadherin in these cells, the adherens junctions were formed but the fibroblastic morphology of the cells remained unchanged. The authors then expressed an E-cadherin-plakoglobin chimeric protein in these cells, which resulted in the formation of stable adherens junctions and desmosomes and induced an epithelioid morphology. Together, these results suggested that E-cadherin-plakoglobin interactions were necessary for the formation of stable adhesive complexes and provided the first indication that plakoglobin served as a molecule involved in the cross-talk between the adherens junctions and desmosomes [28].
Following this study, our laboratory demonstrated the role of plakoglobin in junction formation by expressing plakoglobin in SCC9 cells, a human tongue squamous cell carcinoma cell line that lacks the expression of both plakoglobin and E-cadherin but expresses N-cadherin [29, 30]. Transfectants expressing E-cadherin (SCC9-E) or low/physiological levels of plakoglobin (SCC9-PG) or both, were generated and showed that the independent expression of either E-cadherin or plakoglobin induced a mesenchymal (transformed) to epidermoid (normal) phenotypic transition (MET). This phenotypic transition was associated with decreased cell proliferation and increased cell-cell adhesion, with only SCC9-PG cells capable of forming desmosomes. E-cadherin or plakoglobin expression also coincided with decreased soluble β-catenin levels, while E-cadherin expression downregulated N-cadherin and, plakoglobin expression increased N-cadherin levels and stability [29, 30]. Since then, numerous subsequent studies identified the switch from E- to N-cadherin as a major contributing factor in the epithelial to mesenchymal phenotypic transition (EMT) and metastatic progression. Significantly, our results clearly demonstrated that in the absence of E-cadherin, plakoglobin was able to inhibit N-cadherin tumor promoting activities and that the cadherin switch by itself cannot explain the transformed phenotype of SCC9 cells. Furthermore, the induction of MET by E-cadherin and plakoglobin may occur via a common pathway that also involves β-catenin [29, 30].
Other studies have characterized further the role of plakoglobin in desmosome assembly and function, demonstrating that plakoglobin is essential for the proper assembly of the desmosomal plaque and the efficient binding of desmoplakins to the intermediate filaments [31, 32].
Plakoglobin and embryonic development
While the above studies were aimed at understanding the role of plakoglobin in the formation and regulation of adhesive junctional complexes in tissue culture cells, plakoglobin knockout transgenic mouse models were also generated to study the role of plakoglobin during embryonic development. Work from two independent laboratories demonstrated that the homozygous knockout (double knockout) of plakoglobin resulted in embryonic lethality in mice [33, 34]. Analysis of mouse embryos from different stages of embryogenesis revealed that plakoglobin double knockout embryos were phenotypically similar to wild type and heterozygous embryos until approximately E8.5 and E9.5 [33]. In E10.5 and E12.5 embryos, it was observed that plakoglobin double knockout mice displayed severe heart defects (e.g. thin and weak heart walls). There were considerably fewer desmosomes in these embryos and their plaques were less dense, particularly in the intercalated discs of cardiac muscle [33, 34]. Therefore, these studies demonstrated that the loss of plakoglobin during embryonic development resulted in lethality, which could be attributed to defective desmosome formation and impaired cardiac development.
Similar to plakoglobin, the loss of β-catenin during embryonic development also resulted in lethality. However, β-catenin double knockout mouse embryos began to display defects earlier than plakoglobin double knockout embryos. At E7.0, β-catenin double knockout embryos showed defects in the embryonic ectodermal layer, in which cells became detached from the layer and were dispersed in the proamniotic cavity [35]. Furthermore, at E7.5, β-catenin double knockout embryos were half as large as wild type and heterozygous embryos and histological analyses demonstrated that the three germ cell layers had not formed. Most notably, the normal epithelial organization of the ectodermal layer was completely absent [35]. Together, these studies showed distinct roles for plakoglobin and β-catenin in embryonic development; whereas knockdown of plakoglobin resulted in defects in cardiac structure, β-catenin knockdown resulted in impaired migration of ectodermal cells.
Plakoglobin and cell signaling
i. Overview
As mentioned above, in addition to their roles in regulating cell-cell adhesion, catenin proteins participate in cell signaling through their interactions with various intracellular partners. Catenin-mediated cell signaling has been the focus of many studies, most of which have concentrated on β-catenin and overlooked plakoglobin. Plakoglobin and β-catenin are paralogs and members of the Armadillo family of proteins (Figure 2; [36]). As such, they share common intracellular partners, including classical cadherins, α-catenin, axin, APC and TCF/LEF [18, 37-41]. Despite their structural similarities and their common interacting partners, plakoglobin and β-catenin appear to play opposite roles with respect to cell signaling in tumorigenesis and metastasis. β-catenin has a well-defined oncogenic potential as a component of the Wnt signaling pathway [18, 19, 42, 43], whereas plakoglobin has been typically associated with tumor and metastasis suppressor activities through mechanisms that are beginning to be deciphered [29, 44-58]. In the following sections, we describe experimental evidence, which links plakoglobin to various signaling pathways that regulate tumorigenesis and metastasis.
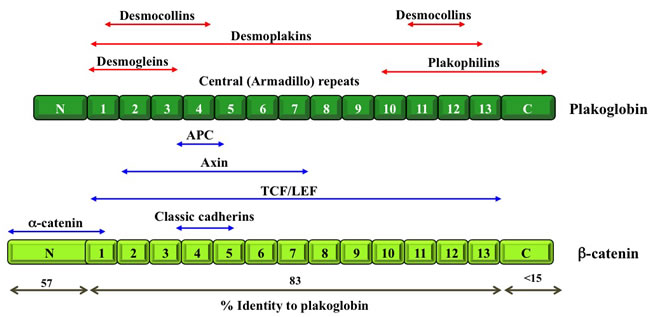
Figure 2: Schematic structure of β-catenin and plakoglobin. Both β-catenin and plakoglobin contain 13 Armadillo repeats that are flanked by N- and C-terminal domains, respectively. The degree of identity between β-catenin and plakoglobin for each domain is indicated. Blue arrows indicate protein partners that interact with both β-catenin and plakoglobin and the domains involved in these interactions. Red arrows indicate proteins that interact with plakoglobin only and the domains involved in these interactions. APC, adenomatous polyposis coli; TCF/LEF, T-cell factor/Lymphoid enhancer factor (see Table 1 for references).
ii. Wnt signaling pathway
The Wnt pathway is a signaling cascade with fundamental roles in the regulation of cell proliferation, cell polarity and cell fate determination during embryonic development and in tissue homeostasis. Deregulation of this pathway results in birth defects and various diseases, including cancer (for reviews, see [19, 43, 59]).
The first indication that plakoglobin might participate in the Wnt pathway (and in turn, cell signaling) was when its levels were increased in PC12 cells following exogenous Wnt-1 expression. In addition to its increased levels, plakoglobin underwent membrane redistribution, suggesting that in addition to β-catenin levels, Wnt-1 can modulate plakoglobin levels and localization [60]. Subsequently, it was demonstrated that the microinjection of mRNAs encoding plakoglobin into fertilized Xenopus embryos resulted in dorsalized gastrulation and anterior axis duplication [61]. In this study, the exogenously expressed plakoglobin localized at both the plasma membrane and in punctate nuclear aggregates. Importantly, when mRNAs encoding plakoglobin and the cytoplasmic domain of desmoglein were co-injected into the embryos, both dorsalized gastrulation and anterior axis duplication were suppressed. In these embryos, plakoglobin was localized primarily to the plasma membrane with some peri-nuclear distribution, suggesting that junction-independent plakoglobin has signaling ability similar to β-catenin.
While this initial study suggested that plakoglobin may have signaling functions similar to β-catenin, several lines of evidence suggest that this is most likely not the case.
The Klymkowsky group has shown that membrane-anchored forms of plakoglobin produced the same axis duplication as the wild type protein [62]. demonstrating that nuclear plakoglobin was inconsequential in inducing a Wnt-like phenotype. The observation that plakoglobin was ineffective in Wnt signaling was also made in Drosophila, when transgenes encoding either β-catenin or plakoglobin were expressed in embryos which lacked functional armadillo protein. Using the ectodermal expression of engrailed as an indicator of Wnt signaling, the authors observed that while zygotic expression of β-catenin resulted in weak engrailed expression, plakoglobin was unable to induce the same the phenotype [63].
Several other groups have shown that compared to β-catenin, plakoglobin has limited signaling activity in the context of the Wnt pathway. Simcha et al. [64] showed that LEF-1 overexpression in MDCK cells resulted in the nuclear translocation of β-catenin but not plakoglobin. In the same study, TOPFLASH reporter activity was considerably higher in HEK293T cells transfected with β-catenin compared to those transfected with plakoglobin expression constructs [64]. Furthermore, expression of a transcriptionally active β-catenin mutant, S37A, induced TOPFLASH reporter activity in 293T cells to a much greater extent than that by the analogous S28A plakoglobin mutant [41]. Consistent with these results, electrophoretic mobility shift assays using in vitro translated β-catenin, plakoglobin, TCF-4 and LEF-1 and radioactively labeled DNA corresponding to TCF/LEF binding sequences showed that β-catenin-TCF-4/LEF-1-DNA complexes were efficiently formed, whereas plakoglobin-TCF-4/LEF-1-DNA complexes were not detected [65, 66]. Taken together, these results suggest that while plakoglobin may have potential signaling activity as part of the Wnt pathway, this activity is minimal, especially when compared to that of β-catenin.
It is important to mention that a small number of in vitro studies have shown that plakoglobin expression resulted in increased cell proliferation, migration and invasion [67, 68], which would be consistent with an oncogenic signaling activity of plakoglobin. It must be noted, however, that in these few cases, plakoglobin was overexpressed in cells that already contained high levels of endogenous β-catenin [69, 70]. Previous work by several groups, including ours, has shown that overexpressed plakoglobin promotes the oncogenic signaling activity of β-catenin by interacting with proteins that normally sequester β-catenin away from the nucleus [64, 71-74]. Therefore, in the few studies that determined that plakoglobin has oncogenic signaling activity, the overexpressed plakoglobin most likely sequestered β-catenin interacting partners, allowing for the liberation of β-catenin and activation of its oncogenic activity (see [18] and the following section on “Inhibition of β-catenin oncogenic signaling”).
iii. Sonic hedgehog signaling pathway
The sonic hedgehog pathway plays an important role in the proper development and patterning of the limbs, brain, musculature, skeleton and lungs, as well as in the renewal of adult stem cells and tumorigenesis [75, 76]. Gli1, a transcription factor that is activated following stimulation of the sonic hedgehog pathway, has been shown to activate the expression of plakoglobin in human rhabdomyosarcoma cells by binding to a Gli1 responsive element in the human plakoglobin gene (JUP) promoter [77]. In addition, Gli1 promotes plakoglobin expression in medulloblastoma, whereas it represses plakoglobin in glioblastoma [78, 79]. These results suggest that plakoglobin, as a target of Gli1 and the sonic hedgehog pathway, may be involved in different signaling pathways that regulate tumorigenesis and metastasis. However, it is not clear what role (if any) plakoglobin plays in the sonic hedgehog pathway.
iv. Src signaling
Experimental evidence from different groups has suggested that there is an inverse relationship between Src signaling and plakoglobin. Specifically, plakoglobin may inhibit cell migration through the regulation of Src signaling [55] and Src kinase may downregulate plakoglobin expression and/or phosphorylation, inhibiting plakoglobin’s tumor suppressor activities, thereby promoting migration [80-82]. Plakoglobin was recently shown to be present in c-Src containing lipid rafts [83] in association with the raft membrane proteins flotillins [84], which are also phosphorylated by c-Src [85]. Prior work on the role of plakoglobin in migration and invasion showed that when MCF-7 cells were treated with human growth hormone (hGH), plakoglobin levels were decreased and cell migration and invasion were increased. Interestingly, this hGH-mediated invasive phenotype was dependent on Src signaling, since chemical inhibitors of Src resulted in increased plakoglobin levels and in turn, decreased invasion and migration [86]. These observations were further confirmed by Shafiei et al. [82], who demonstrated that hGH downregulated plakoglobin expression via the activation of Src and JAK2 kinases. The authors also showed that these kinases stimulated the mRNA and protein expression of DNA methyltransferase 1 (DNMT1), DNMT3A and DNMT3B, which in turn led to the hypermethylation of the plakoglobin (JUP) promoter and its decreased expression [82]. Consistent with these in vitro studies, the GH receptor was shown to be overexpressed in both epithelial and stromal components of axillary lymph node metastasis in breast tumors. This overexpression of the receptor was also associated with decreased plakoglobin expression in nodal metastasis [87]. Another recent study using 28 non-small cell lung cancer (NSCLC) cell lines, and a combination of in vitro and in vivo mouse xenograft experiments showed a significant reduction in tumor growth in 68% of the cell lines upon combined inhibition of the Src and MAPK pathways. The combination drug treatment was shown to induce MET concurrent with the upregulation of plakoglobin and E-cadherin and downregulation of Snail1, FAK and PAX expression [88].
Finally, plakoglobin was shown to regulate cell-extracellular matrix (ECM) adhesion and motility via ECM-dependent Src activation and inhibit the migration of single keratinocyte cells by regulating the deposition of fibronectin and vitronectin, organization of the actin cytoskeleton and RhoGTPases [49, 51, 55].
v. Ras signaling
The phosphorylation of plakoglobin by a Ras-dependent pathway was initially reported by Hegland et al. [89], who showed that the expression of a dominant negative Ras (N17Ras) inhibited the increased expression of plakoglobin in confluent cultures of endothelial cells and inhibited the formation of 3-dimensional vascular structures [89]. These observations were later supported by increased levels of cadherin/catenins (α, β, γ/plakoglobin) in cultures of breast, colon and liver cancer cells treated with FTI-277, which inhibits Ras farnesylation and disrupts MAPK activation [90, 91]. Treatment with FTI-277 also led to a significant reduction in in vivo tumor growth and metastasis in SCID mice, relative to the control animals, when treated for 3 weeks following cancer cell inoculation [91]. Finally, a recent report has suggested that plakoglobin can suppress the oncogenic signaling activity of K-Ras. In this study, the expression of the oncogenic K-Ras (K-Ras12V) in Rat2 cells led to the decreased plakoglobin levels. Furthermore, decreased plakoglobin levels were accompanied by decreased levels of the histone deacetylase HDAC4, and increased cell migration and invasion. The subsequent exogenous expression of plakoglobin, but not β-catenin, in the Rat2-K-Ras12V expressing cells resulted in increased HDAC4 protein and decreased migration and invasion. The increased HDAC4 following plakoglobin expression was dependent on LEF-1, since LEF-1 knockdown in the plakoglobin expressing Rat2-K-Ras12V cells resulted in loss of HDAC4 expression [92]. At this point, however, it is not clear whether plakoglobin itself regulates the expression of HDAC4.
Role of plakoglobin in stem cells, cancer stem cells and circulating tumor cells
i. Plakoglobin and stem/cancer stem cells
A recent study has assessed the role of plakoglobin in the differentiation of mouse embryonic stem cells (mESCs) in the context of the Wnt pathway [93], which is essential for ESC pluripotency [94, 95]. This study compared Wnt stimulated β-catenin+/+ and β-catenin-/- mESCs. While Wnt stimulation in β-catenin-/- mESCs increased plakoglobin levels, the suppression of plakoglobin had no effect on Wnt targets. In contrast, ectopic overexpression of plakoglobin in wild type mESCs, strongly activated Wnt target genes, stabilized the expression of pluripotency markers Sox2, Oct4 and Nanog and inhibited mESCs differentiation. These effects were partially due to the stabilization of the transcriptionally active β-catenin owing to the overexpression of plakoglobin (also see below and Figure 3). Interestingly, it was recently shown that disabled-2 (DAB-2), a negative regulator of Wnt signaling and embryonic development, interacts with plakoglobin. It was further demonstrated that DAB-2 knockdown in mESCs had no effect on pluripotency but inhibited differentiation [96-98]. Together, these results suggest that while βcatenin is involved in the maintenance of mESC pluripotency, plakoglobin may promote mESC differentiation.
Several studies have suggested a role for plakoglobin in the maintenance of stem cell properties in the context of the Wnt/β-catenin signaling pathway and its role in hematopoiesis and hematopoietic malignancies [99, 100]. Interestingly, earlier studies showed that normal multilineage hematopoiesis was independent of β-catenin and plakoglobin [101, 102]. In contrast, subsequent numerous studies have shown the involvement of the canonical Wnt/β-catenin pathway in the regulation of hematopoietic stem cell (HSC) properties, specifically self-renewal vs. differentiation [reviewed in 103]. Both β-catenin and plakoglobin are expressed during normal hematopoietic development and in HSCs and HSPCs (hematopoietic stem progenitor cells) [73]. The same study showed that plakoglobin knockdown induced a decrease in M-CSFR (macrophage colony-stimulating factor receptor, CD115) levels and interfered with monocyte lineage differentiation. Furthermore, overexpression and nuclear localization of plakoglobin increased the expression of the transcription factor PU.1, a master regulator of myeloid differentiation and maintenance of monocyte lineage commitment. The authors also demonstrated that nuclear plakoglobin upregulated PU.1 expression via relieving PU.1 repression by β-catenin/TCF [73].
Among hematopoietic malignancies, the abnormal expression/activity of β-catenin has been well documented in leukemia (reviewed in [104]). Although increased expression of plakoglobin has also been reported, it is not clear whether plakoglobin is involved directly in leukemogenesis or indirectly via the activation of β-catenin. In acute myeloid leukemia (AML), AML-associated translocation products (AATPs) activated the Wnt signaling pathway and self-renewal of HSCs. This Wnt activation was concurrent with the upregulation of plakoglobin expression, increased β-catenin protein levels and activation of β-catenin-LEF target genes including c-Myc and cyclin D1 [105-108]. However, none of these studies examined the role of plakoglobin in the self-renewal of HSCs in the absence of β-catenin. It is well known that increased plakoglobin levels can activate β-catenin signaling in epithelial cells [71, 72]. In the case of leukemia, Niu et al. [109] also showed that knockdown of BCR-ABL in chronic myeloid leukemia (CML) cells downregulated plakoglobin expression and suppressed β-catenin by activating GSK-3β [109]. The same group also reported that Wnt5a enhanced the inhibitory effect of imatinib mesylate on CML cell growth by decreasing the levels of plakoglobin concurrent with increasing JNK and suppressing β-catenin signaling activities, respectively [74]. Yet another study by Morgan et al. [73] reported that in normal CD34+ HSPCs cells, both β-catenin and plakoglobin were expressed and distributed exclusively in the cytoplasm. In contrast, in AML patients and cell lines, plakoglobin was frequently overexpressed and its overexpression was concurrent with β-catenin stabilization and nuclear localization. This study also showed TCF-dependent transcriptional activation of plakoglobin in β-catenin knockdown cells. The authors concluded that the aberrant activation of the Wnt pathway in AML resulted from the overexpression of plakoglobin, stabilization of β-catenin, abnormal nuclear localization of both proteins and activation of the TCF-dependent transcription. The ability of plakoglobin to activate TCF-dependent transcription in the absence of β-catenin was also demonstrated by analysis of the expression of β-catenin and plakoglobin in the chronic phase (CP) and blast crisis (BC) of patients with CML. This study showed a significant negative correlation between β-catenin and plakoglobin in AP/BC cases [73]. The increased expression of plakoglobin in BC cases was concurrent with increased expression of survivin, a β-catenin-TCF-CBP target gene. Furthermore, ICG-001, a specific small molecule inhibitor of CBP, which is currently in early phase of clinical trials for various solid tumors and leukemias, was able to inhibit both β-catenin- and plakoglobin-TCF-CBP activation and survivin expression [110-113].
Together, these studies suggest that, in general, β-catenin is transcriptionally activated in leukemia. However, plakoglobin overexpression may also contribute to hematological malignancies indirectly, by promoting the nuclear localization of β-catenin and activation of β-catenin-TCF transcriptional activity. Furthermore, in the absence of β-catenin, plakoglobin can interact with TCF, activate TCF-dependent transactivation and contribute to the development of leukemia. These possibilities are supported by similar observations in epithelial cells in vitro [71,72]. However, unlike hematopoietic cells that lack junctional complexes, the overexpression of plakoglobin in epithelial cells in vivo has seldom, if ever, been reported. This is due to the sequestration of plakoglobin in adherens junctions and desmosomes via its interaction with both classic and desmosomal cadherins. In contrast, β-catenin only interacts with classic cadherins at the adherens junction and its excess (cadherin-independent pool) has to be strictly regulated by the components of the Wnt pathway. In this pathway, adenomatous polyposis coli (APC), axin, casein kinase 1 (CK1) and glycogen synthase kinase-3β (GSK-3β) form the destruction complex where the phosphorylation of β-catenin by GSK-3β and CK1 marks it for ubiquitination and degradation by the proteasome pathway. Increased β-catenin level is often observed in various carcinomas due to mutations in β-catenin itself or in the components of the Wnt pathway. In contrast, it has been reported that plakoglobin levels are generally decreased in solid tumors. The overexpressed β-catenin protein interacts with TCF and activates the expression of tumorigenic and metastatic genes. Miravet et al. [114] have shown that both proteins can simultaneously interact with TCF and the binding of plakoglobin to TCF that is already in complex with β-catenin hampers β-catenin-TCF transcriptional activity. However, in the absence of β-catenin, plakoglobin can activate TCF-dependent transcription in vitro. Thus, one can envision a scenario (Figure 3) in which β-catenin acts as the primary partner of TCF and promotes TCF transcriptional activity. When present at high levels, plakoglobin can also interact with TCF. However under these conditions, if plakoglobin associates with TCF while it is complexed with β-catenin, it will inhibit β-catenin-TCF transcriptional activation. On the other hand, if plakoglobin associates with TCF independent of β-catenin, it can activate TCF transcriptional activity, albeit at lower levels.
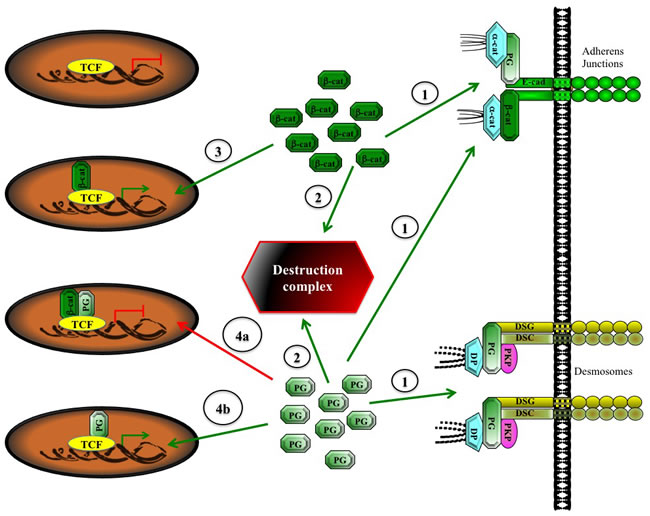
Figure 3: A Potential model of indirect and direct activation of TCF transcriptional activity by plakoglobin. Upon synthesis, β-catenin interacts with cadherins at the adherens junctions, whereas plakoglobin interacts with cadherins at the adherens junctions and desmosomes (1). The excess β-catenin and plakoglobin proteins are recruited to the destruction complex and degraded via the proteasome pathway (2). Excess cytoplasmic β-catenin can translocate to the nucleus, bind to TCF and change its activity from a repressor to an activator (3). The excess cytoplasmic plakoglobin can take one of several fates, depending on the level of β-catenin and its nuclear presence. If the excess plakoglobin remains in the cytoplasm, it can indirectly activate TCF by replacing β-catenin in adherens junctions, allowing for the interaction of the liberated β-catenin with TCF and its transcriptional activation (3). Alternatively, if the excess plakoglobin translocates to the nucleus, it can suppress β-catenin-TCF activation (4a), or in the absence of nuclear β-catenin, it can interact with TCF and directly lead to its transcriptional activation (4b). α-cat, α-catenin; β-cat, β-catenin; E-cad, E-cadherin; PG, plakoglobin, DSG, desmoglein, DSC, desmocollin; PKP, plakophillin, DP, desmoplakin; TCF, T-cell factor.
ii. Plakoglobin and circulating tumor cells
Circulating tumor cells (CTC) are cells that dissociate from primary tumors, enter the circulation and form the seeds for subsequent metastatic growth in distant locations [115, 116]. CTCs are present as single cells or clusters. CTCs are known to have cancer stem cell properties and their presence has been correlated with poor prognosis in various solid tumors [117-119]. The role of cell-cell adhesion proteins E-cadherin, β-catenin and plakoglobin in CTCs has been described in the context of cell-cell adhesion and its role in cell survival. Several studies have shown the overexpression of the E-cadherin/α-catenin/β-catenin complex in lymphovascular emboli (CTC clusters) of inflammatory breast cancers [120-122]. These emboli, known as lymphovascular invasion (LVI), were formed due to the decreased sialyl-Lewis X/A carbohydrate ligand-binding epitopes on MUC1 overexpressed in CTC clusters, distancing them from the endothelial cell layer. The overexpression of the E-cadherin/catenin complex was shown to be necessary for the compaction and maintenance of these clusters [120-122]. E-cadherin overexpression has also been reported in small cell lung carcinoma (SCLC) CTC cell lines and cells isolated from pleural effusion and bone metastases [123]. In breast cancer cells, using xenograft mouse models with tagged mammary tumors, Aceto et al. [124] showed that CTC clusters were multi-clonal collections of 20-50 tumors cells that exhibited significantly higher metastatic potential relative to single CTCs. Additionally, the clusters originated from tumors and not as a result of cell aggregation in the circulation. In this study, single cell RNA sequencing of CTCs and CTC clusters from blood samples of the same patient showed high expression of plakoglobin in the CTC clusters. In addition, the metastasis free survival was significantly lower in patients with primary tumors expressing high plakoglobin [124]. While the authors did not examine the protein levels and distribution of E-cadherin, α/β-catenin or desmosomal cadherins, they suggested that by virtue of its role both in the adherens junctions and desmosomes, high plakoglobin expression increased the stability of CTC clusters as they entered the circulation. In a subsequent report analyzing an open access database of breast cancer patients, Lu et al. [125] found that patients with low plakoglobin expression showed significantly better distant metastasis free and overall survival although they detected very little difference in relapse free survival between patients with high or low/medium plakoglobin expression.
In contrast to the above studies, genetic profiling of tumors from breast cancer patient with (BM+) or without (BM-) bone marrow metastasis has identified a 3-fold reduction in plakoglobin expression in primary tumors from BM+ relative to BM- patients [126]. In another study, using MCF-7 and T47D breast carcinoma cell lines in a combination of in vitro and xenograft mouse models, Holen et al. [54] showed increased cell proliferation, migration and invasion in plakoglobin knockdown cells. They also detected a significantly high number of CTCs in the blood of mice bearing plakoglobin knockdown tumors. In yet another study, PC3 prostate cancer cells were used in an orthotopic mouse model of castration-resistant prostate cancer to generate CTC cell lines (PC3-CTC). PC3-CTCs isolated from the blood of these mice were then compared to PC3 cells with respect to their survival in suspension and their adhesive properties and resistance to anoikis. The results showed decreased E-cadherin, plakoglobin and β4-integrin and increased Bcl-2 expression in PC3-CTCs concurrent with decreased adhesion and increased resistance to anoikis [127].
Collectively, these studies suggest that decreased plakoglobin expression is associated with the shedding of tumor cells in circulation, consistent with its low expression in single CTCs. However, in CTC clusters, the upregulation of plakoglobin, E-cadherin and catenins may increase cell-cell adhesion and provide a survival advantage by stabilizing the clusters as they enter the circulation and are transported to distant locations. In support of this possibility, a recent study has shown that CD133 (prominin-1), a marker of stem cell and CSCs [128, 129], interacted with plakoglobin in ovarian clear cell carcinoma CSCs [130]. The authors showed that the knockdown of CD133 or plakoglobin led to the decreased adhesion and dissociation of CSC clusters. This study did not examine if CD133 interacted with plakoglobin directly or whether this interaction occurred at the adherens junctions or desmosomes.
Plakoglobin tumor and metastasis suppressor activity
i. Overview
Despite the observations that plakoglobin expression induces MET and the suggestion that plakoglobin suppresses tumorigenesis and metastasis, the mechanisms that could account for these activities have remained, until recently, unclear. A number of recent studies have shown plakoglobin’s interaction with an array of intracellular proteins that are either directly or indirectly involved in signaling pathways that regulate tumorigenesis and metastasis (Table 1). Experimental evidence from several groups, including our own, has suggested that there are different mechanisms by which plakoglobin may act as a tumor and metastasis suppressor (Figure 4). First, plakoglobin may compete with β-catenin signaling by inhibiting TCF/β-catenin-DNA interactions and Wnt target genes expression. Second, plakoglobin may interact with various cellular partners involved in signaling and alter their levels, localization and/or functions. Finally, plakoglobin may interact with transcription factors and regulate gene expression independent of β-catenin.
In the following sections, we will discuss the experimental evidence which suggests that plakoglobin acts as a tumor and metastasis suppressor both in the cytoplasm and nucleus, with specific emphasis on the mechanisms by which plakoglobin may accomplish such activities.
Table 1: Plakoglobin intracellular protein partners*
Interacting partner |
Function |
E-/N-/VE-cadherin, PECAM |
|
α-catenin |
Adherens junction [164] |
Desmosomal cadherins, desmogleins (DSG) and desmocollins (DSC) |
|
Desmoplakins |
Desmosomes [168] |
Plakophillins |
|
CPI-17 (PKC-potentiated inhibitory protein of protein phosphatase-1) |
Endothelial junction/barrier function [142] |
Emmprin (Basigin, CD147) |
Endothelial junctions [141] |
PrP (c)-cellular prion protein |
Cell-cell adhesion/barrier function and proliferation [83] |
Flotillins |
Cell-cell adhesion; endocytosis [84] |
MUC1 (mucin1) |
Adhesion, Signaling [172] |
14-3-3γ |
Adaptor protein involved in signaling [173] |
APC |
Tumor suppressor/Wnt signaling [38] |
Axin |
Scaffolding protein/ Wnt signaling [39] |
NPM (nucleophosmin) |
Nuclear proteins chaperone [56] |
Nm23 |
Tumor/metastasis suppressor [53] |
TCF/LEF |
Transcription factor/Wnt signaling [114] |
SOX4 |
Transcription factor [131] |
CBP |
Transcription factor [111] |
p53 |
Transcription factor-tumor/metastasis suppressor [57] |
Presenilin-1 |
Cell-cell adhesion, signaling [174] |
CD133 (prominin-1) |
Cell differentiation, proliferation, apoptosis [130] |
Disabled-2 (Adapter protein that functions as a clathrin-associated sorting protein) |
Cell-cell adhesion, ESC differentiation, Clathrin mediated endocytosis, signaling [98] |
InsR (Insulin receptor) |
|
PI3K-p85- Phosphatidylinositol 3-kinase regulatory subunit alpha |
|
YAP (Yes-associated protein) |
Transcriptional regulator and downstream effector of the Hippo signaling pathway [177] |
* The list includes proteins that have been shown to interact with plakoglobin by co-immunoprecipitation and/or pull down assays. The cited references represent the initial report for each case only. The known functional significance of plakoglobin interaction with each partner is discussed throughout the text.
** Numbers in parentheses indicate references.
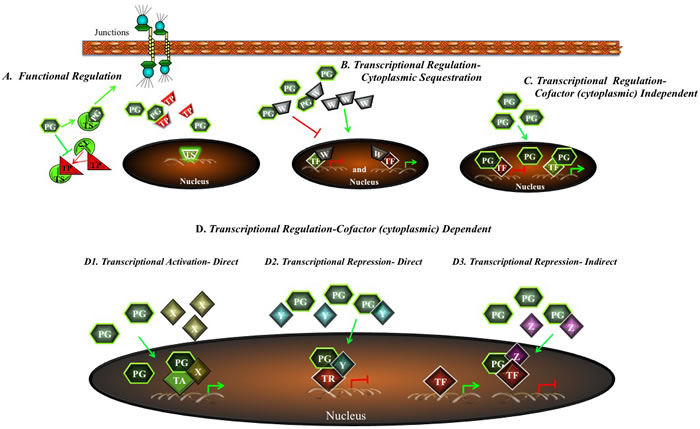
Figure 4: A potential model for regulation of tumorigenesis and metastasis by plakoglobin. Plakoglobin may regulate tumor progression and metastasis both in the cytoplasm and in the nucleus. A. First, plakoglobin may interact with intracellular protein partners involved in tumorigenesis and metastasis and alter their levels, localization or function. It may also regulate gene expression via three concurrent mechanisms: B. Cytoplasmic Sequestration: plakoglobin sequesters a factor (W) in the cytoplasm which, in the nucleus, suppresses the expression of a tumor suppressor gene or activates the expression of an oncogene. C. Cytoplasmic Cofactor Independent: plakoglobin-transcription factor complexes promote the expression of tumor suppressor genes and repress the expression of oncogenes. D. Cytoplasmic Cofactor Dependent: plakoglobin interacts with a cytoplasmic cofactor (X, Y, Z) and this complex moves into the nucleus where it activates tumor suppressor gene expression or represses oncogenic gene expression. PG, plakoglobin; TS, tumor suppressor, TP, tumor promoter; TF, transcription factor.
ii. Inhibition of β-catenin oncogenic signaling
The oncogenic potential of β-catenin and the Wnt signaling pathway has been well- studied and characterized [19, 42, 59]. Following Wnt pathway activation, β-catenin translocates into the nucleus, interacts with TCF/LEF transcription factors and activates the expression of genes that promote cell survival, growth and migration (e.g. Myc, cyclin D1, MMPs, etc.; [19, 42, 59]). Interestingly, one mechanism by which plakoglobin suppresses tumorigenesis is through the inactivation of β-catenin oncogenic activity. Indeed, it was previously shown that in plakoglobin null cell lines, plakoglobin expression resulted in β-catenin displacement from the junctional complexes and its rapid proteasome-mediated degradation [71]. Similarly, we previously showed that expression of plakoglobin in SCC9 cells lacking endogenous plakoglobin expression resulted in decreased β-catenin protein levels and an induction of MET [30].
In addition to the decreasing the levels of β-catenin, plakoglobin expression can also inhibit the signaling activity of β-catenin. Comparison of the nuclear translocation and transactivation abilities of β-catenin and plakoglobin showed a highly efficient formation of β-catenin -TCF complexes, their interactions with DNA and transactivation, while plakoglobin-TCF interactions were formed inefficiently, with significantly weaker signaling activities [64]. Further analysis of the domains of the TCF/LEF transcription factors that are involved in interactions with plakoglobin and β-catenin revealed that plakoglobin and β-catenin bind to consecutive, non-overlapping regions of the TCF-4 protein. Specifically, as mentioned earlier, β-catenin bound to amino acids 1-50 in TCF-4 whereas plakoglobin bound to amino acids 51-80. Additionally, this study showed that plakoglobin can form a complex with TCF-4 and β-catenin, but that plakoglobin inhibits the ability of this complex and TCF-4 alone to interact with DNA [114]. In agreement with these findings, we previously showed that the exclusively nuclear expression of plakoglobin in SCC9 cells resulted in decreased β-catenin -TCF nuclear interactions and in turn, decreased β-catenin oncogenic signaling [72].
Plakoglobin also inhibits Wnt/β-catenin signaling through its interactions with SOX4, a transcription factor that promotes the signaling activity of β-catenin [131]. This study showed that in prostate cancer cells stimulated with Wnt3a, plakoglobin interacted with SOX4 and exported it into the cytoplasm. Furthermore, the formation of the SOX4-plakoglobin complex decreased the expression of Wnt/β-catenin target genes (e.g. Dicer, Axin2). Interestingly, in an earlier study, Zorn et al. [132] demonstrated that in Xenopus embryos, the ectopic expression of XSOX17α, XSOX17β and XSOX3 ventralized embryos by inhibiting the Wnt pathway. They further showed that all three XSOX proteins bind to β-catenin via Armadillo repeats 3-6, that overlap with the TCF binding site (Armadillo repeats 4-9) on β-catenin [132]. Since plakoglobin is able to interact with SOX4, it may be possible that it also interacts with other SOX proteins, thereby inhibiting Wnt signaling. Finally, a recent report demonstrated that knockdown of desmoglein 3 in head and neck cancer cell lines resulted in increased nuclear plakoglobin levels and its interactions with TCF-4, that led to decreased TOPFLASH reporter activity and downregulation of β-catenin -TCF target genes (e.g. c-Myc, cyclin D1, MMP7; [133]). The inhibition of β-catenin-TCF-4 transcriptional activity by plakoglobin was also demonstrated in a recent study that reported the interaction of both catenins with the cellular prion protein PrP(c) [83, 134,135]. PrP(c) was shown to interact with actin, spectrin, annexin A2, plakoglobin, desmoglein 2, desmoplakin and c-Src in epithelial cells. In polarized epithelial cells, PrP(c) showed primarily membranous/junctional distribution, whereas in actively proliferating cells it was localized to the nucleus [83]. This study also detected a pool of PrP(c) in lipid rafts in association with desmoglein 2 and c-Src. Subsequent studies by the same group showed PrP(c) participated in Hippo and Wnt signaling pathways. PrP(c) interacted not only with plakoglobin but also with β-catenin and TCF-4 in both the cytoplasm and nucleus. Futhermore, the interaction of PrP(c) with β-catenin-TCF-4 stimulated its transactivation, whereas PrP(c)-plakoglobin interactions decreased it [134, 135].
iii. Intracellular interactions
Plakoglobin may act as a tumor and metastasis suppressor through its interactions with various intracellular partners, thereby altering their levels, localization or function. In support of this scenario, previous work using Xenopus embryos showed that plakoglobin protein sequestered exogenously-expressed TCF proteins in the cytoplasm, resulting in a decrease in TOPFLASH reporter activity [41].
We recently showed that plakoglobin interacted with nucleophosmin (NPM; [56]), a nucleolar phosphoprotein whose role in tumorigenesis is largely dependent on its subcellular distribution [136, 137]. We showed that plakoglobin expression in MDA-MB-231 cells resulted in increased NPM protein levels and its redistribution from the cytoplasm and nucleoplasm, where it is thought to function as an oncogene, into the nucleolus, where it is typically localized in untransformed cells [56, 136-138]. Therefore, plakoglobin, through its interactions with NPM, altered NPM protein levels and localization concurrent with the decreased growth, invasive and migratory properties of MDA-MB-231-PG cells [56].
In addition, we previously showed that plakoglobin expression in SCC9 and MDA-MB-231 cells resulted in increased protein levels of the metastasis suppressors Nm23-H1 and -H2 [53, 58]. Following expression, plakoglobin interacted with both Nm23-H1 and -H2 leading to a change in the distribution of a significant proportion of Nm23 from cytoplasmic to membrane-associated. Our results suggested that plakoglobin increased the levels of Nm23 proteins via its interactions with Nm23, which potentially led to increased protein stability. A number of subsequent studies showed a critical role for Nm23 in cell-cell adhesion. Nm23 knockdown in hepatoma and colon carcinoma cells led to adherens junction dissociation, β-catenin nuclear translocation, β-catenin-TCF transactivation and upregulation of MMPs, concurrent with increased cell motility and migration [139]. Nm23 was shown to play critical roles in regulating permeability and barrier function of endothelial monolayers [140] via interacting with plakoglobin and EMMPRIN in the stabilization of endothelial junctions [141]. This study further suggested that EMMPRIN-plakoglobin interactions regulated actomyosin-dependent forces that were required for junction formation. Interestingly, plakoglobin also has been shown to interact with CPI-17, which inhibits MLC phosphatase [142]. Together, these studies point to a role for Nm23 and plakoglobin in migration inhibition and endothelial junction stability with implications for metastasis and angiogenesis.
iv. Regulation of gene expression
Plakoglobin has been shown to be a repressor of the c-Myc (MYC) gene. In mouse keratinocytes, plakoglobin suppressed MYC expression in a LEF-1 dependent manner, suggesting that when plakoglobin interacted with LEF-1, this complex was unable to promote gene expression [143]. Furthermore, the plakoglobin-mediated suppression of MYC was similar in both wild type and β-catenin-null keratinocytes, demonstrating that plakoglobin could regulate gene expression independent of β-catenin. Finally, chromatin immunoprecipitation experiments with plakoglobin antibodies demonstrated that plakoglobin and LEF-1 associated with the MYC promoter in keratinocytes undergoing growth arrest, suggesting that the downregulation of c-Myc gene expression was a possible reason for the suppression of cell growth by plakoglobin [143].
Further evidence of a role for plakoglobin in the regulation of gene expression came from a recent study, which demonstrated that plakoglobin regulated the expression of the desmosomal cadherin desmocollin-2 in keratinocytes through interactions with LEF-1 [144]. The plakoglobin-mediated activation of the desmocollin-2 gene (DSC2) promoter was dependent on a functional LEF-1 binding site.
To identify potential plakoglobin target genes, proteins and mechanisms by which plakoglobin may regulate gene expression and in turn, tumorigenesis and metastasis, we performed proteomic and microarray experiments using plakoglobin-null cell lines and their plakoglobin-expressing transfectants. Various transfectants with different levels of expression and subcellular localization of plakoglobin were developed to specifically assess its role at the membrane, in the cytoplasm and in the nucleus [53, 72, 145]. Detailed analyses of these studies have suggested that plakoglobin may regulate gene expression by multiple mechanisms, either in the cytoplasm, through the sequestration of factors involved in regulating gene expression, or in the nucleus, by promoting and/or repressing gene expression itself (Figure 4; also see [18] for details). In the microarray experiments, we identified several p53 target genes that were differentially expressed following plakoglobin expression in various cell lines, suggesting that plakoglobin and p53 may coordinately regulate gene expression. Subsequently, we showed that plakoglobin interacted with p53 both in the cytoplasm and the nucleus, and this interaction was mediated by the DNA-binding domain of p53 and C-terminal transactivation domain of plakoglobin [57, 146]. Using transfectants expressing wild type and various fragments and deletions of p53 and plakoglobin, we demonstrated that wild type p53 and plakoglobin cooperated to decrease cell growth and acted synergistically to reduce migration and invasion [146]. We also have shown that together, p53 and plakoglobin regulated the expression of a number of p53 target genes, including the tumor suppressors 14-3-3σ and Nm23-H1, and the tumor promoter SATB1 [57, 58]. Using subcellular fractionation in conjunction with coimmunoprecipitation experiments, we showed that plakoglobin interacted with both wild type and mutant p53. Chromatin immunoprecipitation experiments showed that the two proteins also associated with the promoter of the SFN gene (which encodes 14-3-3σ). In addition, we showed that mutant p53 protein only associated with its target gene promoters in the presence of plakoglobin. Furthermore, using luciferase reporter assays, we showed that the transcriptional activity of both wild type and mutant p53 proteins was enhanced in the presence of plakoglobin [57]. Consistent with our observations, it was previously shown that plakoglobin promotes the expression of the tumor suppressor PML, a known p53 target gene [147]. More recently, Sechler et al. [148] showed that plakoglobin induced the expression of hepatocyte growth factor activator inhibitor Type I (HAI-1), an upstream inhibitor of c-met, in a p53-dependent manner, to reduce migration in NSCLC cells [148].
We have observed that plakoglobin interacts with a number of different mutant p53 proteins in various cell lines (e.g. SCC9, A431, MDA-231, ES-2; [57, 58, 146, 149]). Furthermore, plakoglobin appeared to help mutant p53 associate with its target gene promoters- an association that is absent when plakoglobin is not expressed. As such, our data suggests that plakoglobin may help promote the wild type tumor suppressor activities of mutant p53 proteins that have otherwise lost these activities, with the end result of a decreased transformed cell phenotype. This is extremely important when considering that approximately 50% of all tumors express a mutant p53 protein [150-153].
We have shown that plakoglobin and p53 associated with and repressed expression from the SATB1 promoter, which encodes SATB1 protein, the oncogenic chromatin remodeling factor [58; 154]. We have also shown that a number of SATB1 target genes were differentially expressed following plakoglobin knockdown in MCF-7 cells [58]. Furthermore, using chromatin immunoprecipitation experiments and luciferase reporter assays we have found that plakoglobin and p53 interacted with and promoted expression from the NME1 promoter, which encodes the metastasis suppressor Nm23-H1 [58]. Our findings are consistent with a previous report that showed Nm23-H1 mRNA levels were decreased following plakoglobin knockdown in breast cancer cells [54]. We further showed that breast, squamous cell, ovarian and non-small cell lung carcinomas that expressed plakoglobin were less proliferative, migratory and invasive compared to those that did not express plakoglobin [29, 58, 146, 149]. These results are in agreement with other studies that have demonstrated that plakoglobin expressing cells are considerably less proliferative and migratory than their non-plakoglobin expressing counterparts [30, 44-52, 54, 55, 155-157]. More importantly, our data also suggests that the plakoglobin-p53 complexes are both activating (in the case of the SFN and NME1 genes) and repressive (SATB1 gene). A summary of our results is shown in Figure 5.
Finally, as mentioned earlier, Todorovic et al. [51] previously showed that plakoglobin regulates cell motility by regulating fibronectin and Rho-dependent Src signaling. In this study, plakoglobin expression also resulted in increased levels of fibronectin mRNA due to its increased stability, as determined by the use of the transcription inhibitor Actinomycin D [51]. This finding suggests that in addition to its role in regulating gene expression at the level of transcription, plakoglobin may also regulate gene expression post-transcriptionally. However, how plakoglobin does so remains unclear.
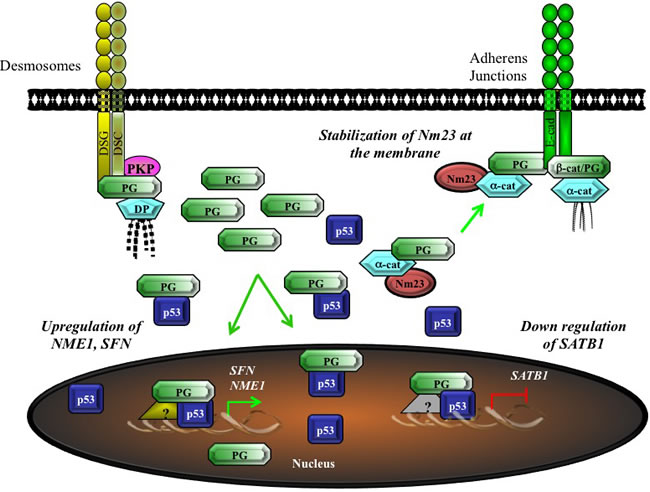
Figure 5: Potential model for the tumor/metastasis suppressor activity of plakoglobin via the regulation of gene expression. Cytoplasmic, non-junctional plakoglobin may regulate tumorigenesis and metastasis by multiple mechanisms. Plakoglobin interacts with the metastasis suppressor Nm23 and increases its protein levels and localization at the membrane. Plakoglobin also interacts with the transcription factor p53 and promotes the expression of tumor and metastasis suppressor genes (e.g. NME1, SFN) and suppresses the expression of oncogenes (e.g. SATB1). α-cat, α-catenin; β-cat, β-catenin; DP, desmoplakin; Nm23, nonmetastatic protein 23; PG, plakoglobin; SFN, stratifin.
Concluding Remarks
Following its initial discovery and early characterization, plakoglobin was primarily regarded as a protein whose function was limited to maintaining proper cell-cell adhesion. However, recent findings have clearly pointed to a more active role for plakoglobin in the regulation of tumorigenesis and metastasis through multiple mechanisms, including the regulation of gene expression. While characterizing the signaling function of plakoglobin in the absence of β-catenin has been a complicated endeavor, there is now ample evidence supporting the role of plakoglobin as an important player in tumorigenesis and metastasis. Plakoglobin interaction with p53 and its apparent ability to restore the wild type transcriptional activity of mutant p53 proteins may have significant therapeutic implications. The identification of the domains in plakoglobin that mediate its interactions with p53 can be used to design small therapeutic peptides/drugs that mimic these interactions and reactivate the wild type activity of mutant p53. These peptides/drugs may potentially have high, specific potency but avoid the low efficacy and toxicity of drugs that have not been derived from a naturally occurring cellular protein. This is particularly important since 50% of all tumors and >80% of metastatic tumors have non-functional p53 and the reactivation of p53 is currently being actively explored as a potentially effective therapeutic intervention in the treatment of various cancers.
Abbreviations
AML-Acute myeloid leukemia; APC-Adenomatous polyposis coli protein; CML-Chronic myeloid leukemia; CTC-Circulating tumor cell; DNMT-DNA methyltransferase; EMT-Epithelial to mesenchymal phenotypic transition; ESC-Embryonic stem cell; HAI-1-Hepatocyte growth factor activator inhibitor Type I; HDAC-Histone deacetylase; hGH-Human growth hormone; HSC-hematopoietic stem cell; HSPC-hematopoietic stem progenitor cell; JUP-Junction plakoglobin; LEF-Lymphoid enhancer factor; M-CSFR-Macrophage colony-stimulating factor receptor; MET-Mesenchymal to epithelial phenotypic transition; Nm23-Nonmetastatic protein 23; NPM-Nucleophosmin; NSCLC-Non-small cell lung carcinoma; PrP(c)-Cellular prion protein; SFN-Stratifin (14-3-3σ); TCF-T-cell factor; TOPFLASH-TCF luciferase reporter vector.
Acknowledgments
The authors apologize to any colleagues whose work they may have overlooked during the preparation of this paper. Work in the authors’ lab has been/is supported by grants from the Canadian Breast Cancer Foundation (Prairies/NWT Chapter), Alberta Cancer Foundation and Graduate Fellowships from Canadian Institute of Health Research, Killam Foundation and University of Alberta Dissertation Award (ZA) and the University of Alberta Faculty of Medicine and Dentistry 75th Anniversary Award and the Cathy and Harold Roozen Entrance Scholarship, Department of Oncology, University of Alberta (MA). We thank Dr. Ellen Shibuya for helpful comments and discussions.
ConflictS of Interest
There is no conflict of interest.
References
1. McCaffrey LM, Macara IG. Epithelial organization, cell polarity and tumorigenesis. Trends Cell Biol. 2011; 21:727–35. doi: 10.1016/j.tcb.2011.06.005.
2. Makrilia N, Kollias A, Manolopoulos L, Syrigos K. Cell adhesion molecules: role and clinical significance in cancer. Cancer Invest. 2009; 27:1023–37. doi: 10.3109/07357900902769749.
3. Yilmaz M, Christofori G. Mechanisms of motility in metastasizing cells. Mol Cancer Res. 2010; 8:629–42. doi: 10.1158/1541-7786.MCR-10-0139.
4. Saito M, Tucker DK, Kohlhorst D, Niessen CM, Kowalczyk AP. Classical and desmosomal cadherins at a glance. J Cell Sci. 2012; 125:2547–52. doi: 10.1242/jcs.066654.
5. David JM, Rajasekaran AK. Dishonorable discharge: the oncogenic roles of cleaved E-cadherin fragments. Cancer Res. 2012; 72:2917–23. doi: 10.1158/0008-5472.CAN-11-3498.
6. Yonemura S. Cadherin-actin interactions at adherens junctions. Curr Opin Cell Biol. 2011; 23:515–22. doi: 10.1016/j.ceb.2011.07.001.
7. Al-Amoudi A, Castaño-Diez D, Devos DP, Russell RB, Johnson GT, Frangakis AS. The three-dimensional molecular structure of the desmosomal plaque. Proc Natl Acad Sci USA. 2011; 108:6480–85. doi: 10.1073/pnas.1019469108.
8. Kowalczyk AP, Green KJ. Structure, function, and regulation of desmosomes. Prog Mol Biol Transl Sci. 2013; 116:95–118. doi: 10.1016/B978-0-12-394311-8.00005-4.
9. Harris TJ, Tepass U. Adherens junctions: from molecules to morphogenesis. Nat Rev Mol Cell Biol. 2010; 11:502–14. doi: 10.1038/nrm2927.
10. Brieher WM, Yap AS. Cadherin junctions and their cytoskeleton(s). Curr Opin Cell Biol. 2013; 25:39–46. doi: 10.1016/j.ceb.2012.10.010.
11. Dusek RL, Attardi LD. Desmosomes: new perpetrators in tumour suppression. Nat Rev Cancer. 2011; 11:317–23. doi: 10.1038/nrc3051.
12. Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. 2004; 23:1739–48. doi: 10.1038/sj.emboj.7600136.
13. Lilien J, Balsamo J. The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta-catenin. Curr Opin Cell Biol. 2005; 17:459–65. doi: 10.1016/j.ceb.2005.08.009.
14. Erez N, Bershadsky A, Geiger B. Signaling from adherens-type junctions. Eur J Cell Biol. 2005; 84:235–44. doi: 10.1016/j.ejcb.2004.12.007.
15. Hatzfeld M. Plakophilins: multifunctional proteins or just regulators of desmosomal adhesion? Biochim Biophys Acta. 2007; 1773:69–77. doi: 10.1016/j.bbamcr.2006.04.009.
16. Watanabe T, Sato K, Kaibuchi K. Cadherin-mediated intercellular adhesion and signaling cascades involving small GTPases. Cold Spring Harb Perspect Biol. 2009; 1:a003020. doi: 10.1101/cshperspect.a003020.
17. Swope D, Li J, Radice GL. Beyond cell adhesion: the role of armadillo proteins in the heart. Cell Signal. 2013; 25:93–100. doi: 10.1016/j.cellsig.2012.09.025.
18. Aktary Z, Pasdar M. Plakoglobin: role in tumorigenesis and metastasis. Int J Cell Biol. 2012; 2012:189521. doi: 10.1155/2012/189521.
19. Kim W, Kim M, Jho EH. Wnt/β-catenin signalling: from plasma membrane to nucleus. Biochem J. 2013; 450:9–21. doi: 10.1042/BJ20121284.
20. Miller RK, Hong JY, Muñoz WA, McCrea PD. Beta-catenin versus the other armadillo catenins: assessing our current view of canonical Wnt signaling. Prog Mol Biol Transl Sci. 2013; 116:387–407. doi: 10.1016/B978-0-12-394311-8.00017-0.
21. Franke WW, Mueller H, Mittnacht S, Kapprell HP, Jorcano JL. Significance of two desmosome plaque-associated polypeptides of molecular weights 75 000 and 83 000. EMBO J. 1983; 2:2211–15.
22. Cowin P, Kapprell HP, Franke WW, Tamkun J, Hynes RO. Plakoglobin: a protein common to different kinds of intercellular adhering junctions. Cell. 1986; 46:1063–73. doi: 10.1016/0092-8674(86)90706-3.
23. Korman NJ, Eyre RW, Klaus-Kovtun V, Stanley JR. Demonstration of an adhering-junction molecule (plakoglobin) in the autoantigens of pemphigus foliaceus and pemphigus vulgaris. N Engl J Med. 1989; 321:631–35. doi: 10.1056/NEJM198909073211002.
24. Vestweber D, Kemler R. Some structural and functional aspects of the cell adhesion molecule uvomorulin. Cell Differ. 1984; 15:269–73. doi: 10.1016/0045-6039(84)90084-8.
25. Ozawa M, Kemler R. Molecular organization of the uvomorulin-catenin complex. J Cell Biol. 1992; 116:989–96. doi: 10.1083/jcb.116.4.989.
26. Pasdar M, Li Z, Chlumecky V. Plakoglobin: kinetics of synthesis, phosphorylation, stability, and interactions with desmoglein and E-cadherin. Cell Motil Cytoskeleton. 1995; 32:258–72. doi: 10.1002/cm.970320403.
27. Navarro P, Lozano E, Cano A. Expression of E- or P-cadherin is not sufficient to modify the morphology and the tumorigenic behavior of murine spindle carcinoma cells. Possible involvement of plakoglobin. J Cell Sci. 1993; 105:923–34.
28. Lewis JE, Wahl JK 3rd, Sass KM, Jensen PJ, Johnson KR, Wheelock MJ. Cross-talk between adherens junctions and desmosomes depends on plakoglobin. J Cell Biol. 1997; 136:919–34. doi: 10.1083/jcb.136.4.919.
29. Parker HR, Li Z, Sheinin H, Lauzon G, Pasdar M. Plakoglobin induces desmosome formation and epidermoid phenotype in N-cadherin-expressing squamous carcinoma cells deficient in plakoglobin and E-cadherin. Cell Motil Cytoskeleton. 1998; 40:87–100. doi: 10.1002/(SICI)1097-0169(1998)40:1<87::AID-CM8>3.0.CO;2-C.
30. Li Z, Gallin WJ, Lauzon G, Pasdar M. L-CAM expression induces fibroblast-epidermoid transition in squamous carcinoma cells and down-regulates the endogenous N-cadherin. J Cell Sci. 1998; 111:1005–19.
31. Palka HL, Green KJ. Roles of plakoglobin end domains in desmosome assembly. J Cell Sci. 1997; 110:2359–71.
32. Acehan D, Petzold C, Gumper I, Sabatini DD, Müller EJ, Cowin P, Stokes DL. Plakoglobin is required for effective intermediate filament anchorage to desmosomes. J Invest Dermatol. 2008; 128:2665–75. doi: 10.1038/jid.2008.141.
33. Bierkamp C, Mclaughlin KJ, Schwarz H, Huber O, Kemler R. Embryonic heart and skin defects in mice lacking plakoglobin. Dev Biol. 1996; 180:780–85. doi: 10.1006/dbio.1996.0346.
34. Ruiz P, Brinkmann V, Ledermann B, Behrend M, Grund C, Thalhammer C, Vogel F, Birchmeier C, Günthert U, Franke WW, Birchmeier W. Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J Cell Biol. 1996; 135:215–25. doi: 10.1083/jcb.135.1.215.
35. Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R. Lack of beta-catenin affects mouse development at gastrulation. Development. 1995; 121:3529–37.
36. Peifer M, McCrea PD, Green KJ, Wieschaus E, Gumbiner BM. The vertebrate adhesive junction proteins beta-catenin and plakoglobin and the Drosophila segment polarity gene armadillo form a multigene family with similar properties. J Cell Biol. 1992; 118:681–91. doi: 10.1083/jcb.118.3.681.
37. Butz S, Stappert J, Weissig H, Kemler R. Plakoglobin and beta-catenin: distinct but closely related. Science. 1992; 257:1142–44. doi: 10.1126/science.257.5073.1142-a.
38. Shibata T, Gotoh M, Ochiai A, Hirohashi S. Association of plakoglobin with APC, a tumor suppressor gene product, and its regulation by tyrosine phosphorylation. Biochem Biophys Res Commun. 1994; 203:519–22. doi: 10.1006/bbrc.1994.2213.
39. Kodama S, Ikeda S, Asahara T, Kishida M, Kikuchi A. Axin directly interacts with plakoglobin and regulates its stability. J Biol Chem. 1999; 274:27682–88. doi: 10.1074/jbc.274.39.27682.
40. Zhurinsky J, Shtutman M, Ben-Ze’ev A. Plakoglobin and beta-catenin: protein interactions, regulation and biological roles. J Cell Sci. 2000; 113:3127–39.
41. Williams BO, Barish GD, Klymkowsky MW, Varmus HE. A comparative evaluation of beta-catenin and plakoglobin signaling activity. Oncogene. 2000; 19:5720–28. doi: 10.1038/sj.onc.1203921.
42. Jamieson C, Sharma M, Henderson BR. Targeting the β-catenin nuclear transport pathway in cancer. Semin Cancer Biol. 2014; 27:20–29. doi: 10.1016/j.semcancer.2014.04.012.
43. Anastas JN. Functional Crosstalk Between WNT Signaling and Tyrosine Kinase Signaling in Cancer. Semin Oncol. 2015; 42:820–31. doi: 10.1053/j.seminoncol.2015.09.020.
44. Simcha I, Geiger B, Yehuda-Levenberg S, Salomon D, Ben-Ze’ev A. Suppression of tumorigenicity by plakoglobin: an augmenting effect of N-cadherin. J Cell Biol. 1996; 133:199–209. doi: 10.1083/jcb.133.1.199.
45. Pantel K, Passlick B, Vogt J, Stosiek P, Angstwurm M, Seen-Hibler R, Häussinger K, Thetter O, Izbicki JR, Riethmüller G. Reduced expression of plakoglobin indicates an unfavorable prognosis in subsets of patients with non-small-cell lung cancer. J Clin Oncol. 1998; 16:1407–13. doi: 10.1200/JCO.1998.16.4.1407.
46. Charpentier E, Lavker RM, Acquista E, Cowin P. Plakoglobin suppresses epithelial proliferation and hair growth in vivo. J Cell Biol. 2000; 149:503–20. doi: 10.1083/jcb.149.2.503.
47. Winn RA, Bremnes RM, Bemis L, Franklin WA, Miller YE, Cool C, Heasley LE. gamma-Catenin expression is reduced or absent in a subset of human lung cancers and re-expression inhibits transformed cell growth. Oncogene. 2002; 21:7497–506. doi: 10.1038/sj.onc.1205963.
48. Rieger-Christ KM, Ng L, Hanley RS, Durrani O, Ma H, Yee AS, Libertino JA, Summerhayes IC. Restoration of plakoglobin expression in bladder carcinoma cell lines suppresses cell migration and tumorigenic potential. Br J Cancer. 2005; 92:2153–59. doi: 10.1038/sj.bjc.6602651.
49. Yin T, Getsios S, Caldelari R, Kowalczyk AP, Müller EJ, Jones JC, Green KJ. Plakoglobin suppresses keratinocyte motility through both cell-cell adhesion-dependent and -independent mechanisms. Proc Natl Acad Sci USA. 2005; 102:5420–25. doi: 10.1073/pnas.0501676102.
50. Närkiö-Mäkelä M, Pukkila M, Lagerstedt E, Virtaniemi J, Pirinen R, Johansson R, Kosunen A, Lappalainen K, Hämäläinen K, Kosma VM. Reduced gamma-catenin expression and poor survival in oral squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 2009; 135:1035–40. doi: 10.1001/archoto.2009.132.
51. Todorović V, Desai BV, Patterson MJ, Amargo EV, Dubash AD, Yin T, Jones JC, Green KJ. Plakoglobin regulates cell motility through Rho- and fibronectin-dependent Src signaling. J Cell Sci. 2010; 123:3576–86. doi: 10.1242/jcs.070391.
52. Bailey CK, Mittal MK, Misra S, Chaudhuri G. High motility of triple-negative breast cancer cells is due to repression of plakoglobin gene by metastasis modulator protein SLUG. J Biol Chem. 2012; 287:19472–86. doi: 10.1074/jbc.M112.345728.
53. Aktary Z, Chapman K, Lam L, Lo A, Ji C, Graham K, Cook L, Li L, Mackey JR, Pasdar M. Plakoglobin interacts with and increases the protein levels of metastasis suppressor Nm23-H2 and regulates the expression of Nm23-H1. Oncogene. 2010; 29:2118–29. doi: 10.1038/onc.2009.495.
54. Holen I, Whitworth J, Nutter F, Evans A, Brown HK, Lefley DV, Barbaric I, Jones M, Ottewell PD. Loss of plakoglobin promotes decreased cell-cell contact, increased invasion, and breast cancer cell dissemination in vivo. Breast Cancer Res. 2012; 14:R86. doi: 10.1186/bcr3201.
55. Franzen CA, Todorović V, Desai BV, Mirzoeva S, Yang XJ, Green KJ, Pelling JC. The desmosomal armadillo protein plakoglobin regulates prostate cancer cell adhesion and motility through vitronectin-dependent Src signaling. PLoS One. 2012; 7:e42132. doi: 10.1371/journal.pone.0042132.
56. Lam L, Aktary Z, Bishay M, Werkman C, Kuo CY, Heacock M, Srivastava N, Mackey JR, Pasdar M. Regulation of subcellular distribution and oncogenic potential of nucleophosmin by plakoglobin. Oncogenesis. 2012; 1:e4. doi: 10.1038/oncsis.2012.4.
57. Aktary Z, Kulak S, Mackey J, Jahroudi N, Pasdar M. Plakoglobin interacts with the transcription factor p53 and regulates the expression of 14-3-3σ. J Cell Sci. 2013; 126:3031–42. doi: 10.1242/jcs.120642.
58. Aktary Z, Pasdar M. Plakoglobin represses SATB1 expression and decreases in vitro proliferation, migration and invasion. PLoS One. 2013; 8:e78388. doi: 10.1371/journal.pone.0078388.
59. Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012; 149:1192–205. doi: 10.1016/j.cell.2012.05.012.
60. Bradley RS, Cowin P, Brown AM. Expression of Wnt-1 in PC12 cells results in modulation of plakoglobin and E-cadherin and increased cellular adhesion. J Cell Biol. 1993; 123:1857–65. doi: 10.1083/jcb.123.6.1857.
61. Karnovsky A, Klymkowsky MW. Anterior axis duplication in Xenopus induced by the over-expression of the cadherin-binding protein plakoglobin. Proc Natl Acad Sci USA. 1995; 92:4522–26. doi: 10.1073/pnas.92.10.4522.
62. Merriam JM, Rubenstein AB, Klymkowsky MW. Cytoplasmically anchored plakoglobin induces a WNT-like phenotype in Xenopus. Dev Biol. 1997; 185:67–81. doi: 10.1006/dbio.1997.8550.
63. White P, Aberle H, Vincent JP. Signaling and adhesion activities of mammalian beta-catenin and plakoglobin in Drosophila. J Cell Biol. 1998; 140:183–95. doi: 10.1083/jcb.140.1.183.
64. Simcha I, Shtutman M, Salomon D, Zhurinsky J, Sadot E, Geiger B, Ben-Ze’ev A. Differential nuclear translocation and transactivation potential of beta-catenin and plakoglobin. J Cell Biol. 1998; 141:1433–48. doi: 10.1083/jcb.141.6.1433.
65. Klymkowsky MW, Williams BO, Barish GD, Varmus HE, Vourgourakis YE. Membrane-anchored plakoglobins have multiple mechanisms of action in Wnt signaling. Mol Biol Cell. 1999; 10:3151–69. doi: 10.1091/mbc.10.10.3151.
66. Zhurinsky J, Shtutman M, Ben-Ze’ev A. Differential mechanisms of LEF/TCF family-dependent transcriptional activation by beta-catenin and plakoglobin. Mol Cell Biol. 2000; 20:4238–52. doi: 10.1128/MCB.20.12.4238-4252.2000.
67. Kolligs FT, Kolligs B, Hajra KM, Hu G, Tani M, Cho KR, Fearon ER. gamma-catenin is regulated by the APC tumor suppressor and its oncogenic activity is distinct from that of beta-catenin. Genes Dev. 2000; 14:1319–31.
68. Pan H, Gao F, Papageorgis P, Abdolmaleky HM, Faller DV, Thiagalingam S. Aberrant activation of gamma-catenin promotes genomic instability and oncogenic effects during tumor progression. Cancer Biol Ther. 2007; 6:1638–43. doi: 10.4161/cbt.6.10.4904.
69. Bommer GT, Jäger C, Dürr EM, Baehs S, Eichhorst ST, Brabletz T, Hu G, Fröhlich T, Arnold G, Kress DC, Göke B, Fearon ER, Kolligs FT. DRO1, a gene down-regulated by oncogenes, mediates growth inhibition in colon and pancreatic cancer cells. J Biol Chem. 2005; 280:7962–75. doi: 10.1074/jbc.M412593200.
70. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997; 275:1787–90. doi: 10.1126/science.275.5307.1787.
71. Salomon D, Sacco PA, Roy SG, Simcha I, Johnson KR, Wheelock MJ, Ben-Ze’ev A. Regulation of beta-catenin levels and localization by overexpression of plakoglobin and inhibition of the ubiquitin-proteasome system. J Cell Biol. 1997; 139:1325–35. doi: 10.1083/jcb.139.5.1325.
72. Li L, Chapman K, Hu X, Wong A, Pasdar M. Modulation of the oncogenic potential of beta-catenin by the subcellular distribution of plakoglobin. Mol Carcinog. 2007; 46:824–38. doi: 10.1002/mc.20310.
73. Morgan RG, Pearn L, Liddiard K, Pumford SL, Burnett AK, Tonks A, Darley RL. γ-Catenin is overexpressed in acute myeloid leukemia and promotes the stabilization and nuclear localization of β-catenin. Leukemia. 2013; 27:336–43. doi: 10.1038/leu.2012.221.
74. Niu CC, Zhao C, Yang ZD, Zhang XL, Wu WR, Pan J, Zhao C, Li ZQ, Ding W, Yang Z, Si WK. Downregulation of γ-catenin inhibits CML cell growth and potentiates the response of CML cells to imatinib through β-catenin inhibition. Int J Mol Med. 2013; 31:453–58.
75. Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properies and tumor mircroenvironment. Mol Cancer. 2016; 15:24. doi: 10.1186/s12943-016-0509-3.
76. Lee RT, Zhao Z, Ingham PW. Hedgehog signalling. Development. 2016; 143:367–72. doi: 10.1242/dev.120154.
77. Yoon JW, Kita Y, Frank DJ, Majewski RR, Konicek BA, Nobrega MA, Jacob H, Walterhouse D, Iannaccone P. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J Biol Chem. 2002; 277:5548–55. doi: 10.1074/jbc.M105708200.
78. Shahi MH, Afzal M, Sinha S, Eberhart CG, Rey JA, Fan X, Castresana JS. Regulation of sonic hedgehog-GLI1 downstream target genes PTCH1, Cyclin D2, Plakoglobin, PAX6 and NKX2.2 and their epigenetic status in medulloblastoma and astrocytoma. BMC Cancer. 2010; 10:614. doi: 10.1186/1471-2407-10-614.
79. Shahi MH, Rey JA, Castresana JS. The sonic hedgehog-GLI1 signaling pathway in brain tumor development. Expert Opin Ther Targets. 2012; 16:1227–38. doi: 10.1517/14728222.2012.720975.
80. Miravet S, Piedra J, Castaño J, Raurell I, Francí C, Duñach M, García de Herreros A. Tyrosine phosphorylation of plakoglobin causes contrary effects on its association with desmosomes and adherens junction components and modulates beta-catenin-mediated transcription. Mol Cell Biol. 2003; 23:7391–402. doi: 10.1128/MCB.23.20.7391-7402.2003.
81. Wang Y, Jin G, Miao H, Li JY, Usami S, Chien S. Integrins regulate VE-cadherin and catenins: dependence of this regulation on Src, but not on Ras. Proc Natl Acad Sci USA. 2006; 103:1774–79. doi: 10.1073/pnas.0510774103.
82. Shafiei F, Rahnama F, Pawella L, Mitchell MD, Gluckman PD, Lobie PE. DNMT3A and DNMT3B mediate autocrine hGH repression of plakoglobin gene transcription and consequent phenotypic conversion of mammary carcinoma cells. Oncogene. 2008; 27:2602–12. doi: 10.1038/sj.onc.1210917.
83. Morel E, Fouquet S, Strup-Perrot C, Pichol Thievend C, Petit C, Loew D, Faussat AM, Yvernault L, Pinçon-Raymond M, Chambaz J, Rousset M, Thenet S, Clair C. The cellular prion protein PrP(c) is involved in the proliferation of epithelial cells and in the distribution of junction-associated proteins. PLoS One. 2008; 3:e3000. doi: 10.1371/journal.pone.0003000.
84. Kurrle N, Völlner F, Eming R, Hertl M, Banning A, Tikkanen R. Flotillins directly interact with γ-catenin and regulate epithelial cell-cell adhesion. PLoS One. 2013; 8:e84393. doi: 10.1371/journal.pone.0084393.
85. Babuke T, Ruonala M, Meister M, Amaddii M, Genzler C, Esposito A, Tikkanen R. Hetero-oligomerization of reggie-1/flotillin-2 and reggie-2/flotillin-1 is required for their endocytosis. Cell Signal. 2009; 21:1287–97. doi: 10.1016/j.cellsig.2009.03.012.
86. Mukhina S, Mertani HC, Guo K, Lee KO, Gluckman PD, Lobie PE. Phenotypic conversion of human mammary carcinoma cells by autocrine human growth hormone. Proc Natl Acad Sci USA. 2004; 101:15166–71. doi: 10.1073/pnas.0405881101.
87. Štajduhar E, Sedić M, Leniček T, Radulović P, Kerenji A, Krušlin B, Pavelić K, Kraljević Pavelić S. Expression of growth hormone receptor, plakoglobin and NEDD9 protein in association with tumour progression and metastasis in human breast cancer. Tumour Biol. 2014; 35:6425–34. doi: 10.1007/s13277-014-1827-y.
88. Chua KN, Kong LR, Sim WJ, Ng HC, Ong WR, Thiery JP, Huynh H, Goh BC. Combinatorial treatment using targeted MEK and SRC inhibitors synergistically abrogates tumor cell growth and induces mesenchymal-epithelial transition in non-small-cell lung carcinoma. Oncotarget. 2015; 6:29991–30005. doi: 10.18632/oncotarget.5031.
89. Hegland DD, Sullivan DM, Rovira II, Li A, Kovesdi I, Bruder JT, Finkel T. Regulation of endothelial cell adherens junctions by a Ras-dependent signal transduction pathway. Biochem Biophys Res Commun. 1999; 260:371–76. doi: 10.1006/bbrc.1999.0919.
90. Lerner EC, Qian Y, Blaskovich MA, Fossum RD, Vogt A, Sun J, Cox AD, Der CJ, Hamilton AD, Sebti SM. Ras CAAX peptidomimetic FTI-277 selectively blocks oncogenic Ras signaling by inducing cytoplasmic accumulation of inactive Ras-Raf complexes. J Biol Chem. 1995; 270:26802–06. doi: 10.1074/jbc.270.45.26802.
91. Nam JS, Ino Y, Sakamoto M, Hirohashi S. Ras farnesylation inhibitor FTI-277 restores the E-cadherin/catenin cell adhesion system in human cancer cells and reduces cancer metastasis. Jpn J Cancer Res. 2002; 93:1020–28. doi: 10.1111/j.1349-7006.2002.tb02479.x.
92. Yim JH, Baek JH, Lee CW, Kim MJ, Yun HS, Hong EH, Lee SJ, Park JK, Um HD, Hwang SG. Identification of HDAC4 as a target of γ-catenin that regulates the oncogenic K-Ras-mediated malignant phenotype of Rat2 cells. Biochem Biophys Res Commun. 2013; 436:436–42. doi: 10.1016/j.bbrc.2013.05.122.
93. Mahendram S, Kelly KF, Paez-Parent S, Mahmood S, Polena E, Cooney AJ, Doble BW. Ectopic γ-catenin expression partially mimics the effects of stabilized β-catenin on embryonic stem cell differentiation. PLoS One. 2013; 8:e65320. doi: 10.1371/journal.pone.0065320.
94. ten Berge D, Kurek D, Blauwkamp T, Koole W, Maas A, Eroglu E, Siu RK, Nusse R. Embryonic stem cells require Wnt proteins to prevent differentiation to epiblast stem cells. Nat Cell Biol. 2011; 13:1070–75. doi: 10.1038/ncb2314.
95. Sokol SY. Maintaining embryonic stem cell pluripotency with Wnt signaling. Development. 2011; 138:4341–50. doi: 10.1242/dev.066209.
96. Huang CL, Cheng JC, Liao CH, Stern A, Hsieh JT, Wang CH, Hsu HL, Tseng CP. Disabled-2 is a negative regulator of integrin alpha(IIb)beta(3)-mediated fibrinogen adhesion and cell signaling. J Biol Chem. 2004; 279:42279–89. doi: 10.1074/jbc.M402540200.
97. Hocevar BA, Mou F, Rennolds JL, Morris SM, Cooper JA, Howe PH. Regulation of the Wnt signaling pathway by disabled-2 (Dab2). EMBO J. 2003; 22:3084–94. doi: 10.1093/emboj/cdg286.
98. Huang CL, Cheng JC, Kitajima K, Nakano T, Yeh CF, Chong KY, Tseng CP. Disabled-2 is required for mesoderm differentiation of murine embryonic stem cells. J Cell Physiol. 2010; 225:92–105. doi: 10.1002/jcp.22200.
99. Sturgeon CM, Ditadi A, Awong G, Kennedy M, Keller G. Wnt signaling controls the specification of definitive and primitive hematopoiesis from human pluripotent stem cells. Nat Biotechnol. 2014; 32:554–61. doi: 10.1038/nbt.2915.
100. McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Abrams SL, Montalto G, D’Assoro AB, Libra M, Nicoletti F, Maestro R, Basecke J, Cocco L, Cervello M, Martelli AM. Multifaceted roles of GSK-3 and Wnt/β-catenin in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leukemia. 2014; 28:15–33. doi: 10.1038/leu.2013.184.
101. Koch U, Wilson A, Cobas M, Kemler R, Macdonald HR, Radtke F. Simultaneous loss of beta- and gamma-catenin does not perturb hematopoiesis or lymphopoiesis. Blood. 2008; 111:160–64. doi: 10.1182/blood-2007-07-099754.
102. Jeannet G, Scheller M, Scarpellino L, Duboux S, Gardiol N, Back J, Kuttler F, Malanchi I, Birchmeier W, Leutz A, Huelsken J, Held W. Long-term, multilineage hematopoiesis occurs in the combined absence of beta-catenin and gamma-catenin. Blood. 2008; 111:142–49. doi: 10.1182/blood-2007-07-102558.
103. Staal FJ, Luis TC. Wnt signaling in hematopoiesis: crucial factors for self-renewal, proliferation, and cell fate decisions. J Cell Biochem. 2010; 109:844–49.
104. Fung TK, Gandillet A, So CW. Selective treatment of mixed-lineage leukemia leukemic stem cells through targeting glycogen synthase kinase 3 and the canonical Wnt/β-catenin pathway. Curr Opin Hematol. 2012; 19:280–86. doi: 10.1097/MOH.0b013e3283545615.
105. Müller-Tidow C, Steffen B, Cauvet T, Tickenbrock L, Ji P, Diederichs S, Sargin B, Köhler G, Stelljes M, Puccetti E, Ruthardt M, deVos S, Hiebert SW, et al. Translocation products in acute myeloid leukemia activate the Wnt signaling pathway in hematopoietic cells. Mol Cell Biol. 2004; 24:2890–904. doi: 10.1128/MCB.24.7.2890-2904.2004.
106. Zheng X, Beissert T, Kukoc-Zivojnov N, Puccetti E, Altschmied J, Strolz C, Boehrer S, Gul H, Schneider O, Ottmann OG, Hoelzer D, Henschler R, Ruthardt M. Gamma-catenin contributes to leukemogenesis induced by AML-associated translocation products by increasing the self-renewal of very primitive progenitor cells. Blood. 2004; 103:3535–43. doi: 10.1182/blood-2003-09-3335.
107. Steinert G, Oancea C, Roos J, Hagemeyer H, Maier T, Ruthardt M, Puccetti E. Sulindac sulfide reverses aberrant self-renewal of progenitor cells induced by the AML-associated fusion proteins PML/RARα and PLZF/RARα. PLoS One. 2011; 6:e22540. doi: 10.1371/journal.pone.0022540.
108. Xu J, Wu W, Shen W, Liu P. The clinical significance of γ-catenin in acute myeloid leukemia. Onco Targets Ther. 2016; 9:3861–71. doi: 10.2147/OTT.S105514.
109. Niu CC, Zhao C, Zhang XL, Pan J, Zhao C, Wu WR, Li ZQ, Liu T, Yang Z, Si WK. Wnt5a enhances the response of CML cells to Imatinib Mesylate through JNK activation and γ-catenin inhibition. Leuk Res. 2013; 37:1532–37. doi: 10.1016/j.leukres.2013.07.013.
110. Henderson WR Jr, Chi EY, Ye X, Nguyen C, Tien YT, Zhou B, Borok Z, Knight DA, Kahn M. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci USA. 2010; 107:14309–14. doi: 10.1073/pnas.1001520107.
111. Kim YM, Ma H, Oehler VG, Gang EJ, Nguyen C, Masiello D, Liu H, Zhao Y, Radich J, Kahn M. The gamma catenin/CBP complex maintains survivin transcription in β-catenin deficient/depleted cancer cells. Curr Cancer Drug Targets. 2011; 11:213–25. doi: 10.2174/156800911794328420.
112. Zhao Y, Masiello D, McMillian M, Nguyen C, Wu Y, Melendez E, Smbatyan G, Kida A, He Y, Teo JL, Kahn M. CBP/catenin antagonist safely eliminates drug-resistant leukemia-initiating cells. Oncogene. 2016; 35:3705–17. doi: 10.1038/onc.2015.438.
113. Gang EJ, Hsieh YT, Pham J, Zhao Y, Nguyen C, Huantes S, Park E, Naing K, Klemm L, Swaminathan S, Conway EM, Pelus LM, Crispino J, et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene. 2014; 33:2169–78. doi: 10.1038/onc.2013.169.
114. Miravet S, Piedra J, Miró F, Itarte E, García de Herreros A, Duñach M. The transcriptional factor Tcf-4 contains different binding sites for beta-catenin and plakoglobin. J Biol Chem. 2002; 277:1884–91. doi: 10.1074/jbc.M110248200.
115. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57–70. doi: 10.1016/S0092-8674(00)81683-9.
116. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74. doi: 10.1016/j.cell.2011.02.013.
117. Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, Concannon KF, Donaldson MC, Sequist LV, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013; 339:580–84. doi: 10.1126/science.1228522.
118. Scatena R, Bottoni P, Giardina B. Circulating tumour cells and cancer stem cells: a role for proteomics in defining the interrelationships between function, phenotype and differentiation with potential clinical applications. Biochim Biophys Acta. 2013; 1835:129–43.
119. Gkountela S, Aceto N. Stem-like features of cancer cells on their way to metastasis. Biol Direct. 2016; 11:33. doi: 10.1186/s13062-016-0135-4.
120. Tomlinson JS, Alpaugh ML, Barsky SH. An intact overexpressed E-cadherin/alpha,beta-catenin axis characterizes the lymphovascular emboli of inflammatory breast carcinoma. Cancer Res. 2001; 61:5231–41.
121. Alpaugh ML, Tomlinson JS, Kasraeian S, Barsky SH. Cooperative role of E-cadherin and sialyl-Lewis X/A-deficient MUC1 in the passive dissemination of tumor emboli in inflammatory breast carcinoma. Oncogene. 2002; 21:3631–43. doi: 10.1038/sj.onc.1205389.
122. Alpaugh ML, Tomlinson JS, Ye Y, Barsky SH. Relationship of sialyl-Lewis(x/a) underexpression and E-cadherin overexpression in the lymphovascular embolus of inflammatory breast carcinoma. Am J Pathol. 2002; 161:619–28. doi: 10.1016/S0002-9440(10)64217-4.
123. Hamilton G, Hochmair M, Rath B, Klameth L, Zeillinger R. Small cell lung cancer: circulating tumor cells of extended stage patients express a mesenchymal-epithelial transition phenotype. Cell Adhes Migr. 2016; 10:360–67. doi: 10.1080/19336918.2016.1155019.
124. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, Brannigan BW, Kapur R, Stott SL, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014; 158:1110–22. doi: 10.1016/j.cell.2014.07.013.
125. Lu L, Zeng H, Gu X, Ma W. Circulating tumor cell clusters-associated gene plakoglobin and breast cancer survival. Breast Cancer Res Treat. 2015; 151:491–500. doi: 10.1007/s10549-015-3416-1.
126. Woelfle U, Cloos J, Sauter G, Riethdorf L, Jänicke F, van Diest P, Brakenhoff R, Pantel K. Molecular signature associated with bone marrow micrometastasis in human breast cancer. Cancer Res. 2003; 63:5679–84.
127. Howard EW, Leung SC, Yuen HF, Chua CW, Lee DT, Chan KW, Wang X, Wong YC. Decreased adhesiveness, resistance to anoikis and suppression of GRP94 are integral to the survival of circulating tumor cells in prostate cancer. Clin Exp Metastasis. 2008; 25:497–508. doi: 10.1007/s10585-008-9157-3.
128. Mimeault M, Batra SK. Molecular biomarkers of cancer stem/progenitor cells associated with progression, metastases, and treatment resistance of aggressive cancers. Cancer Epidemiol Biomarkers Prev. 2014; 23:234–54. doi: 10.1158/1055-9965.EPI-13-0785.
129. Irollo E, Pirozzi G. CD133: to be or not to be, is this the real question? Am J Transl Res. 2013; 5:563–81.
130. Koyama-Nasu R, Takahashi R, Yanagida S, Nasu-Nishimura Y, Oyama M, Kozuka-Hata H, Haruta R, Manabe E, Hoshino-Okubo A, Omi H, Yanaihara N, Okamoto A, Tanaka T, et al. The cancer stem cell marker CD133 interacts with plakoglobin and controls desmoglein-2 protein levels. PLoS One. 2013; 8:e53710. doi: 10.1371/journal.pone.0053710.
131. Lai YH, Cheng J, Cheng D, Feasel ME, Beste KD, Peng J, Nusrat A, Moreno CS. SOX4 interacts with plakoglobin in a Wnt3a-dependent manner in prostate cancer cells. BMC Cell Biol. 2011; 12:50. doi: 10.1186/1471-2121-12-50.
132. Zorn AM, Barish GD, Williams BO, Lavender P, Klymkowsky MW, Varmus HE. Regulation of Wnt signaling by Sox proteins: XSox17 alpha/beta and XSox3 physically interact with beta-catenin. Mol Cell. 1999; 4:487–98. doi: 10.1016/S1097-2765(00)80200-2.
133. Chen YJ, Lee LY, Chao YK, Chang JT, Lu YC, Li HF, Chiu CC, Li YC, Li YL, Chiou JF, Cheng AJ. DSG3 facilitates cancer cell growth and invasion through the DSG3-plakoglobin-TCF/LEF-Myc/cyclin D1/MMP signaling pathway. PLoS One. 2013; 8:e64088. doi: 10.1371/journal.pone.0064088.
134. Besnier LS, Cardot P, Da Rocha B, Simon A, Loew D, Klein C, Riveau B, Lacasa M, Clair C, Rousset M, Thenet S. The cellular prion protein PrPc is a partner of the Wnt pathway in intestinal epithelial cells. Mol Biol Cell. 2015; 26:3313–28. doi: 10.1091/mbc.E14-11-1534.
135. Rousset M, Leturque A, Thenet S. The nucleo-junctional interplay of the cellular prion protein: A new partner in cancer-related signaling pathways? Prion. 2016; 10:143–52. doi: 10.1080/19336896.2016.1163457.
136. Falini B, Martelli MP, Mecucci C, Liso A, Bolli N, Bigerna B, Pucciarini A, Pileri S, Meloni G, Martelli MF, Haferlach T, Schnittger S. Cytoplasmic mutated nucleophosmin is stable in primary leukemic cells and in a xenotransplant model of NPMc+ acute myeloid leukemia in SCID mice. Haematologica. 2008; 93:775–79. doi: 10.3324/haematol.12225.
137. Shandilya J, Swaminathan V, Gadad SS, Choudhari R, Kodaganur GS, Kundu TK. Acetylated NPM1 localizes in the nucleoplasm and regulates transcriptional activation of genes implicated in oral cancer manifestation. Mol Cell Biol. 2009; 29:5115–27. doi: 10.1128/MCB.01969-08.
138. Grisendi S, Mecucci C, Falini B, Pandolfi PP. Nucleophosmin and cancer. Nat Rev Cancer. 2006; 6:493–505. doi: 10.1038/nrc1885.
139. Boissan M, De Wever O, Lizarraga F, Wendum D, Poincloux R, Chignard N, Desbois-Mouthon C, Dufour S, Nawrocki-Raby B, Birembaut P, Bracke M, Chavrier P, Gespach C, Lacombe ML. Implication of metastasis suppressor NM23-H1 in maintaining adherens junctions and limiting the invasive potential of human cancer cells. Cancer Res. 2010; 70:7710–22. doi: 10.1158/0008-5472.CAN-10-1887.
140. Feng Y, Gross S, Wolf NM, Butenschön VM, Qiu Y, Devraj K, Liebner S, Kroll J, Skolnik EY, Hammes HP, Wieland T. Nucleoside diphosphate kinase B regulates angiogenesis through modulation of vascular endothelial growth factor receptor type 2 and endothelial adherens junction proteins. Arterioscler Thromb Vasc Biol. 2014; 34:2292–300. doi: 10.1161/ATVBAHA.114.304239.
141. Moreno V, Gonzalo P, Gómez-Escudero J, Pollán Á, Acín-Pérez R, Breckenridge M, Yáñez-Mó M, Barreiro O, Orsenigo F, Kadomatsu K, Chen CS, Enríquez JA, Dejana E, et al. An EMMPRIN-γ-catenin-Nm23 complex drives ATP production and actomyosin contractility at endothelial junctions. J Cell Sci. 2014; 127:3768–81. doi: 10.1242/jcs.149518.
142. Kim KM, Adyshev DM, Kása A, Zemskov EA, Kolosova IA, Csortos C, Verin AD. Putative protein partners for the human CPI-17 protein revealed by bacterial two-hybrid screening. Microvasc Res. 2013; 88:19–24. doi: 10.1016/j.mvr.2013.04.002.
143. Williamson L, Raess NA, Caldelari R, Zakher A, de Bruin A, Posthaus H, Bolli R, Hunziker T, Suter MM, Müller EJ. Pemphigus vulgaris identifies plakoglobin as key suppressor of c-Myc in the skin. EMBO J. 2006; 25:3298–309. doi: 10.1038/sj.emboj.7601224.
144. Tokonzaba E, Chen J, Cheng X, Den Z, Ganeshan R, Műller EJ, Koch PJ. Plakoglobin as a regulator of desmocollin gene expression. J Invest Dermatol. 2013; 133:2732–40. doi: 10.1038/jid.2013.220.
145. Hakimelahi S, Parker HR, Gilchrist AJ, Barry M, Li Z, Bleackley RC, Pasdar M. Plakoglobin regulates the expression of the anti-apoptotic protein BCL-2. J Biol Chem. 2000; 275:10905–11. doi: 10.1074/jbc.275.15.10905.
146. Alaee M, Padda A, Mehrabani V, Churchill L, Pasdar M. The physical interaction of p53 and plakoglobin is necessary for their synergistic inhibition of migration and invasion. Oncotarget. 2016; 7:26898–915. doi: 10.18632/oncotarget.8616.
147. Shtutman M, Zhurinsky J, Oren M, Levina E, Ben-Ze’ev A. PML is a target gene of beta-catenin and plakoglobin, and coactivates beta-catenin-mediated transcription. Cancer Res. 2002; 62:5947–54.
148. Sechler M, Borowicz S, Van Scoyk M, Avasarala S, Zerayesus S, Edwards MG, Kumar Karuppusamy Rathinam M, Zhao X, Wu PY, Tang K, Bikkavilli RK, Winn RA. Novel Role for γ-Catenin in the Regulation of Cancer Cell Migration via the Induction of Hepatocyte Growth Factor Activator Inhibitor Type 1 (HAI-1). J Biol Chem. 2015; 290:15610–20. doi: 10.1074/jbc.M114.631820.
149. Alaee M, Danesh G, Pasdar M. Plakoglobin Reduces the in vitro Growth, Migration and Invasion of Ovarian Cancer Cells Expressing N-Cadherin and Mutant p53. PLoS One. 2016; 11:e0154323. doi: 10.1371/journal.pone.0154323
150. Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013; 15:2–8. doi: 10.1038/ncb2641.
151. Ferraiuolo M, Di Agostino S, Blandino G, Strano S. Oncogenic Intra-p53 Family Member Interactions in Human Cancers. Front Oncol. 2016; 6:77. doi: 10.3389/fonc.2016.00077.
152. Haupt S, Raghu D, Haupt Y. Mutant p53 Drives Cancer by Subverting Multiple Tumor Suppression Pathways. Front Oncol. 2016; 6:12. doi: 10.3389/fonc.2016.00012.
153. Parrales A, Iwakuma T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front Oncol. 2015; 5:288. doi: 10.3389/fonc.2015.00288.
154. Kohwi-Shigematsu T, Poterlowicz K, Ordinario E, Han HJ, Botchkarev VA, Kohwi Y. Genome organizing function of SATB1 in tumor progression. Semin Cancer Biol. 2013; 23:72–79. doi: 10.1016/j.semcancer.2012.06.009.
155. Zhou L, Pradhan-Sundd T, Poddar M, Singh S, Kikuchi A, Stolz DB, Shou W, Li Z, Nejak-Bowen KN, Monga SP. Mice with Hepatic Loss of the Desmosomal Protein γ-Catenin Are Prone to Cholestatic Injury and Chemical Carcinogenesis. Am J Pathol. 2015; 185:3274–89. doi: 10.1016/j.ajpath.2015.08.019.
156. Fang WK, Liao LD, Gu W, Chen B, Wu ZY, Wu JY, Shen J, Xu LY, Li EM. Down-regulated γ-catenin expression is associated with tumor aggressiveness in esophageal cancer. World J Gastroenterol. 2014; 20:5839–48. doi: 10.3748/wjg.v20.i19.5839.
157. Procházková J, Kabátková M, Šmerdová L, Pacherník J, Sykorová D, Kohoutek J, Šimečková P, Hrubá E, Kozubík A, Machala M, Vondráček J. Aryl hydrocarbon receptor negatively regulates expression of the plakoglobin gene (jup). Toxicol Sci. 2013; 134:258–70. doi: 10.1093/toxsci/kft110.
158. Knudsen KA, Wheelock MJ. Plakoglobin, or an 83-kD homologue distinct from beta-catenin, interacts with E-cadherin and N-cadherin. J Cell Biol. 1992; 118:671–79. doi: 10.1083/jcb.118.3.671.
159. Troyanovsky RB, Chitaev NA, Troyanovsky SM. Cadherin binding sites of plakoglobin: localization, specificity and role in targeting to adhering junctions. J Cell Sci. 1996; 109:3069–78.
160. Aberle H, Butz S, Stappert J, Weissig H, Kemler R, Hoschuetzky H. Assembly of the cadherin-catenin complex in vitro with recombinant proteins. J Cell Sci. 1994; 107:3655–63.
161. Navarro P, Ruco L, Dejana E. Differential localization of VE- and N-cadherins in human endothelial cells: VE-cadherin competes with N-cadherin for junctional localization. J Cell Biol. 1998; 140:1475–84. doi: 10.1083/jcb.140.6.1475.
162. Ilan N, Cheung L, Pinter E, Madri JA. Platelet-endothelial cell adhesion molecule-1 (CD31), a scaffolding molecule for selected catenin family members whose binding is mediated by different tyrosine and serine/threonine phosphorylation. J Biol Chem. 2000; 275:21435–43. doi: 10.1074/jbc.M001857200.
163. Biswas P, Zhang J, Schoenfeld JD, Schoenfeld D, Gratzinger D, Canosa S, Madri JA. Identification of the regions of PECAM-1 involved in beta- and gamma-catenin associations. Biochem Biophys Res Commun. 2005; 329:1225–33. doi: 10.1016/j.bbrc.2005.02.095.
164. Sacco PA, McGranahan TM, Wheelock MJ, Johnson KR. Identification of plakoglobin domains required for association with N-cadherin and alpha-catenin. J Biol Chem. 1995; 270:20201–06. doi: 10.1074/jbc.270.34.20201.
165. Mathur M, Goodwin L, Cowin P. Interactions of the cytoplasmic domain of the desmosomal cadherin Dsg1 with plakoglobin. J Biol Chem. 1994; 269:14075–80.
166. Roh JY, Stanley JR. Plakoglobin binding by human Dsg3 (pemphigus vulgaris antigen) in keratinocytes requires the cadherin-like intracytoplasmic segment. J Invest Dermatol. 1995; 104:720–24. doi: 10.1111/1523-1747.ep12606963.
167. Witcher LL, Collins R, Puttagunta S, Mechanic SE, Munson M, Gumbiner B, Cowin P. Desmosomal cadherin binding domains of plakoglobin. J Biol Chem. 1996; 271:10904–09. doi: 10.1074/jbc.271.18.10904.
168. Kowalczyk AP, Bornslaeger EA, Borgwardt JE, Palka HL, Dhaliwal AS, Corcoran CM, Denning MF, Green KJ. The amino-terminal domain of desmoplakin binds to plakoglobin and clusters desmosomal cadherin-plakoglobin complexes. J Cell Biol. 1997; 139:773–84. doi: 10.1083/jcb.139.3.773.
169. Bornslaeger EA, Godsel LM, Corcoran CM, Park JK, Hatzfeld M, Kowalczyk AP, Green KJ. Plakophilin 1 interferes with plakoglobin binding to desmoplakin, yet together with plakoglobin promotes clustering of desmosomal plaque complexes at cell-cell borders. J Cell Sci. 2001; 114:727–38.
170. Kirchner F, Schuetz A, Boldt LH, Martens K, Dittmar G, Haverkamp W, Thierfelder L, Heinemann U, Gerull B. Molecular insights into arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 missense mutations. Circ Cardiovasc Genet. 2012; 5:400–11. doi: 10.1161/CIRCGENETICS.111.961854.
171. Bonné S, Gilbert B, Hatzfeld M, Chen X, Green KJ, van Roy F. Defining desmosomal plakophilin-3 interactions. J Cell Biol. 2003; 161:403–16. doi: 10.1083/jcb.200303036.
172. Li Y, Yu WH, Ren J, Chen W, Huang L, Kharbanda S, Loda M, Kufe D. Heregulin targets gamma-catenin to the nucleolus by a mechanism dependent on the DF3/MUC1 oncoprotein. Mol Cancer Res. 2003; 1:765–75.
173. Sehgal L, Mukhopadhyay A, Rajan A, Khapare N, Sawant M, Vishal SS, Bhatt K, Ambatipudi S, Antao N, Alam H, Gurjar M, Basu S, Mathur R, et al. 14-3-3γ-Mediated transport of plakoglobin to the cell border is required for the initiation of desmosome assembly in vitro and in vivo. J Cell Sci. 2014; 127:2174–88. doi: 10.1242/jcs.125807.
174. Zhou J, Liyanage U, Medina M, Ho C, Simmons AD, Lovett M, Kosik KS. Presenilin 1 interaction in the brain with a novel member of the Armadillo family. Neuroreport. 1997; 8:1489–94. doi: 10.1097/00001756-199704140-00033.
175. Cohen S, Lee D, Zhai B, Gygi SP, Goldberg AL. Trim32 reduces PI3K-Akt-FoxO signaling in muscle atrophy by promoting plakoglobin-PI3K dissociation. J Cell Biol. 2014; 204:747–58. doi: 10.1083/jcb.201304167.
176. Rivard N. Phosphatidylinositol 3-kinase: a key regulator in adherens junction formation and function. Front Biosci (Landmark Ed). 2009; 14:510–22. doi: 10.2741/3259.
177. Chen SN, Gurha P, Lombardi R, Ruggiero A, Willerson JT, Marian AJ. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ Res. 2014; 114:454–68. doi: 10.1161/CIRCRESAHA.114.302810.