
INTRODUCTION
Aluminium (Al) is widely used as a food and drug additive [1]. Therefore, humans inevitably come into frequent contact with Al. Excessive Al may cause damage to the body’s tissues and organs e.g. renal, bone, lung, heart and central nervous system [2–3]. Previous study showed that the Al accumulation in the liver may be more obvious than brain and other organs [4]. In liver, Al is mainly accumulated in macrophages and lysosomes. Considering that Al in lysosomes can be eliminated by hepatocytes via bile excretion, many scholars believe that Al accumulation does not cause significant liver toxicity [5]. So, hepatotoxicity from long-term exposure to Al has not received sufficient attention. However, recent studies showed that Al administration (50 mg Al per rat, ip) caused significant accumulation of Al in rat hepatic cells, mild hyperplasia of bile duct and liver fatty degeneration [6]. Viezeliene also found that treatment with Al (ip) resulted in a four-fold increase in alanine aminotransferase (ALT) levels and a significant decrease in glutathione (GSH) activity in mice compared to normal controls [7]. Intragastric administration of AlCl3 (34 mg·kg−1 for 30 days) caused a significant increase in ALT, alkaline phosphatase(ALP), aspartate aminotransferase(AST) and lactic dehydrogenase(LDH) levels in rat serum and resulted in obvious damage to hepatic cells, including liver blood sinus expansion, central venous hyperemia, lipid accumulation and lymphocyte infiltration [8]. Furthermore, with a daily injection of 37.5 mg AlCl3·kg−1, degeneration became evident in the livers of rats (e.g., obvious karyopyknosis and cell loss) [9]. These results indicate that Al overload can also cause significant changes in liver morphology and function. However, the mechanism of damage to liver is still unclear.
Trivalent Al could react with water to produce bidentate superoxide coordination spheres [Al (O2) (H2O4)2+ and Al (H2O)63+] that after complexation with O2−, generate Al superoxides [Al(O2−)](H2O5)]2+. Semi reduced AlO2 radicals deplete mitochondrial Fe and promote generation of H2O2, O2 and OH-. So, it is the Al3+-induced formation of oxygen radicals that accounts for the oxidative damage that leads to intrinsic apoptosis [10]. Al induced oxidative stress in tissues like brain, kidney and liver by decreasing intracellular GSH [11]. Various Al salt complexes could provoke macrophage responsibility which promoted an inflammatory cascade [12]. Our previous study also showed that chronic intragastric administration of Al gluconate (Al3+ 200 mg·kg−1 per day, 5 d a week for 20 weeks) causes obvious damage to rat liver and that metal ion imbalance-related oxidative stress may be involved in the mechanism of chronic liver injury caused by Al overload [13]. The relief of oxidative stress could alleviate liver injury induced by Al [14–16]. Those results indicated that the toxicity effect of Al is closely related with inflammation and oxidative stress.
Arachidonic acid (AA) is the precursor of a number of biologically active substances, including prostaglandins(PGs), leukotrienes(LTs), and etc which have important effects on immune and inflammatory systems. 5-lipoxygenase is a metabolic pathway of AA to produce LTs. Our previous research showed that 5-lipoxygenase inhibitors could significantly alleviate inflammation and oxidative stress in chronic Al-overload liver injury [17]. However, cyclooxygenase (COX) pathway is an another very important metabolic pathway of AA. COX includes the structural COX1 and the inducible COX2. The level of COX2 expression is low in normal situation, but it can significantly increase under a variety of pathological conditions [18]. The expression of COX2 of liver tissue was significantly induced by chronic hepatitis C virus and participated in the process of liver inflammation, necrosis, fibrosis, and the hepatocellular carcinoma development caused by the viral infection [19]. COX2 expression was also increased in some non-infectious liver injury such as carbon tetrachloride intoxication, alcoholic and non-alcoholic steatohepatitis [20–21]. Prostaglandin E2 (PGE2) is one of the most abundant and active of the PGs in COX2 downstream. Those results indicated that COX2-PGE2 pathway may participate in pathophysiological process of chronic liver damage and that COX2 may be considered as a novel therapeutic target for liver injury.
However, the relationship between COX2 expression and Al-overload liver damage, and the observation of protective effect of COX2 inhibitor on liver injury caused by chronic Al overload have never been reported. Therefore, the present study was designed to observe the change of COX2 and downstream PGE2 signaling pathway of rat liver and the protective effect of meloxicam, a selective COX2 inhibitor, on liver injury in chronic Al overload rat. The results of this present study will contribute to explore the mechanism of the hepatotoxicity for long-term exposure to Al and to seek new therapeutic strategy for chronic non-infectious liver damage.
RESULTS
Hepatic histopathology
The hepatic lobule structures of the rat livers in the control group were clear and complete. Although the liver cells were arranged and regularly structured, the liver cells in the Al-overload group showed significant vacuolar degeneration, granular degeneration and spotty necrosis. Meloxicam had an obvious protective effect on hepatocyte injury caused by Al overload (Figure 1).
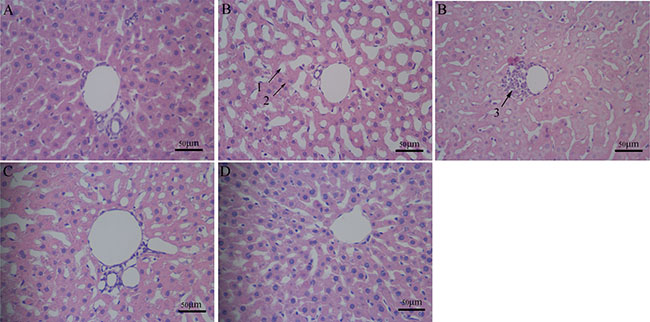
Figure 1: Pathomorphology in rat liver. Hepatocyte pathomorphology in chronic Al overload rat livers (HE × 400, Scale bars = 50 μm). (A) Control group; regular histological features of liver of control group with well formed central vein, cord like arrangement of hepatocytes around the central vein. (B) Model group; regular histological features of liver have changed, section of Al exposed liver with several irregularities: 1. vacuolar degeneration, 2. granular degeneration, 3. spotty necrosis. (C) Al + Meloxicam 0.3 mg.kg−1 group; the administration of meloxicam significantly alleviated the hepatocyte injury of the vacuolar degeneration, spotty necrosis and granular degeneration. (D) Al + Meloxicam 1 mg.kg−1 group; the hepatocyte injury was alleviated and the hepatic lobule structures become clear and complete via the administration of meloxicam.
Changes of liver function in rats
Blood plasma ALT, AST and ALP levels of rats in the model group were significantly increased compared with that in the control group. Administration of meloxicam significantly blunted the increase of ALT, AST and ALP levels caused by Al overload (Figure 2).
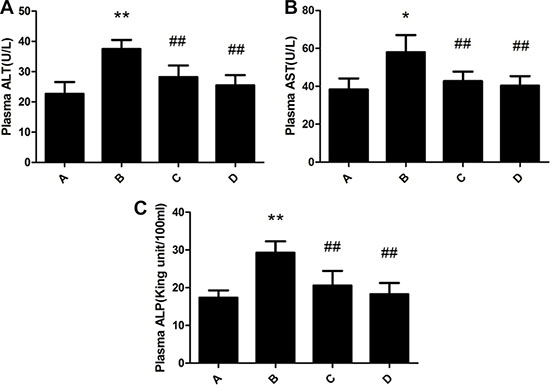
Figure 2: Effect of meloxicam on changes of plasma ALT, AST and ALP in chronic Al-overload rats. (A) Control group; (B) Model group; (C) Al + Meloxicam 0.3 mg.kg−1 group; (D) Al + Meloxicam 1 mg.kg−1 group. (A) The change of ALT. (B) The change of AST. (C) The change of ALP. Data are expressed as the mean ± SD for six individual rats in each group. Compared with that of control group, the level of ALT, AST, and ALP significantly increased in model group. Compared with that of model group, the administration of meloxicam significantly decreased the levels of ALT, AST, and ALP. *P < 0.05 and **P < 0.01 compared with control group, respectively. #P < 0.05 and ##P < 0.01 compared with model group, respectively.
Changes of PGE2 and cAMP contents in rat liver
PGE2 and cAMP content of rat liver in the model group were significantly increased compared with the control group. The administration of meloxicam significantly blunted the increase in PGE2 and cAMP content (Figure 3).
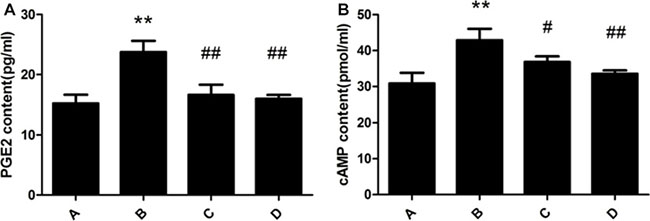
Figure 3: Effect of meloxicam on changes of liver PGE2 and cAMP in chronic Al-overload rats. (A) Control group; (B) Model group; (C) Al + Meloxicam 0.3 mg.kg−1 group; (D) Al + Meloxicam 1 mg.kg−1 group. (A) The change of PGE2 content. (B) The change of cAMP content. Data are expressed as the mean ± SD for six individual rats in each group. Compared with that of control group, the contents of PGE2 and cAMP significantly increased in model group. Compared with that of model group, the administration of meloxicam significantly decreased the contents of PGE2 and cAMP. **P < 0.01 compared with control group. #P < 0.05 and ##P < 0.01 compared with model group, respectively.
Changes of SOD activity and MDA contents in rat liver
SOD activity of rat liver in the model group was significantly decreased compared with the control group. MDA content of rat liver in the model group was significantly increased compared with that in the control group. Meloxicam significantly blunted the decrease of SOD activity and the increase of MDA content caused by Al overload in dose-dependent manner (Figure 4).
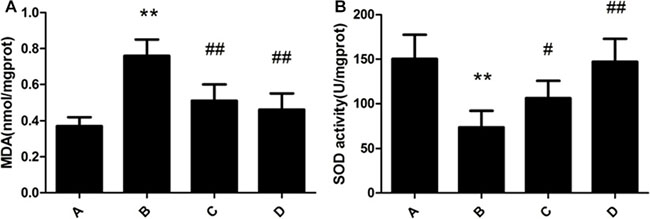
Figure 4: Effect of meloxicam on changes of liver MDA content and SOD activity in chronic Al-overload rats. (A) Control group; (B) Model group; (C) Al + Meloxicam 0.3 mg.kg−1 group; (D) Al + Meloxicam 1 mg.kg−1 group. (A) The change of MDA content. (B) The change of SOD activity. Data are expressed as the mean ± SD for six individual rats in each group. Compared with that of control group, the content of MDA significantly increased and the activity of SOD significantly decreased in model group. Compared with that of model group, the administration of meloxicam significantly decreased the content of MDA and increased the activity of SOD. **P < 0.01 compared with control group. #P < 0.05 and ##P < 0.01 compared with model group, respectively.
Changes of TNF-α, IL-1β and IL-6 contents in rat liver
TNF-α, IL-1β and IL-6 content of rat liver in the model group were significantly increased compared with the control group. The administration of meloxicam, significantly blunted the increase of TNF-α, IL-1β and IL-6 content (Figure 5).
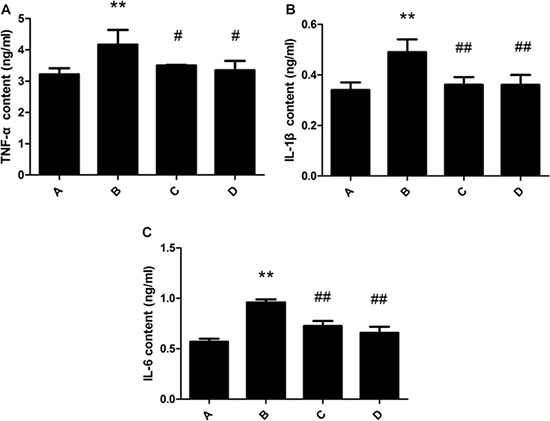
Figure 5: Effect of meloxicam on changes of liver TNF-α, IL-1β and IL-6 content in chronic Al-overload rats. (A) Control group; (B) Model group; (C) Al + Meloxicam 0.3 mg.kg−1 group; (D) Al + Meloxicam 1 mg.kg−1 group. (A) The change of TNF-α content. (B) The change of IL-1β content. (C) The change of IL-6 content. Data are expressed as the mean ± SD for six individual rats in each group. Compared with that of control group, the contents of TNF-α, IL-1β and IL-6 significantly increased in model group. Compared with that of model group, the administration of meloxicam significantly decreased the contents of TNF-α, IL-1β and IL-6. **P < 0.01 compared with control group. #P < 0.05 and ##P < 0.01 compared with model group, respectively.
Changes of COX2 expression in rat liver
COX2 protein was mainly expressed in the cytoplasm of rat liver cell. The expression of COX2 in the model group was significantly increased compared with the control group. The administration of meloxicam significantly blunted the increase of COX2 protein expression caused by Al overload (Figure 6).
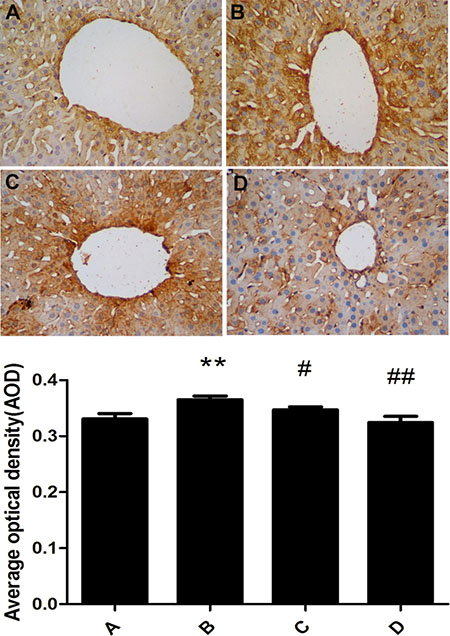
Figure 6: Effect of meloxicam on changes of liver COX2 protein expression in chronic Al-overload rats. (A) Control group; (B) Model group; (C) Al + Meloxicam 0.3 mg.kg−1 group; (D) Al + Meloxicam 1 mg.kg−1 group. Change of COX2 protein expression was measured by immunohistochemistry (400×), the AOD of COX2 were analyzed by IPP. Data are expressed as mean ± SD of three individual experiments. Compared with that of control group, COX2 protein expression significantly increased in model group. Compared with that of model group, the administration of meloxicam significantly decreased COX2 protein expression. **P < 0.01 compared with control group. #P < 0.05 and ##P < 0.01 compared with model group, respectively.
Changes of EP1,2,3,4 expression in rat liver
There was a little expression of EP1,2,3,4 of rat liver in the control group. EP1,2,4 expression of rat liver in the model group was significantly increased and EP3 expression of rat liver in the model group was significantly decreased compared with the control group. The administration of meloxicam significantly increased the expression of EP1,2,3,4 compared with the model group (Figure 7).
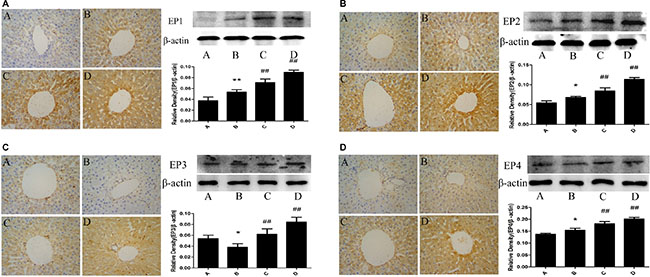
Figure 7: Effect of meloxicam on changes of liver EP1,2,3,4 protein expression in chronic Al-overload rats. (A) Control group; (B) Model group; (C) Al + Meloxicam 0.3 mg.kg−1 group; (D) Al + Meloxicam 1 mg.kg−1 group. Change of EP1(a), EP2(b), EP3(c) and EP4(d) protein expressions were measured by immunohistochemistry (400×) and WB. The relative protein level of EP1,2,3,4 were standardized to endogenous β-actin protein for each sample. Data are expressed as mean ± SD of three individual experiments. Compared with that of control group, EP1,2,4 protein expression significantly increased and EP3 significantly decreased in model group. Compared with that of model group, the administration of meloxicam significantly increased EP1,2,3,4 protein expression. *P < 0.05 and **P < 0.01 compared with control group, respectively. #P < 0.05 and ##P < 0.01 compared with model group, respectively.
Changes of PKA expression in rat liver
The result from the western blotting detection indicated that there is little expression of PKA of rat liver in the control group. The expression of PKA in the model group was significantly increased compared with the control group. The administration of meloxicam significantly blunted the increase of PKA expression caused by Al (Figure 8).
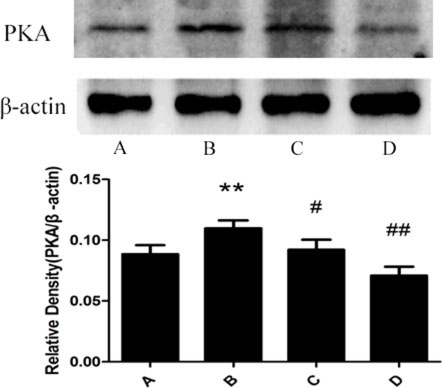
Figure 8: Effect of meloxicam on changes of liver PKA protein expression in chronic Al-overload rats. (A) Control group; (B) Model group; (C) Al + Meloxicam 0.3 mg.kg−1 group; (D) Al + Meloxicam 1 mg.kg−1 group. The protein expression of PKA was measured by Western blot. The relative protein level of PKA was standardized to endogenous β-actin protein for each sample. Data are expressed as mean ± SD of three individual experiments. The expression of PKA in the model group was significantly increased. The administration of meloxicam significantly blunted the increase of PKA expression. **P < 0.01 compared with control group. #P < 0.05 and ##P < 0.01 compared with model group, respectively.
DISCUSSION
Several studies showed similar results that Al overload could cause liver damage [6–9]. Our experiment results also showed obvious vacuolar degeneration, granular degeneration and spotty necrosis in chronic Al-overload rat hepatocytes and the levels of ALT, AST and ALP in plasma were significantly increased in Al-treated rat. These results further confirmed that chronic Al overload could cause liver damage, so the liver function protection should be paid more attention for professional worker subjected to long-term Al exposure [5].
The mechanism of Al producing toxic reaction might be attributed to produce more reactive oxygen species (ROS). Superoxide dismutase (SOD), one type of the antioxidant enzyme, is capable of detoxifying superoxide. Malondialdehyde (MDA) is a product of enzyme reactions which reflects extensive peroxidations [22]. Some studies have shown that Al induced toxicity is mediated by free radical generation and alterations in antioxidant enzymes [23–24]. The conformation of the SOD molecule is altered by the formation of the Al-SOD complex, which might decrease the activity of the SOD [25]. Al easily occupies the binding sites of iron, as a result, the concentration of free Fe2+ is significantly increased to produce more ROS to cause cell injury [26–27]. Our previous study and many other studies have shown that Al may disrupt the homeostasis of metal leads to oxidative stress cause liver damage [13, 28–29]. In the present study, we also found that the activity of SOD was significantly decreased and the content of MDA was significantly increased in Al-overload rats. Those results further indicated that oxidative stress may be involved in chronic liver injury caused by Al. Additionally, oxidative stress also can promote inflammation. Metal ion transporters, such as the divalent metal-ion transporter-1 (DMT-1), it could be activated by inflammation to increase metal ions into the liver to cause oxidative stress [30]. Therefore, oxidative stress and inflammation formed a vicious circle.
Inflammation is also a very important reason involved in the process of liver injury. Some studies have found that Al is mainly accumulated in macrophages [31]. Macrophages are the main source of COX2 in liver [32]. PGs, ROS, cytokines and various kinds of protease were produced in macrophages to release to effect on all cells [33]. Al could decrease the SOD activity to produce more ROS to increase the expression of COX2 in macrophages. So, the activation of macrophages may be the important reason of inflammation in chronic Al-overload rat liver.
ROS could be used as the second messenger activating NF-κB to increase the transcription of genes [34]. There are two NF-κB binding sites in the COX2 promoter, and NF-κB combined with the sites could increase the transcription of COX2 [35]. PGE2 is one of the most abundant and active of the PGs in COX2 downstream [36–37]. PGE2 is also involved in various pathophysiological processes, include inflammation and oxidative stress [38–41]. PGE2 has been found to participate in a variety of liver damage processes [19–21]. PGE2 receptors are designated as EP1, EP2, EP3 and EP4 [42–43]. In the present study, we found that the expression of COX2, protein kinase A(PKA) and EP1,2,4 were significantly increased, the expression of EP3 were significantly decreased, and the content of PGE2 and cyclic adenosine monophosphate (cAMP) were significantly increased in Al-overload rats liver. EP1 could increase the intracellular content of Ca2+. The excessive Ca2+ could injury mitochondria to produce more ROS to damage the liver cells [44–45]. The EP2,4 can stimulate adenylate cyclase(AC) with a subsequent increase of intracellular cAMP which increases PKA. p38 MAPK is involved in the activation of apoptosis because of being activated by cAMP/PKA signaling pathway, and it activates caspase-1, caspase-3 and caspase-11 and causes the produce of TNF-α, IL-1 and IL-6 in parenchymal cells [46]. p38 MAPK is also activated in macrophages to produce more inflammatory cytokines, such as TNF-α, IL-1, IL-6, and others [47]. However, EP3 could reduce the content of cAMP and PKA. The decrease of cAMP and PKA might inhibit the activation of cAMP/PKA signaling pathway. These previous results suggested that EP1,2,4 might promote liver injury and EP3 might improve liver injury. In our study, we also found that the content of TNF-α, IL-1β and IL-6 were significantly increased in Al-overload rats. Considering that inflammatory cytokines had a stimulating effect on the accumulation of neutrophils to aggravate the damage of inflammation in the liver [48], our results indicate that activation of the PGE2-EP1,2,4-cAMP/PKA signaling pathway can cause serious liver injury by stimulation of inflammation and oxidative stress by Al.
Our experimental results showed that the treatment of meloxicam significantly alleviated vacuolar degeneration, granular degeneration and spotty necrosis of rat hepatocyte and decreased the levels of ALT, AST and ALP in Al-treated rats plasma, and it also significantly blunted the decrease of SOD activity and the increase of MDA, TNF-α, IL-1β and IL-6 content in Al-treated rats liver. Moreover, our experimental results also found that meloxicam could significantly decrease the expression of COX2 and PKA, blunt the increase of PGE2 and cAMP, and increase the EP1,2,3,4 expression.
Chronic Al overload could cause the decrease of EP3 expression by the desensitization of EP3 receptor (down-regulation of EP3) long-term exposure to high PGE2 level [49]. This is the normal regulation of receptor [50]. However, the increase of EP1,2,4 expressions in aluminium overload rats suggest that there is a dysregulation of EP1,2,4 expressions long-term exposure to high PGE2 level. But the reason about the dysregulation of EP1,2,4 expressions is not clear. Some studies also have proved that EP1,2,4 would promote inflammation and EP3 would inhibit inflammation [51–53]. Together with our studies, EP1,2,4 expressions could promote liver injury and EP3 expression could protect liver against injury caused by chronic Al overload through their own signaling pathway. Meloxicam treatment inhibited the COX2 activity and decreased the PGE2 level. The decrease of PGE2 level caused the up-regulation of EP1,2,3,4. Perhaps, the effects of the increase of protective EP3 expression exceed the effects of the increase of injury EP1,2,4 expressions. Finally, the cAMP/PKA pathway is inhibited. As a result, the meloxicam protected the liver against the injury caused by Al overload. Moreover, some researchers also have indicated that EP3 expression might inhibit the proinflammatory effect of EP2,4 [54]. These results together indicated that meloxicam can protect liver injury against Al exposure via inhibiting the COX2 to reduce PGE2 and increase the expression of EP3 to alleviate inflammation and oxidative stress through reconstructing the balance of COX2-PGE2-EPs-cAMP/PKA signaling pathway.
In summary, the present results suggested that meloxicam has an obvious protective effect on rat liver damage caused by Al. The protective mechanism of meloxicam on liver injury is attributed to reconstruct the balance of COX2-PGE2-EPs-cAMP/PKA signaling pathway and further to reduce inflammation and oxidative stress in chronic Al-overload rat. These findings pointed out that the COX2-PGE2-EPs-cAMP/PKA signaling pathway which is associated with the inflammation and oxidative stress is a potential therapeutic target for chronic non-infection liver diseases.
MATERIALS AND METHODS
Animals
Rats were housed in the barrier housing facility, and it has in keeping with national standard “Laboratory Animal-Requirements of Environment and Housing Facilities” (GB 14925–2001). The care of laboratory animal and the animal experimental operation have conforming to “Chongqing Administration Rule of Laboratory Animal”. The experimental procedures were approved by the animal laboratory administrative center and the institutional ethics committee of Chongqing Medical University (License number: SYXK YU 2010–0001) and also in accordance with the National Institutes of Health guidelines.
Twenty-four male adult SD rats (obtained from the animal laboratory centre of Chongqing Medical University), weighing 220–250 g, were randomly and equally divided into the following 4 groups: control group, model group, M-0.3 group (Al+Meloxicam 0.3 mg·kg−1 group), and M-1 group (Al+Meloxicam 1 mg·kg−1 group), n = 6 for each groups.
Chemical
AlCl3·6H2O (Sinopharm Chemical Reagent Co., Ltd., China) and sodium gluconate (Beijing Qing Sheng Da Chemical Technology Co., Ltd., China) were of analytical grade. Meloxicam was purchased from Kunshan Rotam Reddy Pharmaceutical Co., Ltd, China. According to previously reported methods [13, 17], an aluminium gluconate solution (20 mg Al3+·ml−1) was prepared on the day of the experiments by adding 17.9 g of AlCl3· 6H2O and 9.9 g of sodium gluconate to 100 ml of double distilled water (ddH2O), and the pH of the solution was adjusted to approximately 6.0.
Establishment of animal models
After 3 days of acclimatisation, the rats were treated intragastrically once a day, 5 d per week for 20 weeks with the following solutions: the model group received 10 ml·kg−1 aluminium gluconate solution (200 mg Al3+·kg−1), and the control group received the same volume of a sodium gluconate solution. The M-0.3 group and M-1 group were treated intragastrically with 0.3 and 1 mg.kg−1 meloxicam, respectively, 30 min after intragastric administration of aluminium gluconate (200 mg Al3 +·kg−1).
Liver function tests
On the second day after the cessation of Al gluconate administration, 6 rats from each group were chosen, and 1 ml blood was collected from each rat orbit. The blood plasma levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST) and alkaline phosphatase (ALP) were measured according to a kit manual (Jiancheng Bioengineering Ltd, Nanjing, China).
Histopathological observation
On the second day after the cessation of Al gluconate or sodium gluconate administration, 3 rats from each group were chosen for histopathological observation. The rats were intraperitoneally anesthetised with 4% chloral hydrate (10 ml·kg-1) and transcardially perfused with heparinised saline (100 ml) followed by 4% paraformaldehyde in phosphate-buffered saline (200 ml). The rat livers were removed and stored in the same fixative solution. The liver tissue was sliced into 5-μm-thick sections for haematoxylin and eosin staining (H.E). Hepatic pathological changes were observed under light microscopy.
Before perfusion, a portion of the fresh liver was taken to prepare for subsequent experiments including measure of MDA content and SOD activity and the test of WB and ELISA.
Immunohistochemical staining test
Immunohistochemistry was performed to investigate the expression of COX2 and EP1,2,3,4 in the rat livers. Briefly, liver sections of 3 rats from each group were dewaxed and rehydrated in decreasing concentrations ethanol. Then the sections were blocked for endogenous peroxidase in 3% H2O2 in methanol for 20 min at room temperature. Slides were washed with PBS for three times and pre-incubated in 1% serum for 30 min at room temperature. Thereafter, the sections were incubated with primary antibodies COX2 (dilution 1:50, Santa, USA) and EP1,2,3,4 (dilution 1:100, Santa, USA) overnight at 4°C. Then, the sections were incubated with biotinylated secondary antibody (dilution 1:100) for 30 min at 37°C, and incubated with streptavidin for 20 min, and then rinsed for another 3 min × 3 with PBS before reaction with DAB solution. The sections were counterstained with hematoxylin and then observed under a microscope. The average optical density (AOD) was used to represent the expression of COX2 in the sections. The AOD of COX2 in the sections were analyzed by Image Pro Plus6.0 (IPP).
Measurement of malondialdehyde (MDA) content and superoxide dismutase (SOD) activity
Rat livers from each group of were removed on the second day after Al gluconate administration was completed (n = 6). The SOD activity and MDA content were detected according to the instruction manual of a kit (Jiancheng Bioengineering Ltd, Nanjing, China). The protein content was measured using a BCA protein assay kit (Beyotime, China).
Enzyme-linked immunosorbent assay (ELISA)
Rat livers from each group of rats (n = 6)were removed on the second day after Al gluconate administration was completed. PGE2 (TaKaRa Japan), cAMP (R&D Systems China shanghai, China), TNF-α (HuaMei Bioengineering Ltd, Wuhan, China), IL-1β (HuaMei Bioengineering Ltd, Wuhan, China) and IL-6 (HuaMei Bioengineering Ltd, Wuhan, China) were detected with ELISA kits.
Western blotting test
Fifty mg of rat liver (n = 3) were added to 0.5 ml of tissue lysate solution for protein extraction and centrifugation at 12,000 × g for 10 min at 4°C, and the supernatant was used for detection of protein concentrations with a BCA protein assay kit (Beyotime, China). A 10 μL sample of protein was separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membranes (Millipore, USA). The membranes were blocked with 5% BSA for 1 h at room temperature and then probed with specific primary antibodies, including anti-EP1, EP2, EP3, EP4 and PKA (1:300; Santa, USA) and β-actin (1:3000; Proteintech, USA) overnight at 4°C. The membranes were washed three times in TBST and incubated with HRP-conjugated secondary antibodies at room temperature for one hour. Following four washes in TBST, protein signals were visualized by ECL (Bio-Rad, USA).
Statistical analysis
The results were expressed as the means ± standard deviation (SD) and were analysed with SPSS 12.0 (SPSS Inc. Chicago, US). Within-group variances were compared using Dennett’s t-test. Statistical significance was represented by P < 0.05.
ACKNOWLEDGMENTS AND FUNDING
This reported study was supported by research grants from the Chongqing science technology commission of China (NO: cstc2013jcyjA10004) and the Chongqing Municipal Health Bureau (NO: 2012-2-047).
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
Authors’ contributions
Junqing Yang made substantial contribution to conception and design and performance of the study. Yang Yang, Qin He, Hong Wang, Xinyue Hu, Ying Luo, Guojuan Liang, Shengnan Kuang, Shaoshan Mai, Jie Ma, Xiaoyan Tian and Qi Chen participated in performance of all experiments and carried out the data analysis. Yang Yang participated in performance of the study and in writing the manuscript. All authors read and approved the final manuscript.
REFERENCES
1. Ochmański W, Barabasz W. Aluminum—occurrence and toxicity for organisms. Przegl Lek. 2000; 57:665–668.
2. Reinke CM, Breitkreutz J, Leuenberger H. Aluminium in over-the-counter drugs: risks outweigh benefits? Drug Saf. 2003; 26:1011–1025.
3. Sushma NJ, Rao KJ. Total ATPases activity in different tissues of albino mice exposed to aluminium acetate. J Environ Biol. 2007; 28:483–484.
4. Kumar V, Gill KD. Aluminium neurotoxicity: neurobehavioural and oxidative aspects. Arch Toxicol. 2009; 83:965–978.
5. Tetsuji N, Kiyokazu K. Aluminium accumulation in the human body and experimental animals. J Health Environ Sci. 2005; 1:12–48.
6. Bogdanović M, Janeva AB, Bulat P. Histopathological changes in rat liver after a single high dose of aluminium. Arh Hig Rada Toksikol. 2008; 59:97–101.
7. Viezeliene D, Jansen E, Rodovicius H, Kasauskas A, Ivanov L. Protective effect of selenium on aluminium-induced oxidative stress in mouse liver in vivo. Envir Toxicol Pharmacol. 2011; 31: 302–306.
8. Türkez H, Yousef MI, Geyikoglu F. Propolis prevents aluminium-induced genetic and hepatic damages in rat liver. Food and Chemical Toxicol. 2010; 48:2741–2746.
9. Agarwal DR, Gupta SB. An alarming hazard in the community using aluminium day to day life on the basis of toxic effects on the liver of abino rats by ingestion of aluminium. National Journal of Community Medicine. 2010; 1:82–84.
10. Willhite CC, Karyakina NA, Yokel RA, Yenugadhati N, Wisniewski TM, Arnold IM, Momoli F, Krewski D. Systematic review of potential health risks posed by pharmaceutical, occupational and consumer exposures to metallic and nanoscale aluminum, aluminum oxides, aluminum hydroxide and its soluble salts. Crit Rev Toxicol. 2014; 4:1–80.
11. Gonzalez MA, Alvarez Mdel L, Pisani GB, Bernal CA, Roma MG, Carrillo MC. Involvement of oxidative stress in the impairment in biliary secretory function induced by intraperitoneal administration of aluminum to rats. Biol Trace Elem Res. 2007; 116:329–348.
12. Jangra A, Kasbe P, Pandey SN, Dwivedi S, Gurjar SS, Kwatra M, Mishra M, Venu AK, Sulakhiya K, Gogoi R, Sarma N, Bezbaruah BK, Lahkar M, et al. Hesperidin and Silibinin Ameliorate Aluminum-Induced Neurotoxicity: Modulation of Antioxidants and Inflammatory Cytokines Level in Mice Hippocampus. Biol Trace Elem Res. 2015; 168:462–71.
13. Yang Y, Wang H, Guo Y, Lei W, Wang J, Hu X, Yang J, He Q. Metal Ion Imbalance-Related Oxidative Stress Is Involved in the Mechanisms of Liver Injury in a Rat Model of Chronic Aluminum Exposure. Biol Trace Elem Res. 2016; 173:126–131.
14. Shati AA, Alamri SA. Role of saffron(Crocus sativus L) and honey syrup on aluminum-induced hepatotoxicity. Saudi medical journal. 2010; 31:1106–1113.
15. El-Sayed WM, Al-Kahtani MA, Abdel-Moneim AM. Prophylactic and therapeutic effects of taurine against aluminum-induced acute hepatotoxicity in mice. Journal of hazardous materials. 2011; 192:880–886.
16. Cheng D, Zhu C, Wang C, Xu H, Cao J, Jiang W. Hepatoprotective effects of apple polypheol extract on aluminum-induced liver oxidative stress in the rat. Canadian joural of physiology and pharmacology. 2014; 92:109–116.
17. Mai S, He Q, Wang H, Hu X, Luo Y, Yang Y, Kuang S, Tian X, Ma J, Yang J. 5-lipoxygenase activation is involved in the mechanisms of chronic hepatic injury in a rat model of chronic aluminum overload exposure. Toxicol Appl Pharmacol. 2016; 305:259–266.
18. Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001; 294:1871–1875.
19. Lu L. NS3 protein of hepatitisC virus regulates cyclooxygenase2 expression through multiple signaling pathways.Virology. 2008; 371: 61–70.
20. Ekor M, Odewabi AO, Kale OE, Adesanoye OA, Bamidele TO. Celecoxib, a selective cyclooxygenase-2 inhibitor, lowers plasma cholesterol and attenuates hepatic lipid peroxidation during carbon-tetrachloride-associated hepatotoxicity in rats. Drug Chem Toxicol. 2013; 36: 1–8.
21. Yu J. COX2 induction in mice with expefimental nutritionaI steatohepatitis: Role as pro-Inflammatory mediator.Hepatology. 2006; 43:826–836.
22. Draper HH, Hadley M. Malondialdhyde determination as index of lipid peroxidation. Methods in enzymology. 1990; 186:421–431.
23. Nanji AA, Jokelainen K, Tipoe GL, Rahemtulla A, Dannenberg AJ. Dietary saturated fatty acids reverse inflammatory and fibrotic changes in rat liver despite continued ethanol administration. J Pharmacol Exp Ther. 2001; 299:638–644.
24. Kutlubay, R. Amelioration of aluminium-induced liver damage by vitamin E. Saudi Med J. 2007; 28:197–200.
25. Di J, Yao K, Han W, Bi S. Study on the interaction of copper-zinc superoxide dismutase with aluminum ions by electrochemical and fluorescent method. Spectrochim Acta A Mol Biomol Spectrosc. 2006; 65:896–900.
26. Nehru B, Anand P. Oxidative damage following chronic aluminium exposure in adult and pup rat brains. J Trace Elem Med Biol. 2005; 19:203–208.
27. Mailloux RJ, Lemire J, Appanna VD. Hepatic response to aluminum toxicity: dyslipidemia and liver diseases. Exp Cell Res. 2011; 317:2231–2238.
28. Harris WR, Berthon G, Day JP, Exley C, Flaten TP, Forbes WF, Kiss T, Orvig C, Zatta PF. Speciation of aluminium in biological systems. J Toxicol Environ Health. 1996; 48:543–68.
29. Takahashi S, Takahashi I, Sato H, Kubota Y, Yoshida S, Muramatsu Y. Determination of major and trace elements in the liver of Wistar rats by inductively coupled plasma-atomic emission spectrometry and mass spectrometry. Lab Anim. 2000; 34:97–105.
30. Ludwiczek S, Aigner E, Theurl I, Weiss G. Cytokine-mediated regulation of iron transport in human monocytic cells. Blood. 2003; 101:4148–4154.
31. Tetsuji N, Kiyokazu K. Aluminium accumulation in the human body and experimental animals. J Health Environ Sci. 2005; 1:12–48.
32. Neyrinck AM, Margagliotti S, Gomez C, Delzenne NM. Kupffer cell-derived prostaglandin E2 is involved in regulation of lipid synthesis in rat liver tissue. Cell Biochem Funct. 2004; 22:327–332.
33. Nanji AA, Miao L, Thomas P, Rahemtulla A, Khwaja S, Zhao S, Peters D, Tahan SR, Dannenberg AJ. Enhanced cyclooxygenase-2 gene expression in alcoholic liver disease in the rat. Gastroenterology. 1997; 112:943–951.
34. Nanji AA, Jokelainen K, Tipoe GL, Rahemtulla A, Dannenberg AJ. Dietary saturated fatty acids reverse inflammatory and fibrotic changes in rat liver despite continued ethanol administration. J Pharmacol Exp Ther. 2001; 299:638–644.
35. Reddy ST, Wadleigh DJ, Herschman HR. Transcriptional regulation of the cyclooxygenase-2 gene in activated mast cells. J Biol Chem. 2000; 275:3107–3113.
36. Sobolewski C, Cerella C, Dicato M, Ghibelli L, Diederich M. The role of cyclooxygenase-2 in cell proliferation and cell death in human malignancies. Int J Cell Biol. 2010; 215158.
37. Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 2001; 41:661–690.
38. Bresell A. Bioinformatic and enzymatic characterization of the MAPEG superfamily. FEBS J. 2005; 272:1688–1703.
39. Siljehav V, Olsson Hofstetter A, Jakobsson PJ, Herlenius E. mPGES-1 and prostaglandin E2:vital role in inflammation, hypoxic response, and survival. Pediatr Res. 2012; 72: 460–467.
40. Andreasson K. Emerging roles of PGE2 receptors in models of neurological disease. Prostaglandins Other Lipid Mediat. 2010; 91:104–112.
41. Ikeda-Matsuo Y. Microsomal prostaglandin E synthase-1 contributes to ischaemic excitotoxicity through prostaglandin E2 EP3 receptors. Br J Pharmacol. 2010; 160:847–859.
42. Boie Y. Molecular cloning and characterization of the four rat prostaglandin E2 prostanoid receptor subtypes. Eur J Pharmacol. 1997; 3400:227–241.
43. Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994; 46:205–229.
44. Lemasters JJ, DiGuiseppi J, Nieminen AL, Herman B. Bl ebbing, free Ca2+ and mitochondrial membrane potential preceding cell death in hepatocytes. Nature. 1987; 325:78–81.
45. Murata M, Monden M, Umeshita K, Nakano H, Kanai T, Gotoh M, Mori T. Role of intracellular calcium in superoxide-induced hepatocyte injury. Hepatology. 1994; 19:1223–1228.
46. Wang M. p38 mitogen activated protein kinase mediates both death signaling and functional depression in the heart. Ann Thorac Surg. 2005; 80:2235–2241.
47. Nguyen G. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002; 109:1417–1427.
48. Olteanu D, Filip A, Mureşan A, Nagy A, Tabaran F, Moldovan R, Decea N, Catoi C, Clichici S. The effects of chitosan and low dose dexamethasone on extrahepatic cholestasis after bile duct ligation in Wistar rats. Acta Physiologica Hungarica. 2012; 99:61–73.
49. Milatovic D, Montine TJ, Aschner M. Prostanoid signaling: dual role for prostaglandin E2 in neurotoxicity. Neurotoxicology. 2011; 32:312–319.
50. Kelly E, Bailey CP, Henderson G. Agonist-selective mechanisms of GPCR desensitization. Br J Pharmacol. 2008;153:S379–88.
51. Roach M, Taniguchi K, Ishikawa T, Kim HS, Flannery PJ, Coffman TM, McNeish JD, Audoly LP. The Prostaglandin E2 EP1 receptor mediates pain perception and regulates blood pressure. J Clin Invest. 2001; 107:325–31.
52. Treffkorn L, Scheibe R, Maruyama T, Dieter P. PGE2 exerts its effect on the LPS-induced release of TNF-alpha, ET-1, IL-1alpha, IL-6 and IL-10 via the EP2 and EP4 receptor in rat liver macrophages. Prostaglandins Other Lipid Mediat. 2004; 74:113–123.
53. Ueta M. Regulation of ocular surface inflammation by prostaglandin E receptor subtype EP3. Cornea. 2010; 29: S57–61.
54. Natura G, Bär KJ, Eitner A, Boettger MK, Richter F, Hensellek S, Ebersberger A, Leuchtweis J, Maruyama T, Hofmann GO, Halbhuber KJ, Schaible HG. Neuronal prostaglandin E2 receptor subtype EP3 mediates antinociception during inflammation. Proc Natl Acad Sci USA. 2013; 110:13648–13653.