
INTRODUCTION
Rac GTPases (Rac1/1b/2/3) have been implicated in cancer cell motility, survival, and proliferation. The three highly homologous Rac proteins are encoded by separate genes (RAC1/2/3), and isoform-specific functions remain to be fully elucidated. Rac1 contains a distinct carboxyl terminus that drives oligomerization and possibly nuclear translocation [1, 2]. Rac1b is a constitutively active splice variant ot Rac1 overexpressed in breast and other cancers [3]. Rac2 is primarily expressed in hematopoetic cells [4]. Rac3 is primarily expressed in brain tissues, and has been shown to be dysregulated in ovarian, breast, gastric, and brain cancers [5, 6]. While activating mutations in RAC1/2/3 are rare, Rac hyperactivation is a common theme in many cancers including breast cancer [7-12]. Aberrant Rac signaling frequently occurs through Rac guanine exchange factor hyperactivation resulting from deregulated upstream signaling events. Rac-activating GEFs such as Tiam1, Trio, Vav3, and PREX-1 are overexpressed in breast tumors [8-11]. Canonical Rac signaling involves activation of p21-activated kinases (PAKs), which in turn activate mitogen-activated protein kinases (MEK1/2 and ERK1/2) to drive proliferation and survival pathways [13]. Mounting evidence suggests that Rac plays a key role in the phosphatidylinositol 3-kinase (PI3K) pathway [14-16]. Class IA PI3Ks are typically activated by receptor tyrosine kinases and G protein-coupled receptors. PI3K phosphorylates phosphatidylinositol (4,5)-bisphosphate (PIP2) to create the 3,4,5-trisphosphate (PIP3) at the plasma membrane, and PIP3 recruits intracellular pleckstrin homology (PH) domain-containing proteins such as AKT for activation. Rac1 directly binds and activates the p110β isoform of PI3K [14]. We recently described a positive feedback loop where Rac signaling drives activation of receptor tyrosine kinase (RTK)/PI3K pathways that activate PREX-1 in breast cancer [15].
The PI3K/AKT/mechanistic target of rapamycin (mTOR) pathway promotes cell growth, proliferation, migration, and survival, and as such, aberrations within this signaling axis occur in the majority of breast and other cancers [17]. Several inhibitors of PI3K and mTOR are in clinical trials for estrogen receptor α-positive (ER+) and HER2-overexpressing (HER2+) breast cancers. mTOR exists in two complexes, mTORC1 and mTORC2, that lie upstream and downstream of AKT, respectively [18, 19]. The mTORC1 inhibitor everolimus is approved for the treatment of advanced ER+ breast cancer. While these drugs have shown encouraging clinical results, efficacy may be limited due to extensive cross-talk and compensatory feedback upregulation of MEK/ERK and RTK signaling, and upregulation of PI3K/AKT signaling by mTORC1 inhibition [20-23]. Preclinical studies testing combinations of PI3K/AKT/mTOR and MEK/ERK pathway-directed inhibitors have shown impressive anti-tumor effects in a variety of cancer subtypes, but these drug combinations have proven toxic in humans [24, 25]. With evidence implicating Rac in both of these key oncogenic signaling pathways, we investigated the therapeutic potential of inhibiting Rac activity as a means to simultaneously target the PI3K and MEK pathways in breast cancer.
RESULTS
Rac inhibition suppresses growth and induces apoptosis in breast cancer cells
The small molecule EHT1864 binds Rac1/1b/2/3 and promotes loss of guanine nucleotide association, locking Rac in an inactive conformation, and inhibiting GTPase activity and engagement of downstream effectors. EHT1864 blocks activation of Rac, but not the related proteins CDC42 or RhoA, at a concentration of 50 μM in glioblastoma cells [26, 27]. We screened 17 human breast cancer cell lines for sensitivity to EHT1864 in growth assays. IC50 values ranged from 2.0 to 39.1 μM (Figure 1A and Supplementary Figure 1). Relative levels of activation of the PI3K/AKT pathway [assessed by phospho-AKTT308 and phospho-AKTS473 as respective markers of phosphatidylinositol 3,4,5-trisphosphate (PIP3) levels and mTORC2 activity] and the MEK/ERK pathway (assessed by P-ERK1/2), or levels of Rac1 and Rac3 did not generally correlate with sensitivity to EHT1864 (Figure 1B). Three tested cell lines harbor RAC3 genomic amplification, but this aberration also did not correlate with EHT1864 sensitivity. Interestingly, cell lines that harbor activating mutations in the gene encoding the p110α catalytic subunit of PI3K (PIK3CA), or amplification of the ERBB2 (HER2) proto-oncogene showed significantly increased sensitivity to EHT1864 (Figure 1C). EHT1864 also induced apoptosis in 4/4 breast cancer cell lines tested in a dose-dependent manner. Notably, Rac inhibition induced a greater degree of apoptosis (compared to baseline) in BT-474 and T47D cells, which had lower IC50 values in growth assays, compared to MDA-MB-415 and CAMA-1 cells (Figure 1D).
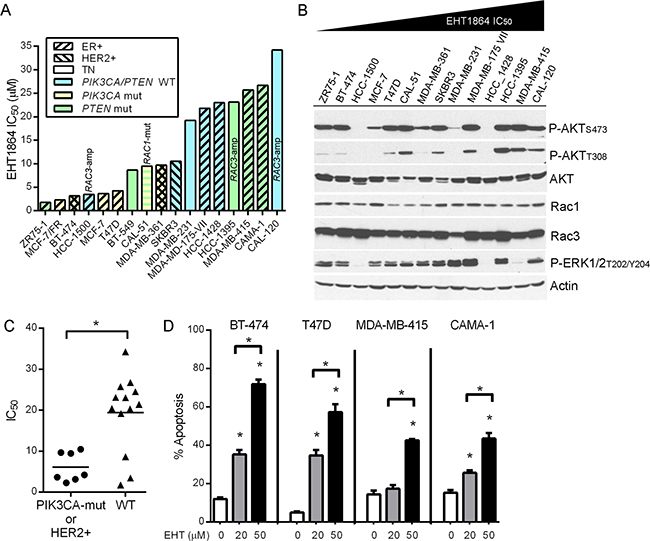
Figure 1: Rac inhibition suppresses growth and induces apoptosis in PIK3CA-mutant and HER2+ breast cancer cells. (A) Breast cancer cells were treated with 0-100 μM EHT1864 for 4-5 d. Relative viable cell numbers were assessed by SRB assay. Mutational and DNA copy number profiles were obtained from ref. [53]. RAC3-amp-RAC3 gene amplification. RAC1-mut- RAC1N39S mutation, predicted to be low-impact per mutationassessor.org [54]. MCF-7/FR- fulvestrant-resistant MCF-7 cells maintained and treated in 1 μM fulv. (B) Cell lysates were analyzed by immunoblot. (C) Comparison of IC50 values between cells harboring a PIK3CA mutation and/or HER2 amplification vs. PIK3CA/HER2-wild-type cells. *p=0.015 by Mann-Whitney U-test. (D) Cells were treated with EHT for 72 h before apoptosis assay. *p<0.0001 by Bonferonni post-hoc test compared to control for each cell line unless otherwise indicated with brackets.
Sensitivity to Rac inhibition is associated with dual inhibition of MEK/ERK and PI3K/AKT/mTOR pathways
We previously reported that Rac inhibition suppresses both the MEK/ERK and PI3K/AKT/mTOR pathways in ER+ breast cancer cells [15]. To identify potential differences in Rac signaling between EHT1864-sensitive vs. -resistant breast cancer cells (Figure 1A), we evaluated effects on these oncogenic pathways. In EHT1864-sensitive cells, but not cells with relative drug resistance, EHT1864 treatment decreased levels of both phospho-AKTT308 and P-ERK1/2 (Figure 2A), suggesting that dual inhibition of the PI3K/AKT and MEK/ERK pathways is required for sensitivity to Rac inhibition.
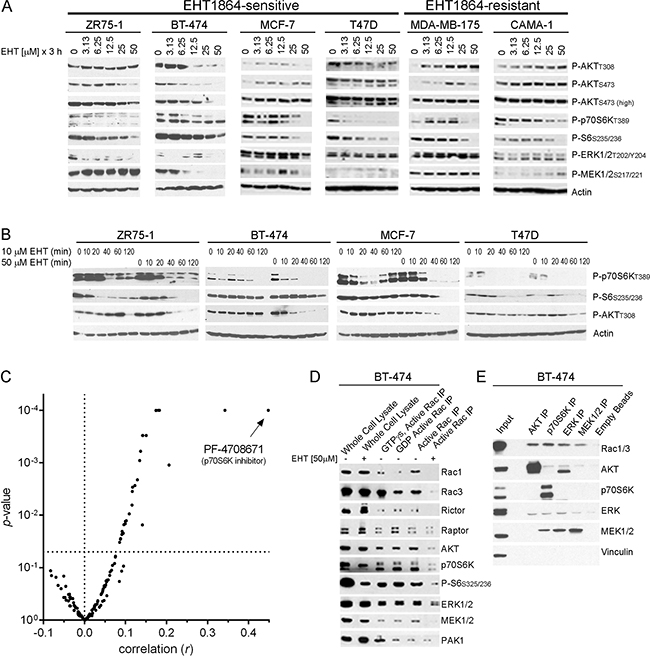
Figure 2: Sensitivity to Rac inhibition is associated with dual inhibition of MEK/ERK and PI3K/AKT/mTORC1 pathways. (A–B) Cells were treated with 0-50 μM EHT1864 for 3 h (A), or 10 or 50 µM EHT1864 for 0-120 min (B), and lysates were analyzed by immunoblot. Cells that are relatively sensitive vs. resistant to EHT1864 (from Figure 1A) are indicated. (C) Mining of sensitivity data from 656 cancer cell lines treated with a panel of 138 drugs [28] revealed that the sensitivity profile of EHT1864 most strongly correlates with the profile of the p70S6K inhibitor PF-4708671. (D–E) In (D), activated Rac was pulled down under control-, GTPγs-, GDP-, and EHT1864-treated conditions. In (E), AKT, p70S6K, ERK, and MEK1/2 were immunoprecipitated from cell lysates. Eluates and lysates were analyzed by immunoblot.
In PIK3CA-mutant and HER2+ breast cancer cells, PI3K/AKT signaling frequently drives mTORC1, which in turn activates p70S6 kinase (p70S6K) (Supplementary Figure 2). We observed that EHT1864 treatment often induced decreases in phospho-p70S6K at lower doses and earlier time points than decreases in phospho-AKT (Figure 2A/2B). This suggests that EHT1864 inhibits mTORC1/p70S6K activation at a node downstream and independent of AKT. Capitalizing on the public availability of drug sensitivity profiles from 656 cancer cell lines to 138 anti-cancer drugs [28], we compared patterns of IC50 values for EHT1864 to each other drug. This analysis revealed that the sensitivity profile of EHT1864 is most strongly correlated with the profile of the p70S6K inhibitor PF-4708671 (Figure 2C), further supporting the notion that the growth-inhibitory effects of Rac inhibition involve p70S6K inhibition.
Active Rac1 has been shown to bind and activate PI3K/p110β [14]. Active Rac1 has been found in complex with mTORC1 and mTORC2, and is thought to direct complex localization [29]. GTP-bound Rac1 has also been shown to bind p70S6K and promote p70S6K activation [30]. To determine whether Rac interacts with proteins in the AKT/mTOR and MEK/ERK signaling cascades in breast cancer cells, we performed Rac pulldown assays: beads coated with protein encoding the p21-binding domain (PBD) of the Rac/CDC42 effector PAK1 are used to capture active GTP-bound Rac and CDC42, which are then detected by immunoblot analysis of bead eluates. Treatment of cell lysates with the non-hydrolyzable nucleotide GTPγs locks Rac in an active conformation, while treatment with GDP stoichiometrically promotes Rac inactivation. In GTPγs-treated lysates of BT-474 cells, increased amounts of Rac1, Rac3, and PAK1 were pulled-down, confirming assay functionality. In untreated lysates, PBD beads pulled-down AKT, the mTORC2 component Rictor, AKT, p70S6K, MEK1/2, and ERK1/2 in a Rac activation-dependent manner, as confirmed by treatment with EHT1864 (Figure 2D). PBD beads also pulled-down Raptor independent of Rac activation. Whether these proteins exist in distinct or overlapping complexes with Rac/CDC42 will require further in-depth study. Reverse immunoprecipitations of AKT, p70S6K, ERK1/2, and MEK1/2 confirmed that these proteins complex with Rac1 and/or Rac3 (Figure 2E). These data also suggested that some proteins exist in unexpected complexes, such as MEK and p70S6K. These observations imply that Rac directly engages components of the PI3K/AKT /mTOR and MEK/ERK axes.
Constitutive AKT activation does not confer resistance to Rac inhibition
Since Rac was found in complex with AKT and p70S6K, and EHT1864 inhibited phosphorylation of p70S6K prior to AKT (Figure 2B/2D/2E), we tested whether AKT inhibition was critical for the growth-suppressive effect of EHT1864. We generated MCF-7 cells stably overexpressing constitutively active AKT (AKTmyr or AKTDD). AKT activation did not alter sensitivity to EHT1864 (Figure 3A/3B and supplementary Figure 3). EHT1864 treatment decreased phosphorylation of p70S6K and the mTORC1 substrate 4EBP1 despite constitutive AKT activation (assessed by phosphorylation of the AKT substrates GSKα/β) (Figure 3C). These data collectively support a model in which Rac activates mTORC1/p70S6K independent of PI3K/AKT.
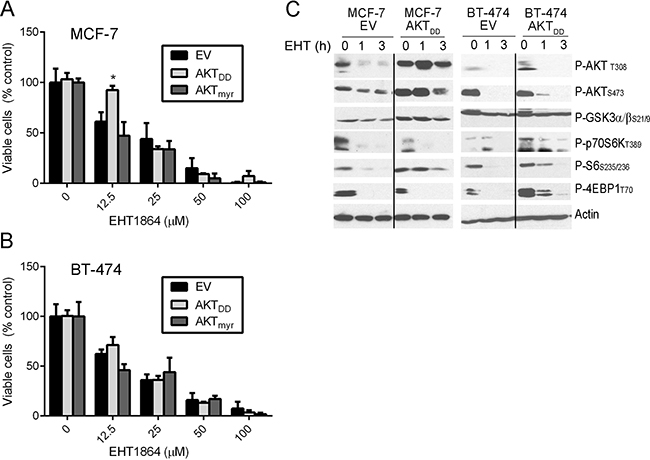
Figure 3: Overexpression of p70S6K confer resistance to EHT1864. (A–B) MCF-7 and BT-474 cells were stably transfected with vectors encoding AKT1DD or AKT1myr constitutively active mutants or EV, and sensitivity to EHT1864 was assessed via growth assay. In (C) lysates were analyzed by immunoblot. *p<0.05 by Bonferroni multiple comparison-adjusted post-hoc test compared to EV control at each dose of EHT.
Duration and magnitude of Rac inhibition affect breast cancer cell growth
We and others have shown that transient interruption of oncogenic kinase signaling is sufficient to elicit robust, delayed anti-cancer effects [31-34]. To determine the duration of Rac inhibition required to induce anti-cancer effects in EHT1864-sensitive cells (from Figure 1A), cells were treated with 0-50 μM EHT1864 for 0-120 h, followed by drug washout. Relative numbers of viable cells were measured after 120 h. A 2- to 4-h exposure to 50 μM EHT1864 decreased cell viability ≥50% (Figure 4). In contrast, a lower concentration of EHT1864 (12.5 μM) required longer durations of exposure (48-60 h) to appreciably decrease viability, reflecting a relationship between duration and magnitude of Rac inhibition, and cell viability.
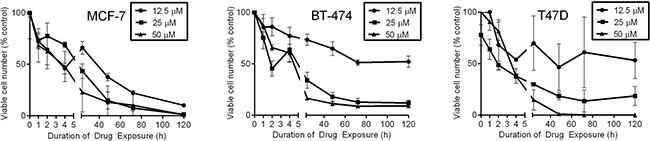
Figure 4: Short-term exposure to EHT1864 elicits prolonged growth-suppressive effects. Cells were treated with 12.5, 25, or 50 μM EHT1864 for 0, 1, 2, 4, 8, 24, 48, 72, or 120 hours, then drug was washed out. Relative viable cell numbers were quantified at the 120-h time point.
Pharmacokinetic analysis of EHT1864 in mice
Pharmacokinetics of EHT1864 in mice
Despite being used as a tool compound in many preclinical studies, the pharmacokinetic properties and anti-tumor efficacy of EHT1864 have not been previously reported. The plasma EHT concentration vs time profile following a single i.p. injection of EHT1864 (100 mg/kg) is shown in Figure 5A. Non-compartmental analysis of the mean plasma EHT1864 concentration versus time data revealed an estimated elimination half-life of 99.2 min (1.65 h).The mean maximum plasma concentration (Cmax) was 125.6 μM (range 107.7-147.8 μM) and the mean Tmax was 5 minutes post injection. After 1 h, the mean plasma concentration of EHT1864 had decreased to 54.1 μM (range 49.1-60 μM), and declined within 4 h to concentrations unlikely to appreciably inhibit cancer cell growth (Figure 5A). Assuming log-linear clearance from plasma, EHT1864 was estimated to be present at ≥10 μM for 1.24 h.
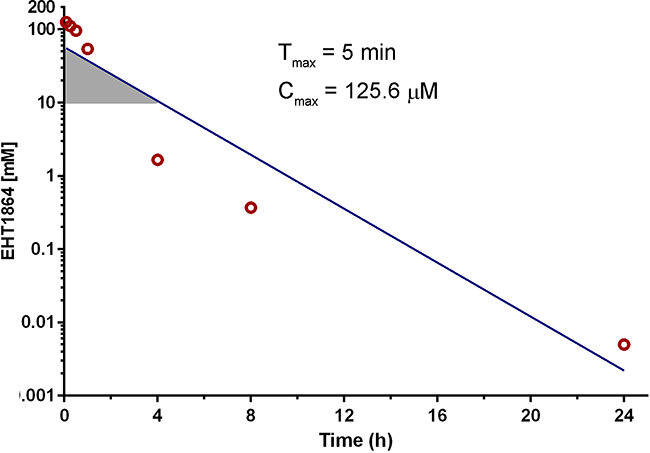
Figure 5: Pharmacokinetic analysis of EHT1864 in mouse plasma. Mice were injected i.p. with a single dose of EHT1864 (100 mg/kg), and blood was collected from 3 mice per time point over the next 24 h. Plasma was separated for EHT1864 concentration measurement. The Tmax- time to maximum concentration; Cmax- maximum concentration; were the observed mean values and the terminal elimination half-life was estimated using non compartmental analysis. (mean elimination t1/2 = 99.2 min /1.65h). The shaded region indicates estimated time that plasma EHT1864 concentration exceeded 10 μM.
Treatment with 100 mg/kg EHT1864 twice daily was well-tolerated, while 150 mg/kg twice daily caused signs of toxicity (i.e., lethargy).
Rac inhibition suppresses breast tumor growth
Mice bearing s.c. ER+/HER2+/PIK3CA-mutant BT-474 xenografts were treated with EHT1864 (100 mg/kg) or vehicle twice daily. EHT1864 significantly slowed tumor growth compared to vehicle control (Figure 6A and Supplementary Figure 4; mean weekly growth rates of 50% vs. 27%). In tumor specimens acquired after 2 wk of treatment, EHT1864 greatly reduced levels of P-ERK1/2, P-AKT, P-p70S6K, and P-Histone H3S10 (marker of mitosis) (Figure 6B).
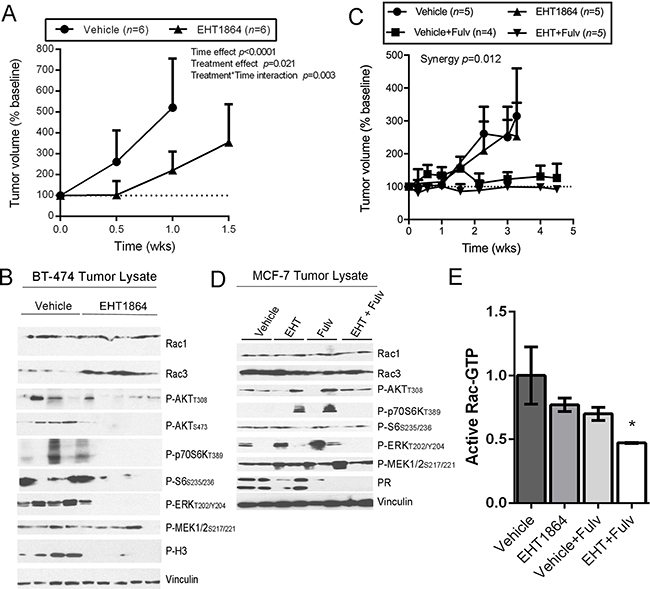
Figure 6: EHT1864 inhibits breast tumor growth. (A, C) Mice bearing BT-474 tumors (A) or MCF-7 tumors (C) were randomized to drug treatments as indicated. Data are presented as % tumor volume relative to baseline (mean + SEM). (B, D, E) After 6 wk of treatment, tumors were harvested at 1 h after the final dose of EHT1864. Lysates were analyzed by immunoblot (B/D), or used for active Rac ELISA (E).
In contrast, single-agent EHT1864 modestly slowed growth of s.c. ER+/HER2-/PIK3CA-mutant MCF-7 xenografts (Figure 6C and Supplementary Figure 5; mean weekly growth rates of 4.32% vs. 3.4%; p=0.035). MCF-7 cells require exogenous estrogen supplementation to form tumors in mice [35]. Estrogen-activated ER is a major driver of MCF-7 tumor growth, and inhibition of oncogenic signaling pathways (e.g., PI3K/AKT) induces upregulation of ER levels and activation. While single-agent therapies targeting oncogenic pathways are often only modestly effective against ER+ breast tumors, combination treatment with anti-estrogens is frequently more effective than anti-estrogens alone [36, 37]. Indeed, treatment with the anti-estrogen fulvestrant significantly inhibited growth of MCF-7 tumors (p<0.0001; mean weekly growth rate of 1.43%), while the combination of EHT1864 and fulvestrant was significantly more effective than either single agent, and provided stable disease (Figure 6C; p<0.0001 vs. vehicle; synergy p=0.012; mean weekly growth rate of 0.97%). In tumor specimens acquired after 3-6 wk of treatment, we observed that fulvestrant downregulated levels of progesterone receptor (PR), which is encoded by an ER-inducible gene (Figure 6D). EHT1864 reduced the levels of active Rac-GTP (Figure 6E and Supplementary Figure 6), but did not appreciably affect ERK, AKT, or p70S6K phosphorylation (Figure 6D); it is possible that tumor cells adapted to Rac inhibition during treatment to maintain oncogenic pathway activation, and/or the time point selected for analysis was outside the window of pathway inhibition. However, EHT1864 in combination with fulvestrant reduced levels of active Rac, MEK1/2, ERK1/2, and AKT (Figure 6D/6E).
DISCUSSION
The role of Rac GTPases in cancer processes has been widely documented. However, Rac has remained an elusive therapeutic target. We demonstrate that, among breast cancer cells, mutations in PIK3CA and/or HER2 are predictive of increased sensitivity to Rac inhibition with EHT1864. Sensitivity to Rac inhibition was associated with EHT1864-induced decreases in activation of both the AKT/mTOR/p70S6K and MEK/ERK pathways, identifying Rac as a key upstream signaling node in both pathways in Rac-dependent cells. Temporal and dose-response analyses revealed that Rac activates mTORC1/p70S6K independently of AKT; when considered in the context of prior findings [14, 15, 29, 30], these observations place Rac both upstream and downstream of AKT/mTORC1. Despite only providing transient Rac inhibition in vivo, EHT1864 significantly inhibited growth of breast tumors in mice.
The PI3K/AKT/mTOR and MEK/ERK pathways are two of the most commonly aberrantly activated pathways in human cancers. Crosstalk and compensatory signaling between these pathways have been widely reported. Although combined therapeutic targeting of these pathways showed impressive preclinical results, such drug combinations elicit considerable toxicity in humans [24, 25], likely because these pathways are essential in many normal cell types. Rac proteins have been directly or indirectly implicated in activation of PI3K/p110β, AKT, mTORC1, mTORC2, p70S6K, MEK1/2, and ERK1/2 [14, 29, 30]. Thus, we considered whether Rac could serve as a single therapeutic target critical to both the PI3K/AKT/mTOR and MEK/ERK pathways. Indeed, Rac inhibition with EHT1864 suppressed growth and induced apoptosis (Figure 1A/1D and Supplementary Figure 1) in cells in which Rac drives both the AKT/mTOR and MEK/ERK pathways (Figure 2A). Among breast cancer cell lines, these features were significantly associated with mutations in PIK3CA or amplification of HER2 (Figure 1C), both of which hyperactivate PI3K. Thus, Rac may drive both the AKT/mTOR and MEK/ERK pathways only in select subtypes of cancer cells, making Rac a promising therapeutic target that could supplant that need for drug combinations targeting individual components of both the PI3K/AKT/mTOR and MEK/ERK pathways. Whether Rac drives activation of the PI3K/AKT/mTOR and MEK/ERK pathways by increasing kinase activity and/or decreasing phosphatase activity requires further study.
A prior study by Katz et al. showed that EHT1864 suppressed cancer cell invasion, proliferation, and survival in a three-dimensional triple-negative breast cancer cell line model, and in patient-derived breast tumor tissue organotypic cultures regardless of ER/HER2 status [38]. EHT1864 decreased levels of STAT3 Ser727 (activating) phosphorylation, survivin, and cyclin D1; the latter two proteins are encoded by STAT3 target genes. These effects were recapitulated by treatment with the STAT3 inhibition Stattic, leading the authors to conclude that EHT1864 effects occurred via STAT3 inhibition. The implication of STAT3, cyclin D1, and survivin in response to Rac inhibition does not necessarily conflict with our findings attributing response to inhibition of the PI3K/AKT/mTOR and MEK/ERK pathways. Cyclin D1 translation is mTORC1-dependent [39]. STAT3 can be phosphorylated at Ser727 by mTORC1 or ERK1/2 [40, 41]. Thus, phospho-STAT3S727 may be a downstream read-out of the PI3K/AKT/mTOR and MEK/ERK pathways.
In addition to EHT1864, other small molecule inhibitors of Rac are in preclinical development. NSC23766 inhibits interactions between of Rac and GEFs, including Trio and Tiam1, inhibiting Rac activation and cancer cell invasion, metastasis, and neoangiogenesis in multiple cancer subtypes [42]. However, concentrations of NSC23766 required to reach efficacious doses limit its therapeutic potential, and NSC23766 does not inhibit the constitutively active Rac1 splice variant Rac1b. EHop-016, a NSC23766 structure-based Rac inhibitor effective at therapeutically achievable doses, slows tumor metastasis and angiogenesis in breast cancer cell lines by blocking Rac interaction with the GEF Vav. However, EHop-016 allowed Rac interaction with Tiam1 and other GEFs, which may ultimately limit its therapeutic utility [43, 44]. In contrast, EHT1864 is a small molecule pan-Rac inhibitor that locks Rac into an inactive conformation by guanine nucleotide displacement rather than inhibition of Rac-GEF interaction. There have been over 70 Rac GEFs reported, many of which are linked to cancer processes. Molecules such as EHT1864 that inhibit Rac in a GEF-independent manner may be a more promising strategy than targeting GEFs [26, 45].
In summary, these results collectively demonstrate that therapeutic targeting of Rac is a promising therapeutic strategy for breast cancer, particularly in cancers harboring activating mutations in PIK3CA or amplification of HER2. Pulsatile treatment studies, combined with pharmacokinetic and tumor growth studies in mice, suggest that transient Rac inhibition elicits significant growth-inhibitory effects. These results warrant further development of Rac inhibitors to improve pharmacologic properties prior to clinical testing.
MATERIALS AND METHODS
Cell Culture
CAL-51 and CAL-120 cells were obtained from DSMZ (Braunschweig, Germany). Other parental cell lines were obtained from ATCC. All cell lines were cultured in DMEM with 10% FBS (Hyclone). Fulvestrant-resistant MCF-7/FR cells were obtained from Matthew Ellis (Washington Univ., St. Louis, MO) and maintained in 1 μM fulvestrant (Tocris Biosciences). EHT1864 [27] was generously provided by Diaxonhit (Paris, France). Cells were stably transfected with viral vectors as described in Supplemental Methods.
Immunoblotting
Immunoblotting of protein extracts from cells and frozen tumor fragments was performed as previously described [46].
Sulforhodamine B (SRB) growth assay
Cells were plated at 5,000/well in 96-well plates and treated in triplicate as indicated. Relative numbers of adherent cells were determined by SRB staining as previously described [47].
Apoptosis assay
Cells were plated at 50,000/well in 12-well plates and treated with EHT1864 for 72 h. Twelve hours before analysis, positive and negative control wells were treated with or without 5 µM BKM120 (PI3K inhibitor, Selleck Chemicals), respectively. Floating and adherent cells (dislodged by trypsinization) were processed using ApoScreen Annexin Apoptosis kit (Southern Biotech) and analyzed by flow cytometry. Cells staining positively for Annexin-V were considered apoptotic.
Active Rac assay
Measurement of active Rac in cell lysates was performed using Rac1 Pull-down Activation Assay Biochem Kit (Cytoskeleton). Cells treated +/- 50 μM EHT1864 for 2 h were lysed on ice in lysis buffer, adjusted for equal protein content (determined by BCA assay, Pierce), and incubated with 10 μL of GST-tagged PAK-PBD beads for 1 h at 4°C per manufacturer’s instructions. Positive and negative control lysates were incubated with GTPγS (non-hydrolysable GTP) and GDP, respectively, for 15 min at room temperature prior to incubation with beads. Beads were then washed, and protein was eluted with 1x NuPAGE LDS Sample Buffer (Life Technologies) with 5% β-mercaptoethanol. Eluates and whole-cell lysates were analyzed by immunoblotting.
Measurement of active Rac in tumor lysates was performed using the colorimetric Rac1 G-LISA Activation Assay Kit (Cytoskeleton). Frozen tumor fragments were homogenized in lysis buffer, and protein content was quantified by BCA assay. Active Rac in lysates was bound to wells during a 30-min incubation at 4°C. Recombinant active Rac protein and lysis buffer were used as positive and negative controls, respectively. Wells were then washed and serially incubated with antigen-presenting buffer, anti-Rac1 primary antibody, HRP-conjugated secondary antibody, and HRP-detection reagents per manufacturer’s instructions. Relative amounts of active Rac were determined through spectrophotometric readings at 490 nm.
Pharmacokinetic analyses
All animal studies were approved by the Dartmouth College IACUC. Female NOD-scid/IL2Rγ−/− (NSG; NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice (6-7 wks old; obtained from the Norris Cotton Cancer Center Transgenic & Genetic Construct Shared Resource) were treated with 100 mg/kg EHT1864 i.p. Blood was collected by cardiac puncture from 3 mice per time point for up to-24 h post i.p injection. Plasma was separated and stored at −80°c until analyzed. Plasma EHT1864 concentrations were measured using a liquid chromatography (LC) tandem mass spectrometry (MS/MS) assay with technical triplicates (detailed in Suppl. Methods). The pharmacokinetic data analysis (modeling) was undertaken using non compartmental modeling (model 200) in WinNonLn (Pharsight, Mountain View, CA)
Xenograft studies
Female NSG mice were s.c. injected with 5-10x106 BT-474 or MCF-7 cells. Mice injected with MCF-7 cells were s.c. implanted on the same day with a 17β-estradiol (1 mg) beeswax pellet [48]. When average tumor volume reached 200 mm3, mice were randomized to treatment with vehicle, EHT1864 (100 mg/kg, i.p. BID), fulvestrant (5 mg/wk, s.c; clinical formulation kindly provided by AstraZeneca), or the combination. Tumor volumes were measured twice weekly using calipers (volume = length2 x width/2). Tumors were harvested for snap-freezing.
Statistical analyses
In vitro cell growth and apoptosis data were analyzed by ANOVA followed by Bonferroni multiple comparison-adjusted post-hoc test between groups. To estimate progression/regression of tumors, the following linear mixed model was employed: log10(tumor volumeit) = ai + b*t + eit, where i represents the i-th mouse and t represents the time of tumor volume measurement, ai represents the mouse-specific log tumor volume at the baseline (t=0), slope b represents the rate of tumor volume growth (or reduction), and eit represents the deviation of measurements from the model over time (refs. [49-51]). The variance of ai is interpreted as mouse heterogeneity and b*loge(10)*100 estimates the percent tumor volume increase per week. The computation was carried out in statistical package R [52], using function ‘lme’ from library ‘nlme.’ Treatment groups were compared using Z-test for slopes with standard error derived from lme. Synergy (Figure 6) was assessed using the difference of slopes (b1-b0)+(b2-b0) - (b12-b0) where b1, b2, b12, and b0 are the slopes from the treatment groups 1 and 2, the combined treatment, and control group.
ACKNOWLEDGMENTS
We thank the following Norris Cotton Cancer Center Shared Resources for assistance: Transgenic & Genetic Construct, Clinical Pharmacology, Pathology Translational Research, and Immunoassays & Flow Cytometry.
CONFLICTS OF INTEREST
L. Désiré and B. Leblond are employees of Diaxonhit. No potential conflicts of interest were disclosed by any of the other authors.
GRANT SUPPORT
Financial support was provided by the NIH (R00CA142899 to TWM; Dartmouth College Norris Cotton Cancer Center Support Grant P30CA023108) and the American Cancer Society (RSG-13-292-01-TBE to TWM).
REFERENCES
1. Lanning CC, Daddona JL, Ruiz-Velasco R, Shafer SH, Williams CL. The Rac1 C-terminal polybasic region regulates the nuclear localization and protein degradation of Rac1. The Journal of biological chemistry. 2004; 279: 44197-210.
2. Zhang B, Gao Y, Moon SY, Zhang Y, Zheng Y. Oligomerization of Rac1 gtpase mediated by the carboxyl-terminal polybasic domain. The Journal of biological chemistry. 2001; 276: 8958-67.
3. Schnelzer A, Prechtel D, Knaus U, Dehne K, Gerhard M, Graeff H, Harbeck N, Schmitt M, Lengyel E. Rac1 in human breast cancer: overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene. 2000; 19: 3013-20.
4. Ambruso DR, Knall C, Abell AN, Panepinto J, Kurkchubasche A, Thurman G, Gonzalez-Aller C, Hiester A, deBoer M, Harbeck RJ, Oyer R, Johnson GL, Roos D. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc Natl Acad Sci U S A. 2000; 97: 4654-9.
5. Haataja L, Groffen J, Heisterkamp N. Characterization of RAC3, a novel member of the Rho family. The Journal of biological chemistry. 1997; 272: 20384-8.
6. Walker MP, Zhang M, Le TP, Wu P, Laine M, Greene GL. RAC3 is a pro-migratory co-activator of ERalpha. Oncogene. 2011; 30: 1984-94.
7. Jordan P, Brazao R, Boavida MG, Gespach C, Chastre E. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene. 1999; 18: 6835-9.
8. Sosa MS, Lopez-Haber C, Yang C, Wang H, Lemmon MA, Busillo JM, Luo J, Benovic JL, Klein-Szanto A, Yagi H, Gutkind JS, Parsons RE, Kazanietz MG. Identification of the Rac-GEF P-Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol Cell. 2010; 40: 877-92.
9. Lane J, Martin TA, Watkins G, Mansel RE, Jiang WG. The expression and prognostic value of ROCK I and ROCK II and their role in human breast cancer. Int J Oncol. 2008; 33: 585-93.
10. Lee K, Liu Y, Mo JQ, Zhang J, Dong Z, Lu S. Vav3 oncogene activates estrogen receptor and its overexpression may be involved in human breast cancer. BMC Cancer. 2008; 8: 158.
11. Minard ME, Kim LS, Price JE, Gallick GE. The role of the guanine nucleotide exchange factor Tiam1 in cellular migration, invasion, adhesion and tumor progression. Breast Cancer Res Treat. 2004; 84: 21-32.
12. Walch A, Seidl S, Hermannstadter C, Rauser S, Deplazes J, Langer R, von Weyhern CH, Sarbia M, Busch R, Feith M, Gillen S, Hofler H, Luber B. Combined analysis of Rac1, IQGAP1, Tiam1 and E-cadherin expression in gastric cancer. Modern pathology. 2008; 21: 544-52.
13. Eblen ST, Slack JK, Weber MJ, Catling AD. Rac-PAK signaling stimulates extracellular signal-regulated kinase (ERK) activation by regulating formation of MEK1-ERK complexes. Mol Cell Biol. 2002; 22: 6023-33.
14. Miller TW, Nigg JT, Miller RL. Attention deficit hyperactivity disorder in African American children: what can be concluded from the past ten years? Clinical psychology review. 2009; 29: 77-86.
15. Ryu HW, Oh SR, Curtis-Long MJ, Lee JH, Song HH, Park KH. Rapid Identification of Cholinesterase Inhibitors from the Seedcases of Mangosteen Using an Enzyme Affinity Assay. J Agr Food Chem. 2014; 62: 1338-43.
16. Ebi H, Costa C, Faber AC, Nishtala M, Kotani H, Juric D, Della Pelle P, Song Y, Yano S, Mino-Kenudson M, Benes CH, Engelman JA. PI3K regulates MEK/ERK signaling in breast cancer via the Rac-GEF, P-Rex1. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110: 21124-9.
17. Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011; 13: 224.
18. Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nature cell biology. 2004; 6: 1122-8.
19. Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002; 110: 163-75.
20. Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, Papa A, Nardella C, Cantley LC, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. Journal of Clinical Investigation. 2008; 118: 3065-74.
21. Miller TW, Forbes JT, Shah C, Wyatt SK, Manning HC, Olivares MG, Sanchez V, Dugger TC, Granja ND, Narasanna A, Cook RS, Kennedy JP, Lindsley CW, et al. Inhibition of Mammalian Target of Rapamycin Is Required for Optimal Antitumor Effect of HER2 Inhibitors against HER2-Overexpressing Cancer Cells. Clin Cancer Res. 2009; 15: 7266-76.
22. Jacinto GS, Sotto LP, Senal MI, San Diego-McGlone ML, Escobar MT, Amano A, Miller TW. Hypoxia in Manila Bay, Philippines during the northeast monsoon. Marine pollution bulletin. 2011; 63: 243-8.
23. Serra V, Scaltriti M, Prudkin L, Eichhorn PJ, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M, Rodriguez S, Gili M, Russillo M, Parra JL, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011; 30: 2547-57.
24. Saini KS, Loi S, de Azambuja E, Metzger-Filho O, Saini ML, Ignatiadis M, Dancey JE, Piccart-Gebhart MJ. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat Rev. 2013; 39: 935-46.
25. Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, Gunn S, Smetzer L, Mays TA, Kaiser B, Wick MJ, Alvarez C, Cavazos A, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012; 18: 2316-25.
26. Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F, Der CJ. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. The Journal of biological chemistry. 2007; 282: 35666-78.
27. Desire L, Bourdin J, Loiseau N, Peillon H, Picard V, De Oliveira C, Bachelot F, Leblond B, Taverne T, Beausoleil E, Lacombe S, Drouin D, Schweighoffer F. RAC1 inhibition targets amyloid precursor protein processing by gamma-secretase and decreases Abeta production in vitro and in vivo. J Biol Chem. 2005; 280: 37516-25.
28. Fiegl M, Samudio I, Mnjoyan Z, Korchin B, Fritsche H, Andreeff M. Physiological hypoxia promotes lipid raft and PI3K-dependent activation of MAPK 42/44 in leukemia cells. Leukemia. 2010; 24: 1364-7.
29. Saci A, Cantley LC, Carpenter CL. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Molecular cell. 2011; 42: 50-61.
30. Chou MM, Blenis J. The 70 kDa S6 kinase complexes with and is activated by the Rho family G proteins Cdc42 and Rac1. Cell. 1996; 85: 573-83.
31. Yang W, Hosford SR, Dillon LM, Shee K, Liu SC, Bean JR, Salphati L, Pang J, Zhang X, Nannini MA, Demidenko E, Bates D, Lewis LD, et al. Strategically Timing Inhibition of Phosphatidylinositol 3-Kinase to Maximize Therapeutic Index in Estrogen Receptor Alpha-Positive, PIK3CA-Mutant Breast Cancer. Clin Cancer Res. 2016.
32. Amin DN, Sergina N, Ahuja D, McMahon M, Blair JA, Wang D, Hann B, Koch KM, Shokat KM, Moasser MM. Resiliency and vulnerability in the HER2-HER3 tumorigenic driver. Sci Transl Med. 2010; 2: 16ra7.
33. Shah NP, Kasap C, Weier C, Balbas M, Nicoll JM, Bleickardt E, Nicaise C, Sawyers CL. Transient potent BCR-ABL inhibition is sufficient to commit chronic myeloid leukemia cells irreversibly to apoptosis. Cancer Cell. 2008; 14: 485-93.
34. Will M, Qin AC, Toy W, Yao Z, Rodrik-Outmezguine V, Schneider C, Huang X, Monian P, Jiang X, de Stanchina E, Baselga J, Liu N, Chandarlapaty S, et al. Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS-ERK signaling. Cancer Discov. 2014; 4: 334-47.
35. Cameron D, Brown J, Dent R, Jackisch C, Mackey J, Pivot X, Steger GG, Suter TM, Toi M, Parmar M, Laeufle R, Im YH, Romieu G, et al. Adjuvant bevacizumab-containing therapy in triple-negative breast cancer (BEATRICE): primary results of a randomised, phase 3 trial. Lancet Oncology. 2013; 14: 933-42.
36. Miller TW, Balko JM, Fox EM, Ghazoui Z, Dunbier A, Anderson H, Dowsett M, Jiang A, Smith RA, Maira SM, Manning HC, Gonzalez-Angulo AM, Mills GB, et al. ERalpha-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer discovery. 2011; 1: 338-51.
37. Haas NB, Manola J, Uzzo RG, Flaherty K, Pins M, Messing EM, DiPaola RS. A multivariate analysis of prognostic factors from assure (E2805): Adjuvant sorafenib or sunitinib for unfavorable renal carcinoma. J Clin Oncol. 2016; 34.
38. Katz E, Sims AH, Sproul D, Caldwell H, Dixon MJ, Meehan RR, Harrison DJ. Targeting of Rac GTPases blocks the spread of intact human breast cancer. Oncotarget. 2012; 3: 608-19. doi: 10.18632/oncotarget.520.
39. Averous J, Fonseca BD, Proud CG. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene. 2008; 27: 1106-13.
40. Kim JH, Yoon MS, Chen J. Signal transducer and activator of transcription 3 (STAT3) mediates amino acid inhibition of insulin signaling through serine 727 phosphorylation. J Biol Chem. 2009; 284: 35425-32.
41. Chung J, Uchida E, Grammer TC, Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol. 1997; 17: 6508-16.
42. Bid HK, Roberts RD, Manchanda PK, Houghton PJ. RAC1: an emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol Cancer Ther. 2013; 12: 1925-34.
43. Castillo-Pichardo L, Humphries-Bickley T, De La Parra C, Forestier-Roman I, Martinez-Ferrer M, Hernandez E, Vlaar C, Ferrer-Acosta Y, Washington AV, Cubano LA, Rodriguez-Orengo J, Dharmawardhane S. The Rac Inhibitor EHop-016 Inhibits Mammary Tumor Growth and Metastasis in a Nude Mouse Model. Transl Oncol. 2014; 7: 546-55.
44. Montalvo-Ortiz BL, Castillo-Pichardo L, Hernandez E, Humphries-Bickley T, De la Mota-Peynado A, Cubano LA, Vlaar CP, Dharmawardhane S. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. The Journal of biological chemistry. 2012; 287: 13228-38.
45. Onesto C, Shutes A, Picard V, Schweighoffer F, Der CJ. Characterization of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. Methods Enzymol. 2008; 439: 111-29.
46. Dillon LM, Bean JR, Yang W, Shee K, Symonds LK, Balko JM, McDonald WH, Liu S, Gonzalez-Angulo AM, Mills GB, Arteaga CL, Miller TW. P-REX1 creates a positive feedback loop to activate growth factor receptor, PI3K/AKT and MEK/ERK signaling in breast cancer. Oncogene. 2014.
47. Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006; 1: 1112-6.
48. Hossain T, Islam MA, Pal R, Saha A. Exploring structural requirement and binding interactions of beta-amyloid cleavage enzyme inhibitors using molecular modeling techniques. Med Chem Res. 2013; 22: 4766-74.
49. Demidenko E. The assessment of tumour response to treatment. Applied Statistics. 2006; 55: 365-77.
50. Demidenko E. Three endpoints of in vivo tumour radiobiology and their statistical estimation. Internat J Radiat Biol. 2010; 86: 164-73.
51. Demidenko E. Mixed Models: Theory and Applications with R. 2nd Edition ed. New York: Wiley; 2013.
52. Team RC. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2014.
53. Garner F, Brown J, Katzenellenbogen J, Lyttle CR, Hattersley G. Abstract P3-05-07: RAD1901, a novel oral, selective estrogen receptor degrader (SERD) with single agent efficacy in an ER+ primary patent derived ESR1 mutant xenograft model. Cancer Res. 2016; 76: Abstract nr P3-05-7.
54. Khaleel SS, Andrews EH, Ung M, DiRenzo J, Cheng C. E2F4 regulatory program predicts patient survival prognosis in breast cancer. Breast Cancer Res. 2014; 16.