
INTRODUCTION
Each year over 500,000 new cases of head and neck cancers (HNCs) are reported worldwide [1], 40,000 of which occur in the United States [2]. There are two biologically distinct subtypes of these malignancies: human papillomavirus positive (HPV+) and negative (HPV-) [3]. HPV- disease predominates in older HNC patients with a history of long-term alcohol and tobacco use and is declining in prevalence alongside a decrease in smoking habits [4, 5]. This translates to a decrease in HNC at anatomical sites that predominately harbor HPV- tumors. However, the incidence for HPV+ HNC, particularly in oropharyngeal squamous cell carcinomas (OPSCC), has increased by 58% in the past two decades [6–9]. Even with the generous assumption that all eligible people receive immunization against HPV, this upward trend is still expected to continue until the vaccinated generation comes of age in 30–40 years [9]. Meanwhile, developing new innovations to characterize and treat OPSCC, the form of HNC most likely to harbor HPV, will be a necessity.
In general, patients with HPV+ malignancies have a decreased risk of disease progression, respond better to therapy, and have overall better survival compared to HPV- malignances [10–12]. However, HPV+ disease occurs in younger patients and the morbidities associated with aggressive surgical and chemoradiation therapy can reduce their quality of life dramatically [13, 14]. Because of their more favorable prognosis, there are efforts to de-escalate treatment regimens to avoid these morbidities, but it is unclear which patients will respond optimally [12]. Additionally, a sub-population of patients with advanced T4 or N3 stage HPV+ OPSCCs do poorly with standard treatment and have a 5-year survival rate of 54% [15]. In order to identify high-risk HPV+ OPSCCs, further stratification of their clinical and biological characteristics is needed, together with the identification of new biomarkers indicative of more aggressive disease.
HPV is the most prevalent sexually transmitted virus, well-known to induce cervical cancer [16], and the high-risk HPV16 serotype is the cause of almost 90% of HPV+ OPSCC [17, 18]. While just over 50% of all OPSCC are HPV+ [8], the mechanism for how HPV+ tumors respond more favorably to treatment [10, 11] is still incompletely understood [19]. However, inhibition of p53 [20] and retinoblastoma (RB) pocket proteins [21] by the HPV E6 and E7 oncoproteins, respectively, are likely at play as the p53 and RB tumor suppressors often remain intact during malignant transformation [22]. Because of the sustained expression of E6 and E7, HPV+ OPSCC require fewer somatic mutations to develop tumors [23–25], may not have developed the complement of mutations necessary for chemoradiation therapy resistance, and can still respond to treatment by activating p53, RB family members, and other interacting tumor suppressors [26]. In both HPV+ and HPV- OPSCC, early stage non-invasive tumors can be resected with good outcomes (> 80% survival) [27]. Unfortunately, the majority of cases are diagnosed at advanced, locally invasive stages where chemoradiotherapy response and overall survival decrease [28]. Generally, patients with HPV+ disease respond better than their HPV– counterparts even at these advanced stages; however, HPV+ tumors with large primary (T4) and/or large tumor extensions into lymph node (N3) still carry a relatively poor survival prognosis compared to early stage disease [29].
One method of determining the HPV status of OPSCC is by immunohistochemical staining for the presence of p16 (CDKN2A). In OPSCC there is a high degree of correlation between p16 and HPV infection [30], but there are false positives which can be ruled out with high-sensitivity in-situ hybridization (ISH) or polymerase chain reaction for viral nucleic acids [30–33]. The p16 gene product is a well-known tumor suppressor capable of inducing senescence in both OPSCC and primary keratinocytes, the normal epidermal cell type from which OPSCC tumors originate [34–36]. In these systems, p16 drives senescence by stabilizing and preventing the inactivation of the RB pocket proteins RB1, p107, and p130. Should these factors be inactivated, such as by E7 expression during HPV infection, then p16 expression can rise dramatically in the absence of senescence induction and even acquires oncogenic functions [37–39]. In HPV– OPSCC disease, p16 is commonly mutated, deleted, or silenced through promoter methylation [35, 40]. In general, p16 is considered a good surrogate marker for HPV infection. However, there is a subset of HPV– OPSCC that are p16 positive, whose clinical and biological characteristics are not well studied [30, 32, 33].
Inactivation of the retinoblastoma proteins by HPV also drives the expression of other oncogenic factors through E2F-mediated transcription [22, 41]. One important oncogene in many tumors upregulated in this manner is DEK [42]. This oncogene was originally described as a DEK-CAN (NUP214) fusion protein in t(6:9) acute myeloid leukemia [43] and is a highly conserved DNA binding protein in vertebrates with no known paralogs. In normal cells, this protein has functions in DNA replication [44], mRNA splicing [45], chromatin remodeling [44, 46, 47], and DNA repair [48, 49]. In normal keratinocytes, DEK overexpression has been shown to promote hyperplasia and proliferation [50, 51], inhibit differentiation [51], induce mitotic defects and chromosome abnormalities [52], block apoptosis [53], and drive transformation in cells expressing HPV E6 and E7 [53]. While the molecular mechanisms whereby this oncogene promotes these phenotypes remain surprisingly unclear, it is thought that DEK functions through chromatin binding/modification [54]. Due to the importance of DEK for these varied oncogenic phenotypes, the near-ubiquity of high DEK expression in most cancers [55], the similarity of phenotypes observed across tumor types [55], and the ability of the protein to be secreted by cells [56], DEK is currently being evaluated as a biomarker for bladder carcinoma and other malignancies [57].
Compared to normal tissue, DEK expression is upregulated in most surveyed tumor types, including breast [58, 59], hepatocellular carcinoma [60], colorectal cancer [61], and, recently, OPSCC [50]. Our previous study found that DEK protein was highly expressed in all of a small subset of 21 HPV+ and HPV– OPSCC samples that were analyzed [50]. To validate these initial results and establish a more refined relationship between DEK expression and HPV status, we surveyed a large population of OPSCC patients using an established set of primary OPSCC tissue microarrays (TMAs). We first examined the association of DEK with HPV based on p16 (CDKN2A) status and HPV16 genome in-situ hybridization (ISH). Following this analysis, the association of DEK with IL6 expression was also tested. IL6 is a pro-inflammatory interleukin that is strongly associated with poor overall OPSCC patient survival [62] and increased risk of metastasis [63]. Even though IL6 was found to be transcriptionally downregulated following loss of DEK in HNC tissue culture models [64], a link between DEK and IL6 expression in patient OPSCC tumors has yet to be elucidated.
In this study, we found that elevated DEK expression associates with IL6 expression, higher stage tumors, and worse prognosis in HPV16+ OPSCC. These findings support further work into developing DEK as a biomarker for HPV16+ disease and are in agreement with the DEK biomarker literature. Surprisingly, however, our data do not support similar conclusions for HPV16 negative OPSCC as there was no association between DEK expression and survival or tumor stage. This potentially marks HPV16- OPSCC as one of few solid tumors where DEK is not useful as a biomarker, and may indicate distinct biological activities for this protein in the development and progression of HPV16+ versus HPV16- disease.
RESULTS
For this retrospective case study, 194 patients were enrolled, and the cohort represented the expected demographics of OPSCC disease (Table 1). Approximately 57% of this cohort had HPV16+ disease, as determined by genomic HPV in-situ hybridization (ISH), which was expected from a previous epidemiological study [8]. In line with the prognostic focus of this work, the collected tumor specimens were treatment naïve and obtained from initial surgery with curative intent. Overall the cohort experienced a 56.0% survival rate with 34.9% and 71.8% in the HPV16- and HPV16+ groups, respectively (Supplementary Table 1).
Table 1: Patient and tumor sample characteristics
Patient Characteristics |
Mean |
SD |
|---|---|---|
Age (years) |
57.6 |
9.8 |
n |
% |
|
Gender |
||
Male |
158 |
81.4 |
Female |
36 |
18.6 |
Race |
||
African American/Black |
9 |
4.6 |
White |
185 |
95.4 |
Marital Status |
||
Single/Divorced/Widowed |
80 |
45.2 |
Married |
97 |
54.8 |
HPV16 Status |
||
Negative |
83 |
43.0 |
Positive |
110 |
57.0 |
Smoking Status: Pack Years |
||
10 pack years or less |
47 |
25.3 |
More than 10 pack years |
139 |
74.7 |
Node Stage |
||
N0/N1 |
70 |
36.1 |
N2/N3 |
124 |
63.9 |
Tumor Stage |
||
T1/T2 |
122 |
62.9 |
T3/T4 |
72 |
37.1 |
AJCC Stage |
||
I |
4 |
2.1 |
II |
10 |
5.1 |
IIII |
47 |
24.2 |
IV |
133 |
68.6 |
Recurrence Status |
||
No Recurrence |
131 |
72.0 |
Recurrence |
51 |
28.0 |
Recurrence Type |
||
Distant |
19 |
37.3 |
Locoregional |
32 |
62.7 |
Extranodal Extension |
||
No |
110 |
58.2 |
Yes |
79 |
41.8 |
Perineural Invasion |
||
No |
143 |
74.1 |
Yes |
50 |
25.9 |
DEK is most highly expressed in HPV16+/p16+ OPSCC tumors
DEK protein expression levels and HPV16/p16 status were determined using tissue microarrays (TMAs). For each patient sample, three non-adjacent tumor tissue cores and one normal tissue core were used, and DEK staining was consistent across the three tumors cores in each case. Figure 1A–1D shows representative DEK staining patterns and quantification of stain intensity, proportion of tumor cells stained, and the calculated quick score for each image. All quantified samples were grouped based on HPV16 and p16 status; representative positive p16 stains are shown in Figure 1E–1F. There were significant differences in DEK staining across the HPV16+/p16+ (n = 109), HPV16-/p16+ (n = 36), and HPV16–/p16– (n = 46) OPSCC subtypes (HPV16+/p16- not shown because n = 1). HPV16+/p16+ OPSCC had significantly higher average DEK stain intensity (Figure 1G), stain proportion (Figure 1H), and quick score (Figure 1I) compared to double-negative HPV16–/p16– subjects (p < 0.01 in all cases after Bonferroni correction), while the HPV16-/p16+ group only had higher stain proportion (p = 0.01) and quick score (p = 0.03) relative to HPV16–/p16– subjects. HPV16+/p16+ and HPV16-/p16+ groups were not statistically significantly different for any of the three DEK measures after Bonferroni correction.
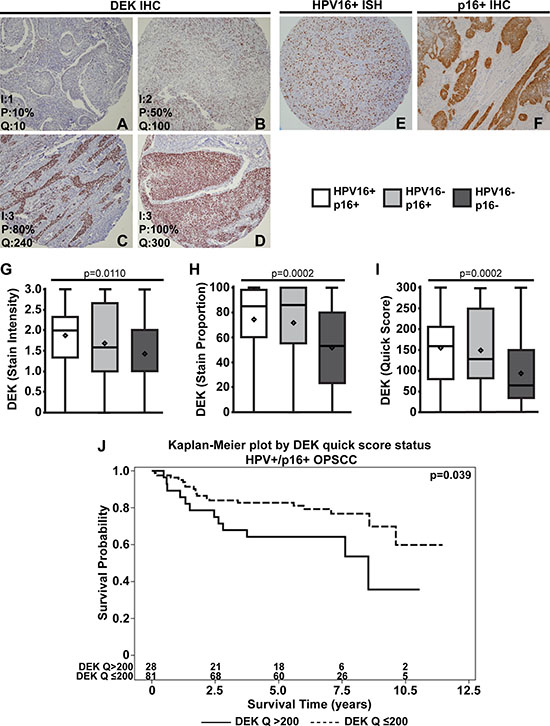
Figure 1: DEK is most highly expressed in HPV16+/p16+ OPSCC. Tumors were scored based on DEK stain intensity (0: none, 1: low, 2: moderate, 3: high), and the proportion of tumor cells stained for DEK (0–100%). Representative images of DEK staining and quantification are as follows: low DEK staining (A), moderate staining (B), high DEK staining (C), and high DEK staining with complete tumor coverage (D) (100x magnification). Scores for intensity (I), proportion of tumor cells stained (P), and the quick score (Q, Q = IxP) are shown in the bottom left of each image. Representative images of positive stains for HPV16 ISH (E) and p16 IHC (F) are shown. After quantification, tumors were separated based on HPV16/p16 status and analyzed for differences in DEK stain intensity (G), proportion of cells stained (H), and quick score (I). DEK quick score dichotomized at > 200 vs. ≤ 200 is a predictor of survival in HPV16+/p16+ OPSCC (hazard ratio (HR) = 2.1, 95% CI = (1.02, 4.3), p = 0.039). The number at risk for each group (i.e., the number remaining for each group at a given time point) is given at the bottom of the graph. (J).
DEK expression in tumors predicts advanced disease and poorer survival in HPV16+, but not in HPV16- OPSCC
While our previous report indicated consistently high DEK staining in 21 OPSCC tumors, 17 of these were AJCC stage IV disease [50]. This current work sought to use a larger sample size, with well-represented stage I-IV disease to further assess the utility of DEK as a biomarker in OPSCC (Table 1). In this cohort, a wider range of DEK staining was noted compared to our previous study [50], indicating that DEK expression was more dynamic than initially predicted (Figure 1, Supplementary Table 1), and some of the increased variability may be due to the increased representation of early stage OPSCCs (Table 1). Tumors bearing high DEK staining and a larger percentage of DEK positive cells, as indicated by the quick score (p = 0.039), had an increase in the hazard of death for patients with HPV16+/p16+ malignancies (Table 2). To better clinically understand the association, Kaplan-Meier curves based on categorizing patients into two groups using the 75th percentile of the DEK quick score (DEK Q > 200 vs. DEK Q ≤ 200) are given (Figure 1J, hazard ratio (HR) = 2.1, 95% CI = (1.02, 4.3), p < 0.039). When the malignancies where separated based on HPV16 status alone, patients with HPV16+ tumors were much more likely to bear higher DEK quick scores than their negative counterparts (Figure 2A, Supplementary Table 2, p < 0.005) and maintained the increased hazard of death (Supplementary Table 3, p < 0.039). These results suggest DEK is a marker of worse prognosis within the subset of HPV16+ OPSCC. This hypothesis is supported by the finding that a high burden of DEK positive cells in HPV16+/p16+ specimens correlates with advanced tumor stage (Table 3, Supplementary Table 4, p = 0.02). In stark contrast, HPV16- OPSCCs did not show a similar correlation between high DEK expression and tumor stage (Table 3) or hazard of death (Table 2, Supplementary Table 3). DEK instead predicted a lesser degree of perineural invasion, the scientific basis of which is unknown. Importantly, the survival data mark HPV16- OPSCC, both p16+ and p16-, as a minority of solid tumors where high DEK expression does not correlate with disease stage or outcome.
Table 2: DEK expression is associated with an increased hazard of death in HPV16+/p16+ but not in HPV16- disease (survival univariate models)
Predictor |
Hazard Ratio |
95% CI |
p-value |
N |
|
|---|---|---|---|---|---|
HPV+/p16+ |
|||||
DEK Stain Intensity |
1.518 |
0.964 |
2.392 |
0.0718 |
109 |
DEK Stain Proportion |
1.013 |
0.998 |
1.028 |
0.0859 |
109 |
DEK Quick Score |
1.004 |
1.000 |
1.008 |
0.0388 |
109 |
HPV–/p16+ |
|||||
DEK Stain Intensity |
0.918 |
0.589 |
1.430 |
0.7043 |
36 |
DEK Stain Proportion |
1.002 |
0.990 |
1.015 |
0.7217 |
36 |
DEK Quick Score |
0.999 |
0.995 |
1.003 |
0.6319 |
36 |
HPV–/p16– |
|||||
DEK Stain Intensity |
0.650 |
0.421 |
1.006 |
0.0531 |
46 |
DEK Stain Proportion |
0.993 |
0.982 |
1.004 |
0.2063 |
46 |
DEK Quick Score |
0.997 |
0.993 |
1.002 |
0.2015 |
46 |
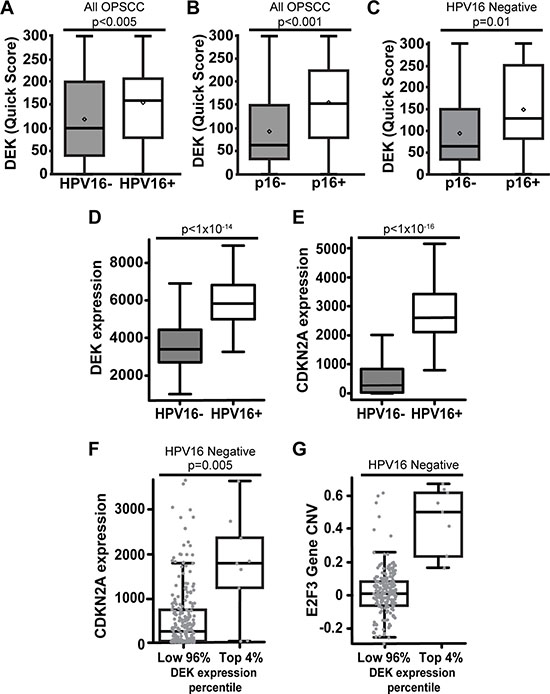
Figure 2: DEK expression correlates with p16+ status in both HPV+ and HPV– OPSCC. Comparing all 194 OPSCC tumors, the proportion and intensity of DEK expression was significantly higher in HPV16+ (A) and p16+ (B) tumors. Quartiles and statistical significance of DEK staining intensity and tissue proportion for (A) and (B) are depicted in Supplementary Table 2. DEK expression correlated with p16 status in HPV16- tumors (C). D-G are an analysis of published Cancer Genome Atlas Network RNA-Seq data for HNCs [22]. DEK mRNA expression was significantly elevated in HPV+ disease (D). While p16 (CDKN2A) message was significantly elevated in HPV16+ disease as expected, there was substantial variability in p16 expression in HPV- tumors (E). The highest 4% of HPV- DEK expressing tumors (9 out of 242) were significantly increased for p16 expression (F). DEK was highly expressed in these tumors in correlation with amplification of DEK and the nearby E2F3 locus on 6p22.3 (G).
Table 3: High DEK expression is associated with higher tumor stage in HPV+/p16+ OPSCC, and reduced perineural invasion in HPV- OPSCC
HPV16+/p16+, DEK (Stain Proportion) |
|||||||
|---|---|---|---|---|---|---|---|
Tumor Stage |
N |
Minimum |
25th Pctl |
Median |
75th Pctl |
Maximum |
p-value |
T1/T2 |
76 |
0.00 |
53.33 |
75.00 |
98.33 |
100.00 |
0.0227 |
T3/T4 |
33 |
23.33 |
80.00 |
93.33 |
100.00 |
100.00 |
|
Perineural Invasion |
N |
Minimum |
25th Pctl |
Median |
75th Pctl |
Maximum |
p-value |
No |
90 |
0.00 |
60.00 |
80.00 |
98.33 |
100.00 |
0.2786 |
Yes |
19 |
11.67 |
83.33 |
90.00 |
100.00 |
100.00 |
|
HPV16–/p16+, DEK (Stain Proportion) |
|||||||
Tumor Stage |
N |
Minimum |
25th Pctl |
Median |
75th Pctl |
Maximum |
p-value |
T1/T2 |
24 |
0.00 |
41.67 |
81.67 |
98.33 |
100.00 |
0.2399 |
T3/T4 |
12 |
5.00 |
65.00 |
92.50 |
100.00 |
100.00 |
|
Perineural Invasion |
N |
Minimum |
25th Pctl |
Median |
75th Pctl |
Maximum |
p-value |
No |
26 |
0.00 |
63.33 |
89.17 |
100.00 |
100.00 |
0.1189 |
Yes |
10 |
0.00 |
43.33 |
78.33 |
90.00 |
100.00 |
|
HPV16–/p16–, DEK (Stain Proportion) |
|||||||
Tumor Stage |
N |
Minimum |
25th Pctl |
Median |
75th Pctl |
Maximum |
p-value |
T1/T2 |
19 |
0.00 |
23.33 |
53.33 |
76.67 |
98.33 |
0.5842 |
T3/T4 |
27 |
0.00 |
20.00 |
53.33 |
83.33 |
100.00 |
|
Perineural Invasion |
N |
Minimum |
25th Pctl |
Median |
75th Pctl |
Maximum |
p-value |
No |
25 |
0.00 |
50.00 |
66.67 |
86.67 |
100.00 |
0.0144 |
Yes |
20 |
0.00 |
11.67 |
33.33 |
51.67 |
100.00 |
|
Statistics for other clinical characteristics listed in Supplementary Table 4.
DEK expression is associated with p16+ status in both HPV16+ and HPV16- disease
While DEK expression did not correlate with survival in either p16+ or p16- OPSCC patients (Supplementary Tables 1, 5), DEK expression by quick score was significantly increased in p16+ tumors (p < 0.001, Figure 2B, Supplementary Table 2). Considering that HPV E7 disrupts and inactivates the entire RB pocket protein family, a correlation between the expression of DEK and p16 was expected as both are upregulated upon pocket protein loss of function [37, 38]. This correlation between p16 and DEK quick score was conserved in HPV16-/p16+ OPSCCs (Figure 2C). Since the percentage of HPV16-/p16+ tumors in this study (24.8% of p16+ tumors) was similar to what has been reported previously (25.7%) [32, 33], we sought to validate the relationship between p16 and DEK using published HPV– HNC Cancer Genome Atlas data [23]. HPV status was rigorously validated through a combination of whole genome, whole exome, and RNA sequencing for viral sequences, as well as ISH for HPV16, 18, 33, 35, 39, 45, 51, 52, 56, 58, and 66 serotypes. DEK and HPV status correlated with p16 (CDKN2A) expression (Figure 2D–2E), and the majority of p16+ tumors were also HPV16+, as expected (Figure 2E). However, a sizable subgroup of HPV16- tumors was identified that expressed moderate to high levels of p16 (Figure 2E–2F), with the top 4% of DEK-expressing HPV– HNCs harboring significant p16 mRNA over-expression (Figure 2F). To provide a potential mechanism for how some HPV– OPSCCs may induce p16 overexpression, we re-analyzed the TCGA data, shown in Figure 2E–2F, and found that chromosome region 6p22.3 was amplified in tumors where DEK was most highly expressed (Figure 2G). This amplified region encodes both the DEK and E2F3 genes; the latter has been implemented in driving expression of p16 [65].
DEK correlates with IL6 expression in HPV16+ OPSCC
In addition to promoting tumor growth and invasion, DEK has potential pro-inflammatory properties. These include autoantigen properties when secreted or present in body fluids [56, 66] and control of robust expression of inflammatory pathway members such as IRAK1 [64]. In the latter report, RNA-sequencing of HPV+ and HPV– HNC cells identified shared decreased IL6 expression in response to DEK knockdown. To determine whether DEK expression correlated with IL6 status in OPSCC, we quantified IL6 expression in the TMAs (Figure 3A). HPV16+ and HPV+/p16+ OPSCC tumors showed a correlation between high DEK stain intensity and positive IL6 status (Figure 3B–3C, p < 0.04 and p = 0.05 respectively). While this relationship was inverse in total HPV16- OPSCC (Supplementary Figure 1), this study lacked the power to confirm this association in either the HPV–/p16+ or HPV–/p16- subgroup as no significance was observed in DEK stain intensity, stain proportion, or quick score (Figure 3C, data not shown). A role for HPV16 in independently regulating both IL6 and DEK expression in HPV16+ disease cannot be ruled out by this study, and the relationship between DEK and IL6 in HPV16- OPSCC is subtle.
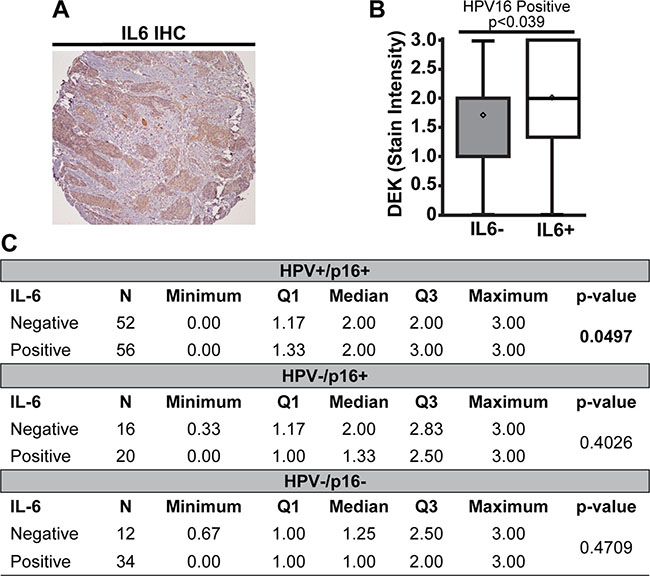
Figure 3: High DEK expression was associated with IL6 expression in HPV16+ tumors. A representative IL6+ staining section is shown (A). HPV16+ tumors that were also IL6+ stained darker for DEK than tumors not expressing IL6 (B). There was no association between DEK and IL6 status in HPV16-/p16+ or HPV16–/p16– tumors (C).
DISCUSSION
Most clinical studies of DEK across human tumors have correlated high expression of the oncogene with advanced tumor stages and poor outcomes [59, 61, 67–69]. We found that this relationship holds true in HPV16+ OPSCC (Figure 1J, Table 2 and Supplementary Table 3). Considering that HPV16 accounts for approximately 90% of HPV+ OPSCCs [17, 18], these data indicate that DEK has potential as a prognostic biomarker for the vast majority of these tumors. However, this study was a medium-sized retrospective cohort designed to determine the potential for DEK as a biomarker in different OPSCC groups. To rigorously ascertain the clinical significance and prognostic value of DEK in HPV16+ disease, a large multi-institutional prospective study is now warranted.
Importantly, our data do not support DEK as a biomarker in OPSCC identified as HPV16-, HPV16-/p16+, or HPV16–/p16– (Table 2 and Supplementary Table 3). This is a surprising finding given the extensive literature on DEK expression in other HPV– solid tumor types, and suggests that HPV16- OPSCC is one of few wherein DEK does not have prognostic utility. Our data thus strongly suggests that DEK is not prognostic in all subgroups of a given malignancy, and must therefore be carefully examined as an appropriate biomarker in each case. Interestingly, DEK was significantly associated with a decreased risk of perineural invasion in the double negative group. The scientific basis of this latter finding is unknown, and should warrant further investigation as high DEK expression is usually associated with invasion and migration phenotypes [70]. With regard to the HPV16-/p16+ group, the combination of limited sample number (n = 36) and heterogeneity of HPV+ and HPV– tumors likely precluded any significant clinical findings. To address these issues and more deeply study HPV16-/p16+ OPSCC, a significantly larger patient population would be required.
DEK mutations cannot explain the observed disparities between HPV16+ versus HPV16- disease. Such mutations are rare in all tumors, and found in only 0.4% of OPSCC. Specifically, we analyzed the TCGA provisional head and neck cancer study, and only 2 out of 530 tumors harbored mutations within the DEK gene. The first was an E20V substitution, and the second was a nonsense frameshift at amino acid 11. No DEK mutations were identified in the 279 samples previously published by TCGA [23].
Alongside survival analysis, this report determined that the average DEK expression was highest in HPV16+ OPSCC by TMA (Figure 1G–1I) and in published TCGA data [23] (Figure 2D). This is likely due to early increases in DEK expression. DEK is upregulated by E2F transcriptional activator family members, which in turn are activated by HPV E7 disruption of RB family members [41, 42]. The additional increase in DEK protein levels which correlated with advanced stage HPV16+ tumors (Table 3) is most likely a result of HPV E7 gene amplification and increases in E7 transcriptional activation and mRNA stability. These are all common occurrences in HPV+ malignancies [71, 72]. Apart from HPV16+ tumors, the next highest DEK-expressing group was the HPV16-/p16+ cohort. This group is partially comprised of tumors bearing other high-risk HPV serotypes which would over-express DEK in the same manner as HPV16+ OPSCCs. True HPV–/p16+ tumors likely comprise the remainder of this group, and we have found that a significant fraction of HPV– tumors express p16 in the TCGA cohort (Figure 3E–3F). Other reports have also described this HPV–/p16+ population [30, 32, 33], but their clinical and biological characteristics are the least studied of OPSCC subtypes. We found that p16+ status was significantly correlated with the highest DEK-expressing HPV– tumors (Figure 2F). Biologically, this may be a consequence of chromosome 6p22.3 amplification, a common occurrence in multiple tumor types [73], but one that has not been reported in HNC until now. This region contains both the DEK and E2F3 genes, and was amplified in top DEK-expressing HPV– malignancies (Figure 2G). Currently, it is thought that one mechanism of p16 upregulation may occur through activator E2F genes, including E2F3, although the exact mechanism for this relationship is unknown [65]. It is thus possible that the amplification of E2F3 drives p16 and DEK expression.
As the field moves towards incorporating personalized medicine for HNC care, identifying and characterizing distinct subtypes of HPV+ and HPV– OPSCC will become paramount. This study contributes to this endeavor by identifying DEK as a potential prognostic biomarker for further study in HPV16+ OPSCC, and does not support its use for HPV16- disease. Prior to this current study, we and others have argued for the development of therapeutic DEK inhibiting molecules to take advantage of broad solid tumor dependency on high DEK expression for survival [53, 68, 74–76]. While DEK targeting strategies will undoubtedly be useful in the treatment of most solid tumor types, our study also suggests for the first time that use of these agents will need to be carefully tailored in the care of OPSCC patients.
MATERIALS AND METHODS
Study population
This retrospective case study was approved by the Ohio State University Institutional Review Board and a waiver of HIPAA authorization was obtained. All patient OPSCC specimens were requested from samples obtained by The Ohio State University James Cancer Hospital and Solove Research Institute from 2002 to 2009, and were treatment naïve at the time of collection. During the enrollment period, all patients were given the option of primary (C)RT or primary surgery with follow-up (C)RT as necessary. The majority of patients opted for initial surgery, and this is the population that was included in this study. All samples were chemo- and radiotherapy naïve, and post-surgical standard of care treatment was followed as necessary. There was no bias in selecting patient samples based on size or stage. The following patient attributes were accessed: age, race, gender, marital status, smoking status, tumor size, nodal status, AJCC stage, presence of local metastasis, presence of perineural invasion, survival time and tumor recurrence. In this report, survival time was defined as the time from the patient’s primary surgical resection of OPSCC to death. The date of the last living observation was censored. Recurrence is defined as any occurrence of a new suspicious head and neck mass that is confirmed by radiology or pathology as squamous cell carcinoma within five years of surgical resection.
Tissue microarray (TMA), immunohistochemistry (IHC), and HPV in-situ hybridization
The Ohio State University Department of Pathology Histology Core Laboratory generated master TMA blocks from selected archived paraffin-embedded tissue. Briefly, the distribution of tumor and normal tissue was determined via hematoxlin and eosin staining by a pathologist, and the TMA master blocks were created from 0.6-mm punch cores of 3 representative tumor tissues and 1 normal tissue for comparison in each specimen. TMA slides were stained for IL6 and p16, by IHC, and for HPV16, by in-situ hybridization (GenPoint, Dako), following previously published methods [62]. IHC staining for p16 was performed using the CINtec mtm antibody (E6H4 clone). DEK IHC staining followed standard xylene deparaffination and rehydration in decreasing ethanol concentrations. Antigens were retrieved using a Biocare Medical LLC (Concord, CA, USA) decloaking chamber with Dako antigen unmasking buffer for 20 minutes at 120ºC. After returning to room temperature, slides were incubated for 10 minutes at ambient conditions with Dako dual endogenous enzyme block. This was followed with blocking in PBS/serum solution corresponding to species of the secondary antibody. Following this, BD-Pharmingen mouse anti-DEK primary antibody was applied to the TMA and incubated for 1 hour at 37ºC. After washing, a room temperature 30 minute incubation was performed using biotinylated donkey anti-mouse secondary antibody (Vectastain Elite Kit). After washing, slides were incubated with an avidin-biotin complex for 30 minutes (Vector Laboratories, Burlingame, CA, USA) prior to the addition of 3,3′-diaminobenzidine (Sigma). The reaction was quenched in water, counterstained with Mayer’s hematoxylin, and coverslipped with Permount.
IHC scoring
A treatment-blinded pathologist interpreted the slides and scored for stain intensity (0: none, 1: low, 2: moderate, 3: high), and stain proportion (0–100%). A descriptive quick score (0–300) was acquired by multiplying these two dimensions. To be considered positive for p16, strong and diffuse nuclear and cytoplasmic staining in ≥ 50% of the tumor cells was required.
Statistical methods
Descriptive statistics were used to summarize the study population, including means for the continuous variables and frequencies for the categorical variables. Cox proportional hazards models were used to assess univariate associations of DEK expression and the risk of death for the overall study population, and also stratified by HPV status, p16 status, and a combination of HPV and p16 status. Unadjusted hazard ratios and confidence intervals (CI) are reported. Mann-Whitney tests were used to assess associations between biomarkers/demographic/clinical characteristics and DEK expression (quantitative). Analyses were conducted in SAS, version 9.3 (SAS Institute, Cary, North Carolina).
ACKNOWLEDGMENTS
We would like to thank Drs. Lisa Privette-Vinnedge and Trisha Wise-Draper for their assistance in proof-reading this manuscript.
CONFLICTS OF INTEREST
The authors do not report any potential conflicts of interest.
GRANT SUPPORT
This work was supported by the National Institutes of Health [R01-CA116316 to S. Wells, T32 GM063483-13, T32-ES007250, CA178649 to P. Kumar] and The Ohio State University Comprehensive Cancer Center.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87–108.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65:5–29.
3. Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, Zahurak ML, Daniel RW, Viglione M, Symer DE, Shah KV, Sidransky D. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. Journal of the National Cancer Institute. 2000; 92:709–720.
4. Marur S, Forastiere AA. Head and neck cancer: changing epidemiology, diagnosis, and treatment. Mayo Clin Proc. 2008; 83:489–501.
5. Sturgis EM, Cinciripini PM. Trends in head and neck cancer incidence in relation to smoking prevalence: an emerging epidemic of human papillomavirus-associated cancers? Cancer. 2007; 110:1429–1435.
6. Stein AP, Saha S, Yu M, Kimple RJ, Lambert PF. Prevalence of human papillomavirus in oropharyngeal squamous cell carcinoma in the United States across time. Chem Res Toxicol. 2014; 27:462–469.
7. Marur S, Forastiere AA. Head and Neck Squamous Cell Carcinoma: Update on Epidemiology, Diagnosis, and Treatment. Mayo Clin Proc. 2016; 91:386–396.
8. Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E, Jiang B, Goodman MT, Sibug-Saber M, Cozen W, Liu L, Lynch CF, Wentzensen N, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. Journal of clinical oncology. 2011; 29:4294–4301.
9. Gillison ML, Chaturvedi AK, Anderson WF, Fakhry C. Epidemiology of Human Papillomavirus-Positive Head and Neck Squamous Cell Carcinoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015; 33:3235–3242.
10. Vidal L, Gillison ML. Human papillomavirus in HNSCC: recognition of a distinct disease type. Hematology/oncology clinics of North America. 2008; 22:1125–1142, vii.
11. Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, Forastiere A, Gillison ML. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. Journal of the National Cancer Institute. 2008; 100:261–269.
12. Masterson L, Moualed D, Liu ZW, Howard JE, Dwivedi RC, Tysome JR, Benson R, Sterling JC, Sudhoff H, Jani P, Goon PK. De-escalation treatment protocols for human papillomavirus-associated oropharyngeal squamous cell carcinoma: a systematic review and meta-analysis of current clinical trials. Eur J Cancer. 2014; 50:2636–2648.
13. Sharma A, Mendez E, Yueh B, Lohavanichbutr P, Houck J, Doody DR, Futran ND, Upton MP, Schwartz SM, Chen C. Human papillomavirus-positive oral cavity and oropharyngeal cancer patients do not have better quality-of-life trajectories. Otolaryngol Head Neck Surg. 2012; 146:739–745.
14. Baxi SS, Salz T, Xiao H, Atoria CL, Ho A, Smith-Marrone S, Sherman EJ, Lee NY, Elkin EB, Pfister DG. Employment and return to work following chemoradiation in patient with HPV-related oropharyngeal cancer. Cancers of the Head & Neck. 2016; 1:1–8.
15. Huang SH, Xu W, Waldron J, Siu L, Shen X, Tong L, Ringash J, Bayley A, Kim J, Hope A, Cho J, Giuliani M, Hansen A, et al. Refining American Joint Committee on Cancer/Union for International Cancer Control TNM stage and prognostic groups for human papillomavirus-related oropharyngeal carcinomas. Journal of clinical oncology. 2015; 33:836–845.
16. Bosch FX, Lorincz A, Munoz N, Meijer CJ, Shah KV. The causal relation between human papillomavirus and cervical cancer. Journal of clinical pathology. 2002; 55:244–265.
17. Melkane AE, Auperin A, Saulnier P, Lacroix L, Vielh P, Casiraghi O, Msakni I, Drusch F, Temam S. Human papillomavirus prevalence and prognostic implication in oropharyngeal squamous cell carcinomas. Head Neck. 2014; 36:257–265.
18. Liu SZ, Zandberg DP, Schumaker LM, Papadimitriou JC, Cullen KJ. Correlation of p16 expression and HPV type with survival in oropharyngeal squamous cell cancer. Oral oncology. 2015; 51:862–869.
19. Wen Z, Pyeon D, Wang Y, Lambert P, Xu W, Ahlquist P. Orphan nuclear receptor PNR/NR2E3 stimulates p53 functions by enhancing p53 acetylation. Mol Cell Biol. 2012; 32:26–35.
20. Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990; 63:1129–1136.
21. Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996; 56:4620–4624.
22. Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989; 243:934–937.
23. Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015; 517:576–582.
24. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, Shefler E, Ramos AH, Stojanov P, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011; 333:1157–1160.
25. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, Zhang N, El-Naggar AK, Jasser SA, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011; 333:1154–1157.
26. Xie X, Piao L, Bullock BN, Smith A, Su T, Zhang M, Teknos TN, Arora PS, Pan Q. Targeting HPV16 E6-p300 interaction reactivates p53 and inhibits the tumorigenicity of HPV-positive head and neck squamous cell carcinoma. Oncogene. 2014; 33:1037–1046.
27. Kalavrezos N, Bhandari R. Current trends and future perspectives in the surgical management of oral cancer. Oral oncology. 2010; 46:429–432.
28. Hauswald H, Simon C, Hecht S, Debus J, Lindel K. Long-term outcome and patterns of failure in patients with advanced head and neck cancer. Radiation oncology. 2011; 6:70.
29. Gillison ML. Human Papillomavirus and Oropharyngeal Cancer Stage. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2016; 34:1833–1835.
30. Seiwert TY. Ties that bind: p16 as a prognostic biomarker and the need for high-accuracy human papillomavirus testing. Journal of clinical oncology. 2014; 32:3914–3916.
31. Smeets SJ, Hesselink AT, Speel EJ, Haesevoets A, Snijders PJ, Pawlita M, Meijer CJ, Braakhuis BJ, Leemans CR, Brakenhoff RH. A novel algorithm for reliable detection of human papillomavirus in paraffin embedded head and neck cancer specimen. International journal of cancer Journal international du cancer. 2007; 121:2465–2472.
32. Lewis JS, Jr., Thorstad WL, Chernock RD, Haughey BH, Yip JH, Zhang Q, El-Mofty SK. p16 positive oropharyngeal squamous cell carcinoma:an entity with a favorable prognosis regardless of tumor HPV status. Am J Surg Pathol. 2010; 34:1088–1096.
33. Rietbergen MM, Snijders PJ, Beekzada D, Braakhuis BJ, Brink A, Heideman DA, Hesselink AT, Witte BI, Bloemena E, Baatenburg-De Jong RJ, Leemans CR, Brakenhoff RH. Molecular characterization of p16-immunopositive but HPV DNA-negative oropharyngeal carcinomas. International journal of cancer Journal international du cancer. 2014; 134:2366–2372.
34. Enders GH, Koh J, Missero C, Rustgi AK, Harlow E. p16 inhibition of transformed and primary squamous epithelial cells. Oncogene. 1996; 12:1239–1245.
35. Cody DT, 2nd, Huang Y, Darby CJ, Johnson GK, Domann FE. Differential DNA methylation of the p16 INK4A/CDKN2A promoter in human oral cancer cells and normal human oral keratinocytes. Oral oncology. 1999; 35:516–522.
36. Darbro BW, Schneider GB, Klingelhutz AJ. Co-regulation of p16INK4A and migratory genes in culture conditions that lead to premature senescence in human keratinocytes. J Invest Dermatol. 2005; 125:499–509.
37. Simpson DS, Mason-Richie NA, Gettler CA, Wikenheiser-Brokamp KA. Retinoblastoma family proteins have distinct functions in pulmonary epithelial cells in vivo critical for suppressing cell growth and tumorigenesis. Cancer Res. 2009; 69:8733–8741.
38. Kotake Y, Cao R, Viatour P, Sage J, Zhang Y, Xiong Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes & development. 2007; 21:49–54.
39. Munger K, Gwin TK, McLaughlin-Drubin ME. p16 in HPV-associated cancers. Oncotarget. 2013; 4:1864–1865. doi: 10.18632/oncotarget.1523.
40. Loughran O, Malliri A, Owens D, Gallimore PH, Stanley MA, Ozanne B, Frame MC, Parkinson EK. Association of CDKN2A/p16INK4A with human head and neck keratinocyte replicative senescence: relationship of dysfunction to immortality and neoplasia. Oncogene. 1996; 13:561–568.
41. Dyson N. The regulation of E2F by pRB-family proteins. Genes & development. 1998; 12:2245–2262.
42. Carro MS, Spiga FM, Quarto M, Di Ninni V, Volorio S, Alcalay M, Muller H. DEK Expression is controlled by E2F and deregulated in diverse tumor types. Cell Cycle. 2006; 5:1202–1207.
43. von Lindern M, Fornerod M, Soekarman N, van Baal S, Jaegle M, Hagemeijer A, Bootsma D, Grosveld G. Translocation t(6;9) in acute non-lymphocytic leukaemia results in the formation of a DEK-CAN fusion gene. Baillieres Clin Haematol. 1992; 5:857–879.
44. Alexiadis V, Waldmann T, Andersen J, Mann M, Knippers R, Gruss C. The protein encoded by the proto-oncogene DEK changes the topology of chromatin and reduces the efficiency of DNA replication in a chromatin-specific manner. Genes & development. 2000; 14:1308–1312.
45. Soares LM, Zanier K, Mackereth C, Sattler M, Valcarcel J. Intron removal requires proofreading of U2AF/3’ splice site recognition by DEK. Science. 2006; 312:1961–1965.
46. Ko SI, Lee IS, Kim JY, Kim SM, Kim DW, Lee KS, Woo KM, Baek JH, Choo JK, Seo SB. Regulation of histone acetyltransferase activity of p300 and PCAF by proto-oncogene protein DEK. FEBS letters. 2006; 580:3217–3222.
47. Kappes F, Waldmann T, Mathew V, Yu J, Zhang L, Khodadoust MS, Chinnaiyan AM, Luger K, Erhardt S, Schneider R, Markovitz DM. The DEK oncoprotein is a Su(var) that is essential to heterochromatin integrity. Genes & development. 2011; 25:673–678.
48. Kavanaugh GM, Wise-Draper TM, Morreale RJ, Morrison MA, Gole B, Schwemberger S, Tichy ED, Lu L, Babcock GF, Wells JM, Drissi R, Bissler JJ, Stambrook PJ, et al. The human DEK oncogene regulates DNA damage response signaling and repair. Nucleic Acids Res. 2011; 39:7465–7476.
49. Kappes F, Fahrer J, Khodadoust MS, Tabbert A, Strasser C, Mor-Vaknin N, Moreno-Villanueva M, Burkle A, Markovitz DM, Ferrando-May E. DEK is a poly(ADP-ribose) acceptor in apoptosis and mediates resistance to genotoxic stress. Mol Cell Biol. 2008; 28:3245–3257.
50. Adams AK, Hallenbeck GE, Casper KA, Patil YJ, Wilson KM, Kimple RJ, Lambert PF, Witte DP, Xiao W, Gillison ML, Wikenheiser-Brokamp KA, Wise-Draper TM, Wells SI. DEK promotes HPV-positive and -negative head and neck cancer cell proliferation. Oncogene. 2015; 34:868–877.
51. Wise-Draper TM, Morreale RJ, Morris TA, Mintz-Cole RA, Hoskins EE, Balsitis SJ, Husseinzadeh N, Witte DP, Wikenheiser-Brokamp KA, Lambert PF, Wells SI. DEK proto-oncogene expression interferes with the normal epithelial differentiation program. Am J Pathol. 2009; 174:71–81.
52. Matrka MC, Hennigan RF, Kappes F, DeLay ML, Lambert PF, Aronow BJ, Wells SI. DEK over-expression promotes mitotic defects and micronucleus formation. Cell Cycle. 2015:0.
53. Wise-Draper TM, Mintz-Cole RA, Morris TA, Simpson DS, Wikenheiser-Brokamp KA, Currier MA, Cripe TP, Grosveld GC, Wells SI. Overexpression of the cellular DEK protein promotes epithelial transformation in vitro and in vivo. Cancer Res. 2009; 69:1792–1799.
54. Privette Vinnedge LM, Kappes F, Nassar N, Wells SI. Stacking the DEK: from chromatin topology to cancer stem cells. Cell Cycle. 2013; 12:51–66.
55. Pease NA, Wise-Draper T, Privette Vinnedge L. Dissecting the Potential Interplay of DEK Functions in Inflammation and Cancer. J Oncol. 2015; 2015:106517.
56. Mor-Vaknin N, Punturieri A, Sitwala K, Faulkner N, Legendre M, Khodadoust MS, Kappes F, Ruth JH, Koch A, Glass D, Petruzzelli L, Adams BS, Markovitz DM. The DEK nuclear autoantigen is a secreted chemotactic factor. Mol Cell Biol. 2006; 26:9484–9496.
57. Datta A, Adelson ME, Mogilevkin Y, Mordechai E, Sidi AA, Trama JP. Oncoprotein DEK as a tissue and urinary biomarker for bladder cancer. BMC cancer. 2011; 11:234.
58. Ying G, Wu Y. DEK: A novel early screening and prognostic marker for breast cancer. Mol Med Rep. 2015; 12:7491–7495.
59. Liu S, Wang X, Sun F, Kong J, Li Z, Lin Z. DEK overexpression is correlated with the clinical features of breast cancer. Pathology international. 2012; 62:176–181.
60. Yu L, Huang X, Zhang W, Zhao H, Wu G, Lv F, Shi L, Teng Y. Critical role of DEK and its regulation in tumorigenesis and metastasis of hepatocellular carcinoma. Oncotarget. 2016; 7:26844–26855. doi: 10.18632/oncotarget.8565.
61. Lin L, Piao J, Gao W, Piao Y, Jin G, Ma Y, Li J, Lin Z. DEK over expression as an independent biomarker for poor prognosis in colorectal cancer. BMC cancer. 2013; 13:366.
62. Kumar B, Brown NV, Swanson BJ, Schmitt AC, Old M, Ozer E, Agrawal A, Schuller DE, Teknos TN, Kumar P. High expression of myoferlin is associated with poor outcome in oropharyngeal squamous cell carcinoma patients and is inversely associated with HPV-status. Oncotarget. 2016; 7:18665–77. doi: 10.18632/oncotarget.7625.
63. Yadav A, Kumar B, Datta J, Teknos TN, Kumar P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Molecular cancer research : MCR. 2011; 9:1658–1667.
64. Adams AK, Bolanos LC, Dexheimer PJ, Karns RA, Aronow BJ, Komurov K, Jegga AG, Casper KA, Patil YJ, Wilson KM, Starczynowski DT, Wells SI. IRAK1 is a novel DEK transcriptional target and is essential for head and neck cancer cell survival. Oncotarget. 2015; 6:43395–43407. doi: 10.18632/oncotarget.6028.
65. Li J, Poi MJ, Tsai MD. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry. 2011; 50:5566–5582.
66. Mor-Vaknin N, Kappes F, Dick AE, Legendre M, Damoc C, Teitz-Tennenbaum S, Kwok R, Ferrando-May E, Adams BS, Markovitz DM. DEK in the synovium of patients with juvenile idiopathic arthritis: characterization of DEK antibodies and posttranslational modification of the DEK autoantigen. Arthritis Rheum. 2011; 63:556–567.
67. Lin LJ, Chen LT. The role of DEK protein in hepatocellular carcinoma for progression and prognosis. Pak J Med Sci. 2013; 29:778–782.
68. Martinez-Useros J, Rodriguez-Remirez M, Borrero-Palacios A, Moreno I, Cebrian A, Gomez del Pulgar T, del Puerto-Nevado L, Vega-Bravo R, Puime-Otin A, Perez N, Zazo S, Senin C, Fernandez-Acenero MJ, et al. DEK is a potential marker for aggressive phenotype and irinotecan-based therapy response in metastatic colorectal cancer. BMC cancer. 2014; 14:965.
69. Liu X, Qi D, Qi J, Mao Z, Li X, Zhang J, Li J, Gao W. Significance of DEK overexpression for the prognostic evaluation of non-small cell lung carcinoma. Oncology reports. 2016; 35:155–162.
70. Privette Vinnedge LM, McClaine R, Wagh PK, Wikenheiser-Brokamp KA, Waltz SE, Wells SI. The human DEK oncogene stimulates beta-catenin signaling, invasion and mammosphere formation in breast cancer. Oncogene. 2011; 30:2741–2752.
71. Jeon S, Lambert PF. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc Natl Acad Sci U S A. 1995; 92:1654–1658.
72. Akagi K, Li J, Broutian TR, Padilla-Nash H, Xiao W, Jiang B, Rocco JW, Teknos TN, Kumar B, Wangsa D, He D, Ried T, Symer DE, et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome research. 2014; 24:185–199.
73. Santos GC, Zielenska M, Prasad M, Squire JA. Chromosome 6p amplification and cancer progression. Journal of clinical pathology. 2007; 60:1–7.
74. Khodadoust MS, Verhaegen M, Kappes F, Riveiro-Falkenbach E, Cigudosa JC, Kim DS, Chinnaiyan AM, Markovitz DM, Soengas MS. Melanoma proliferation and chemoresistance controlled by the DEK oncogene. Cancer Res. 2009; 69:6405–6413.
75. Wise-Draper TM, Allen HV, Jones EE, Habash KB, Matsuo H, Wells SI. Apoptosis inhibition by the human DEK oncoprotein involves interference with p53 functions. Mol Cell Biol. 2006; 26:7506–7519.
76. Yamazaki H, Iwano T, Otsuka S, Kagawa Y, Hoshino Y, Hosoya K, Okumura M, Takagi S. SiRNA knockdown of the DEK nuclear protein mRNA enhances apoptosis and chemosensitivity of canine transitional cell carcinoma cells. Vet J. 2015; 204:60–65.