
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most prevalent malignancies worldwide, and is the 5th and 7th most common cancer in men and women, respectively [1]. As HCC is characteristically refractory to current drug treatments, it is imperative to identify effective therapies for the treatment of advanced HCC.
Sorafenib was the first multikinase inhibitor to improve the overall survival of patients with advanced HCC. This drug inhibits tumor cell proliferation and angiogenesis and increases tumor cell apoptosis in HCC [2]. However, sorafenib only increases the survival time by a few months [3], and has a variety of adverse effects, including dermatological, digestive and cardiovascular toxicity [4]. These toxic effects may prompt the modification or discontinuation of sorafenib treatment. Thus, agents that can potentiate the anti-tumor activity of sorafenib without significant systemic toxicity need to be identified.
Fluvastatin, a member of the statin family, is widely used as a lipid-lowering drug. In addition to its cholesterol-lowering activities, fluvastatin has selective anti-cancer effects, such as the induction of tumor cell apoptosis in specific cancer cell lines (e.g., glioma and breast cancer cell lines) [5, 6]. Fluvastatin has also been suggested to have therapeutic potential for the treatment of HCC [7]. Notably, fluvastatin was shown to increase the cytotoxicity of sorafenib in melanoma cells [8].However, the effects of a combination of sorafenib and fluvastatin in HCC have not been reported.
We hypothesized that co-administration of sorafenib and fluvastatin would have synergistic anti-tumor efficacy against HCC compared to treatment with either drug alone. In the present study, we investigated whether simultaneous treatment with low-dose sorafenib and fluvastatin would have synergistic anti-tumor effects in HCC cells and in an animal HCC model. The molecular pathways underlying these effects were also explored.
RESULTS
Synergistic anti-tumor effects of sorafenib and fluvastatin in vivo
Low doses of sorafenib (10 mg/kg) and fluvastatin (10 mg/kg) were used in a diethylnitrosamine (DEN)-induced HCC rat model. The visible tumor nodules were reduced in the livers of rats of the combination group (P < 0.05, Figure 1A and 1B), but not of either of the single-treatment groups, compared with the control group (vehicle treated HCC rats). Survival analysis indicated that the survival time were longer in rats given the dual therapy than in those given the single-agent treatments (P < 0.05, Figure 1C). The serum alpha-fetoprotein (AFP) levels were also lower in the combination group than in the other groups (P < 0.05, Figure 1D). None of the rats displayed weight loss, bleeding in the skin or major organs, or significant hepatic or kidney injury (Figure 1E and 1F). These data suggested that the combination treatment more effectively inhibited tumor development in the rat HCC model than monotherapy.
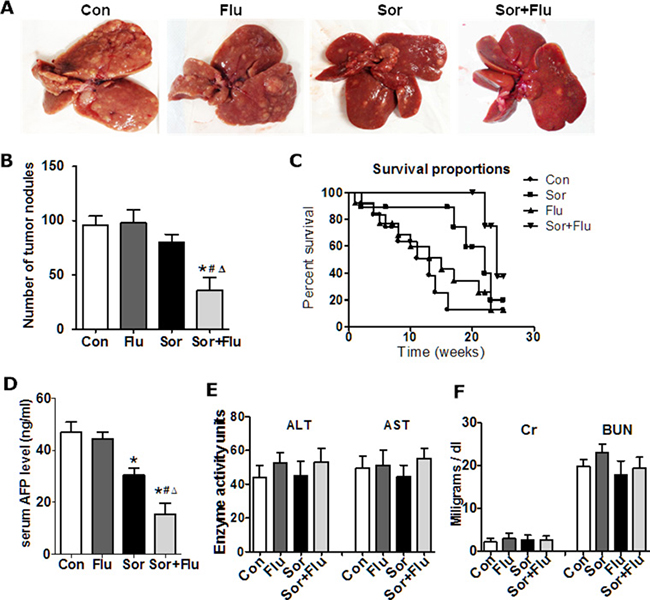
Figure 1: Sorafenib and fluvastatin reduced the progression of DEN-induced HCC. (A) Representative photos of livers from the rat HCC model (n = 6). (B) Number of tumors larger than 3 mm in the rat livers. (C) Survival analysis for each treatment group (n = 12). (D) Serum AFP levels detected by ELISA. (E) Safety analysis was carried out. Liver toxicity was estimated based on AST and ALT levels. Kidney toxicity was evaluated by creatinine and blood urea nitrogen levels (F). The data are expressed as the mean ± SEM of three independent experiments. *P < 0.05 vs. Con.#P < 0.05 vs. Flu. ΔP < 0.05 vs. Sor.
The dual treatment is superior in inhibiting tumor proliferation and increasing apoptosis
The effects of sorafenib and fluvastatin on the viability and apoptosis of HCC cells were evaluated both in vivo and in vitro. In the rat HCC model, a TUNEL assay revealed greater tumor necrosis in the combination group than in the other groups (P < 0.05) (Figure 2A). Cell proliferation was significantly lower in the combination group than in the control and monotherapy groups (P < 0.05, Figure 2B), as indicated by assays of proliferating cell nuclear antigen (PCNA)-positive cells. In HCC cell lines, sorafenib reduced cell viability in a dose-dependent manner (Figure 2C), but fluvastatin at 1 μM had no significant anti-tumor effects (Supplementary Figure 1). The combination of sorafenib and 1μM fluvastatin reduced cell proliferation to a greater extent than sorafenib alone (P < 0.05, Figure 2C). Low doses of sorafenib (1 μM) or fluvastatin (1 μM) had no effect on the apoptosis of HepG2 cells, as assessed by Annexin V/propidium iodide (PI), while the combination treatment significantly increased the level of apoptosis (Figure 2D). These results indicated that the dual therapy was more effective than monotherapy in inhibiting proliferation and inducing apoptosis in HCC.
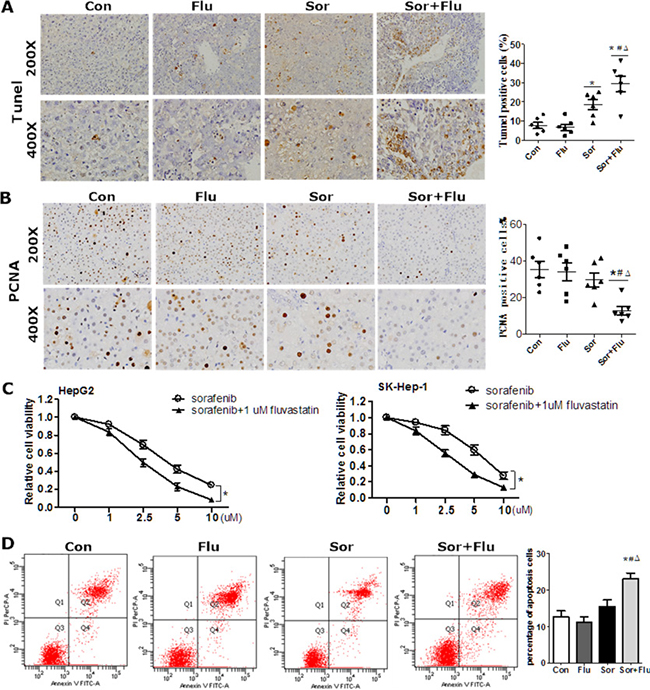
Figure 2: Synergistic cytotoxicity of sorafenib and fluvastatin against HCC. (A) TUNEL assays of rat liver tissues. (B) IHC staining for PCNA in the rat liver (magnification, 200 × & 400 ×). (C) HepG2 and SK-Hep-1 cells were incubated various doses of sorafenib plus 1 μM fluvastatin or vehicle for 72 h. Cell viability was evaluated by CCK8 assays. *P < 0.05 vs. sorafenib group. (D) HepG2 and SK-Hep-1 cells were incubated with vehicle, 1μM sorafenib, 1 μM fluvastatin, or both agents for 48 h. Apoptosis was measured by Annexin V/PI. Annexin V-fluorescein isothiocyanate+ PI- apoptotic cells are shown in the upper section. Data are expressed as the mean ± SEM of three independent experiments. *P < 0.05 vs. Con. #P < 0.05 vs. Flu. ΔP < 0.05 vs. Sor.
Effects of sorafenib plus fluvastatin on fibrosis and activation of hepatic stellate cells (HSCs) in HCC
Masson staining was performed to detect liver fibrosis in the rat HCC model. As shown in Figure 3A, DEN treatment induced notable accumulation of collagen in the livers of control rats compared with normal rats (P < 0.05). The accumulation of collagen in the liver was not reduced by monotreatment, but was significantly reduced by the co-treatment (Figure 3A). High α-smooth muscle actin (α-SMA) expression in the liver was observed in the control and monotherapy groups, while co-treatment significantly reduced α-SMA expression (Figure 3B). Similar results were found for the hepatic expression of COL1A1, COL3A1 and Fibronectin (Figure 3C).
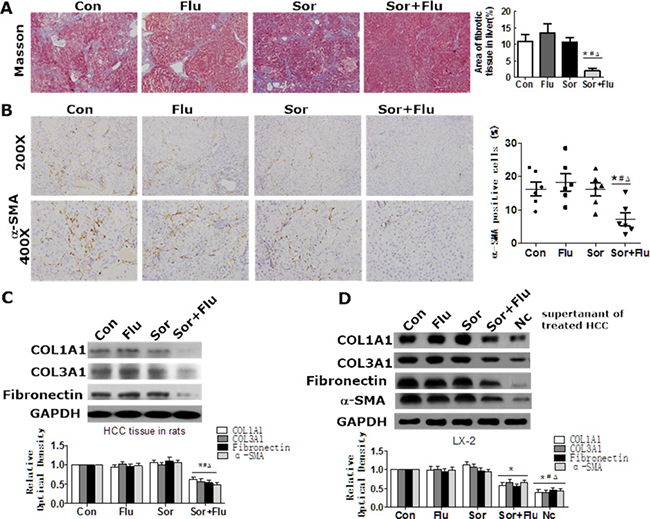
Figure 3: Effects of sorafenib and fluvastatin on liver fibrosis and HSCs activation. (A) Representative Masson trichrome staining of rat liver sections. The fibrotic area was quantified with a computerized grid. (B) IHC of α-SMA in rat liver (magnification, 200 × & 400 ×). (C) The protein levels of COL1A1, COL3A1, and Fibronectin in the liver tissues of rats were detected by Western blotting. (D) HepG2 cells were treated with 1 μM sorafenib, 1 μM fluvastatin, 1 μM sorafenib plus 1 μM fluvastatin, or the vehicle. LX-2 cells were cultured in the supernatants of HepG2 cells or control medium (Nc) for 48 h. Western blotting was used to detect the levels of COL1A1, COL3A1, Fibronectin and α-SMA in LX-2 cells. Values are expressed as the mean ± SEM of at least three independent experiments. *P < 0.05 vs. Con. #P < 0.05 vs. Flu. ΔP < 0.05 vs. Sor.
We next assessed the in vitro effects of sorafenib and fluvastatin on the activation of HSCs. Low doses of sorafenib and fluvastatin had no effect on the proliferation, apoptosis or activation of HSCs (data not shown). However, the supernatants of control HCC cells significantly stimulated the activation of HSCs, while the supernatants of dual-treated HCC cells inhibited the activation of HSCs (Figure 3D). These results indicated that the combination treatment attenuated liver fibrosis and inhibited the activation of HSCs in HCC to a greater extent than monotherapy.
Combination treatment significantly inhibits the expression of TLR4 in HCC
Toll-like receptor 4 (TLR4) contributes to HCC initiation and progression, as well as liver fibrosis [9]. TLR4 was overexpressed in human HCC tissues compared to normal liver tissues (P < 0.05, Supplementary Figure 2A). The mRNA and protein levels of TLR4 in HCC tissues were significantly lower in co-treated rats than in singly-treated rats (P < 0.05, Figure 4A and 4B). Moreover, TLR4 expression in HCC tissues correlated positively with PCNA-positivity in the rat model (Figure 4C). In HCC cell lines (HepG2 and SK-Hep-1 cells), lipopolysaccharide(LPS) stimulated the expression of TLR4 in a dose-dependent manner (Supplementary Figure 2B). TLR4 expression was significantly lower in HCC cell lines administered the combination treatment than in cells treated with a single agent (P < 0.05, Figure 4D). Dual treatment also reduced NF-κB p65 subunit binding to the TLR4 promoter, as determined by a binding activity assay (Figure 4E). Further analysis revealed that ectopic TLR4 expression reversed the reduced cell viability following co-treatment (Figure 4F). These data suggested that sorafenib plus fluvastatin significantly inhibited expression of TLR4 in HCC.
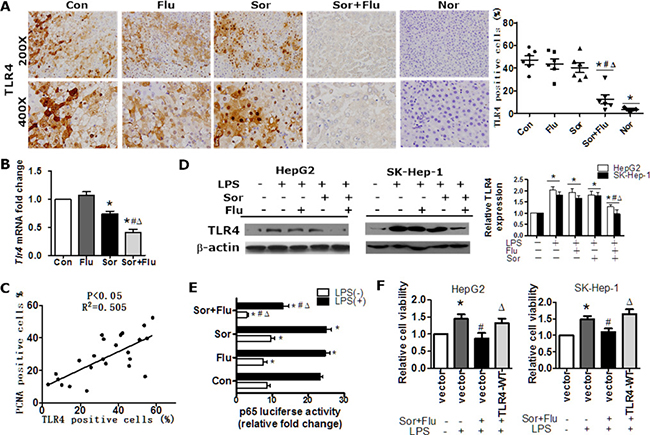
Figure 4: The combination treatment significantly inhibited TLR4 expression in HCC. (A) IHC staining of TLR4 in rat liver tissues (magnification, 200 × & 400 ×). (B) The transcription of Tlr4 in rat liver tissues was detected by qRT-PCR. *P < 0.05 vs. Con. #P < 0.05 vs. Flu. ΔP < 0.05 vs. Sor. (C) The correlation between TLR4 and PCNA expression in rat liver tumor tissues was determined by Pearson’s χ2 test. (D) HepG2 and SK-Hep-1 cell lines were pre-treated with DMSO, sorafenib (1 μM) or fluvastatin (1 μM) for 2 h and then stimulated with 10 ug/mL LPS for 48 h. TLR4 expression was determined by Western blot analysis. *P < 0.05 vs. control. **P < 0.05 vs. LPS. #P < 0.05 vs. LPS+Flu. ΔP < 0.05 vs. LPS+Sor. (E) HepG2 cells were treated with 1 μM sorafenib, 1 μM fluvastatin, 1 μM sorafenib plus 1 μM fluvastatin, or the vehicle for 30 min, and then were incubated with vehicle or LPS (10 ug/mL) for 24 h. A luciferase assay was performed to detect p65 transcription factor binding activity. *P < 0.05 vs. Con. #P < 0.05 vs. Flu. ΔP < 0.05 vs. Sor. (F) HepG2 and SK-Hep-1 cells were transfected with the control vector or TLR4-WT, treated with DMSO or sorafenib (1 μM) plus fluvastatin (1 μM) 30 min prior to the addition of LPS (10 μg/mL), and cultured for another 72 h. Cell viability was determined by CCK8 assays. *P < 0.05 vs. control. #P < 0.05 vs. LPS alone. ΔP < 0.05 vs. LPS+Sor+Flu. Data are shown as the mean ± SEM of three independent experiments.
Sorafenib and fluvastatin coordinately inhibit the activation of the NF-κB and MAPK pathways by TLR4 in HCC
NF-κB and MAPK are important proteins downstream of TLR4 that are critical stimulators of proliferation and inhibitors of apoptosis in HCC [10]. We further investigated whether sorafenib and fluvastatin would inhibit the NF-κB and MAPK pathways in HCC. In the rat HCC model, the phosphorylation of ERK and the nuclear translocation of p65 in liver tissues were significantly lower following co-treatment than following single drug administration (Figure 5A and 5B). In HCC cell lines, low doses of sorafenib or fluvastatin had no effect on p65, whereas co-treatment significantly suppressed the nuclear protein level of p65 (Figure 5C). The activity of the NF-κB p65 subunit in the nucleus exhibited similar results in an enzyme-linked immunosorbent assay (ELISA; Supplementary Figure 3A). The phosphorylation of ERK and p38 were also significantly inhibited in the co-treatment group (Figure 5D). Moreover, ectopic TLR4 expression reversed the inhibition of the MAPK and NF-κB pathways caused by co-treatment (Figure 5E, 5F and Supplementary Figure 3B). These data suggested that sorafenib and fluvastatin synergistically inhibited the activation of the NF-κB and MAPK pathways by TLR4 in HCC.
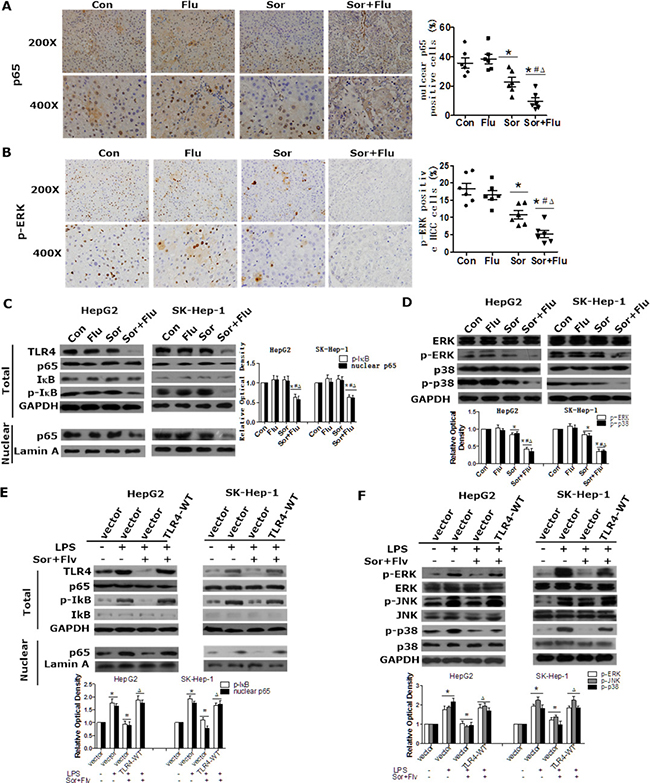
Figure 5: Sorafenib and fluvastatin inhibit the activation of the NF-κB and MAPK pathways in HCC. The levels of p65 (A) and p-ERK (B) in rat liver tissues were detected by IHC (magnification, 200 × & 400 ×). (C)HepG2 and SK-Hep-1 cells were pretreated with sorafenib(1 μM), fluvastatin (1 μM) or both drugs for 24 h. Western blot analysis was performed to determine the protein level of p65. (D) Cells were treated as indicated in (C) for 30 min or 24 h. The levels of p-ERK, p-p38 (30 min), ERK and p38 (24 h) were examined by Western blot analyses. *P < 0.05 vs. Con. #P < 0.05 vs. Flu. ΔP < 0.05 vs. Sor. (E) HepG2 and SK-Hep-1 cells were transfected with the control vector or TLR4-WT. Twenty-four hours after transfection, cells were pre-treated with DMSO or sorafenib (1 μM) plus fluvastatin (1 μM) for 30 min and then induced by 10 ng/mL LPS for 24 h or 2 h. Western blotting was used to analyze the protein levels of p65, IκB (24 h) and p-IκB (2 h). The levels of p-ERK, p-JNK, p-p38 (2h), ERK, JNK and p38 (24 h) were also detected by Western blotting (F). *P < 0.05 vs. control. #P < 0.05 vs. LPS alone. ΔP < 0.05 vs. LPS+Sor+Flu. Data are shown as the mean ± SEM. The experiments were repeated three times.
Dual treatment inhibited the activation of HSCs by reducing stromal cell-derived factor (SDF)-1α expression in HCC
The SDF-1α/CXCR4 axis has been shown to directly stimulate HSCs differentiation to myofibroblasts in HCC by activating the MAPK pathway [11]. Inhibition of ERK with U0126 prevented the recombinant-SDF-1α (rSDF-1α)-induced upregulation of α-SMA and Fibronectin in HSCs, which confirmed that SDF-1α/MAPK can stimulate HSCs (Supplementary Figure 4A). We then investigated whether sorafenib and fluvastatin influenced SDF-1α levels. In the rat model, SDF-1α expression was increased in HCC tissues of the control group, while co-treatment significantly diminished the upregulation of SDF-1α (Figure 6A and 6B). In HCC cell lines, the secretion of SDF-1α was also significantly inhibited by co-treatment (Figure 6C). Chromatin immunoprecipitation (ChIP) assays demonstrated that fluvastatin and sorafenib synergistically inhibited the binding of NF-κB p65 subunit to the SDF-1α promoter (Figure 6D). These data suggested that sorafenib plus fluvastatin significantly inhibited SDF-1α expression in HCC cells.
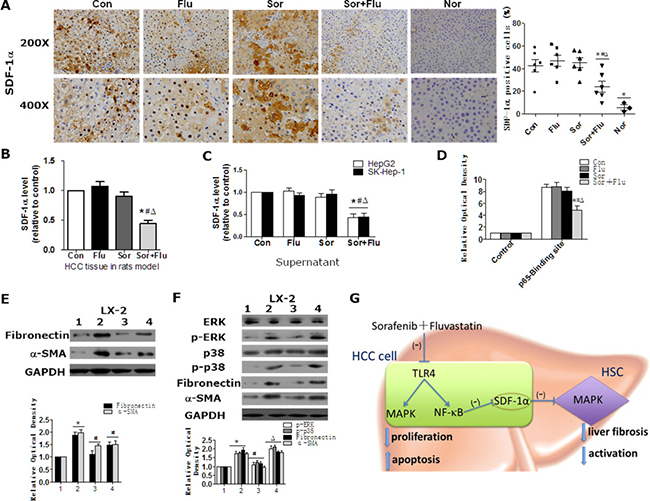
Figure 6: Sorafenib and fluvastatin inhibit the activation of HSCs by blocking the SDF1α/MAPK pathway. SDF-1α expression in rat liver tissues was detected by IHC (A) and ELISA (B). (C) HepG2 and SK-Hep-1 cells were treated with sorafenib (1 μM), fluvastatin (1 μM) or both drugs for 24 h. ELISA was performed to determine the protein levels of SDF-1α in the culture supernatants. (D) HepG2 cells were treated as indicated in (C). The binding of p65 to SDF-1α was detected through ChIP assays. *P < 0.05 vs. Con. #P < 0.05 vs. Flu. ΔP < 0.05 vs. Sor. (E) LX-2 cells were treated with control medium (lane 1), supernatants from HepG2 cells transfected with control siRNA (lane 2), or supernatants from HepG2 cells transfected with SDF-1α siRNA-1 (lane 3) and SDF-1α siRNA-2 (lane 4) for 24 h. Western blot analysis was performed to detect the expression of Fibronectin and α-SMA. *P < 0.05 vs. lane 1. #P < 0.05 vs. lane 2. (F) LX-2 cells were treated with control medium (lane 1), control supernatants (lane 2), or supernatants from dual-agent-treated (1 μM sorafenib+1 μM fluvastatin) HepG2 cells with (lane 3) or without rSDF1-α (40 ng/mL) (lane 4) for 30 min or 24 h. Western blot analysis was performed to detect the expression of p-ERK, p-p38 (30 min), ERK, p38, Fibronectin and α-SMA (24 h). *P < 0.05 vs. lane 1. #P < 0.05 vs. lane 2. ΔP < 0.05 vs. lane 3. Data are shown as the mean ± SEM of three independent experiments. (G) Schematic diagram describing the mechanism of combined sorafenib and fluvastatin in the treatment of HCC.
The expression of SDF-1α correlated positively with α-SMA expression in rat HCC tissues (Supplementary Figure 4B). Culture supernatants from treated HCC cells were used to culture HSCs. The supernatants of HepG2 cells activated the MAPK pathway as well as the expression of Fibronectin and α-SMA in HSCs, whereas blocking SDF-1α with neutralizing antibody inhibited these effects (Supplementary Figure 4C). In addition, HSCs cultured with supernatants from specific SDF-1α-silenced HepG2 cells had significantly lower Fibronectin and α-SMA expression compared to those cultured with supernatants from control HepG2 cells (Figure 6E and Supplementary Figure 4D). Similar results were obtained in SK-Hep-1cells (data not shown). These data confirmed the involvement of SDF-1α in the activation of HSCs. The supernatants of dual-agent-treated HCC cells inhibited ERK phosphorylation and Fibronectin and α-SMA expression in HSCs, while rSDF-1α reversed these inhibitory effects (Figure 6F). Our data suggested that sorafenib plus fluvastatin inhibited the expression and secretion of SDF-1α, which subsequently blocked the MAPK pathway and suppressed the activation of HSCs.
DISCUSSION
Drug resistance to sorafenib is common in patients with HCC. There is a growing need to explore combination treatments with sorafenib to enhance the anti-tumor effects on HCC. Fluvastatin has been suggested to have therapeutic potential for the treatment of HCC [7]. In the present study, we hypothesized that combining sorafenib and fluvastatin would maximize the therapeutic effects of these treatments. We used a low dose of sorafenib to assess its potential synergistic effects with fluvastatin [12]. We demonstrated that the combination treatment was superior to either drug treatment alone in promoting apoptosis and inhibiting proliferation in HCC, both in vivo and in vitro. In combination, these drugs also exhibited clear synergism in inhibiting liver fibrosis and activating HSCs. To our knowledge, this is the first report disclosing the synergistic effects of co-treatment with sorafenib and fluvastatin in HCC.
As an essential cell surface protein for LPS recognition, TLR4 can activate HSCs and promote liver fibrosis [13].TLR4 activation is central to the progression of hepatic fibro-inflammatory disease and carcinoma [14–16]. High expression of TLR4 in HCC tissues has been shown to be strongly associated with early recurrence and poor survival in patients [17].Consistent with a previous study [18], we found that TLR4 expression was greater in human HCC samples than in normal liver samples. The combination of sorafenib with fluvastatin significantly reduced Tlr4 mRNA and protein levels in HCC cells, both in vitro and in vivo. These synergistic effects may have been due to the inhibition of p65 binding to the TLR4 promoter.
The NF-κB and MAPK pathways are major stimulators of proliferation and inhibitors of apoptosis in HCC. Consistent with previous studies [15, 19, 20], we found that LPS/TLR4 signaling activated the NF-κB/MAPK pathways. Moreover, co-treatment synergistically inhibited the LPS/TLR4-activated NF-κB/MAPK pathways in HCC cells.
The interaction between HCC cells and HSCs is central to tumor progression. HCC cells secrete cytokines and chemokines to promote the activation of HSCs, while activated HSCs also produce chemokines to promote tumor cell proliferation and migration [21]. A recent study demonstrated that HSCs induce HCC resistance to sorafenib [22].This indicates the potential involvement of tumor fibrosis in the development of resistance to sorafenib in HCC. Sorafenib has dual effects on HSCs and liver fibrosis in HCC progression. Despite its inhibitory effect on hepatic fibrogenesis, sorafenib may activate the SDF-1/CXCR4 pathway and increase tumor-associated fibrosis in HCC [11, 23]. Fluvastatin has been shown to attenuate hepatic steatosis-induced liver fibrosis by inhibiting the paracrine effects of hepatocytes on HSCs [24]. However, the effect of fluvastatin on liver fibrosis in HCC has never been determined. In our study, low doses of sorafenib or fluvastatin alone could not attenuate fibrogenesis in HCC, while dual treatment significantly alleviated fibrosis by inhibiting the activation of HSCs both in vivo and in vitro.
SDF-1α is a potent chemoattractant for fibrocytes and thus contributes to liver fibrosis. The SDF-1α/CXCR4 pathway directly induces HSC differentiation and proliferation by activating MAPK to increase tumor-associated fibrosis [11]. Consistent with recently reported findings, we found that SDF-1α was overexpressed in HCC and could activate HSCs by stimulating the MAPK pathway. Sorafenib has been shown to elevate SDF-1α/CXCR4 in a mouse HCC model [23]. However, in our DEN-induced rat HCC model, sorafenib treatment did not alter the expression of SDF-1α in HCC cells. This difference may have been due to the low dose of sorafenib used in our study. Single treatment with fluvastatin also had no effect on SDF-1α expression in HCC. However, dual treatment significantly inhibited the expression of SDF-1α by preventing the binding of p65 to the SDF-1α promoter in HCC cells. Our data indicated that sorafenib plus fluvastatin inhibited the expression and secretion of SDF-1α in HCC cells and subsequently reduced the activation of HSCs by inhibiting the MAPK pathway.
In conclusion, the combination of sorafenib and fluvastatin significantly inhibited cellular proliferation and promoted apoptosis in HCC by inhibiting the TLR4-activated NF-κB and MAPK pathways. Dual treatment also significantly alleviated tumor-associated fibrosis and inhibited the activation of HSCs by blocking the expression and secretion of SDF-1α in HCC cells, and subsequently inhibited the MAPK pathway in HSCs (Figure 6G). Our results indicate that the combination of sorafenib and fluvastatin may be a potential therapeutic strategy for patients with advanced HCC.
MATERIALS AND METHODS
Chemicals
For in vitro experiments, sorafenib (Selleck Chemicals, Houston, TX, USA) and fluvastatin (Sigma, St. Louis, MO, USA) were dissolved in DMSO. For in vivo experiments, sorafenib was dissolved in a Cremophor EL-ethanol solution (50:50, Sigma Cremophor EL :95% ethyl alcohol) and diluted with distilled water [7], while fluvastatin was dissolved in distilled water.
Animal studies
All protocols for animal care and use were approved by the Animal Care Committee of the Southern Medical University. Male Wistar rats received intraperitoneal injections of DEN (Sigma) at 50 mg/kg weekly for 12 weeks. At the end of three months of DEN injection, three rats were randomly chosen and killed to ensure the development of hepatocellular carcinoma, as confirmed by pathological analysis. Rats were then submitted to daily gavage administration of either 10 mg/kg sorafenib (Group Sor), 10 mg/kg fluvastatin (Group Flu), a combination of both substances (Group Sor+Flu), or vehicle (Group Con). Vehicle-injected normal rats were used as controls (Group Nor). After 8 weeks of drug treatment, rats were sacrificed for analysis (n = 6) or continuously treated as indicated for survival analysis(n = 12). Malignant liver nodules ≥ 3 mm in diameter were counted by two independent investigators. Liver tissue and serum samples were collected for later analysis.
Human tissue samples
The use of human samples was approved by the patients and the ethics committee of Nanfang Hospital, Southern Medical University. Fifty liver tumor tissue samples from patients with HCC at Nanfang Hospital were confirmed by pathology. Thirty normal liver tissue samples were also collected. The patients’ characteristics are shown in Table 1.
Table 1: Characteristics of patients with HCC
Characteristic |
No. of patients |
Percentage |
|
|---|---|---|---|
Gender |
Male |
32 |
64 |
Female |
18 |
36 |
|
Age |
≤ 45 |
23 |
46 |
>45 |
27 |
54 |
|
Pathology |
High differentiated |
15 |
30 |
Moderately differentiated |
24 |
48 |
|
Low differentiated |
11 |
22 |
|
AFP |
≤ 400 |
18 |
36 |
> 400 |
32 |
64 |
|
Tumor size |
≤ 5 cm |
25 |
50 |
> 5 cm |
25 |
50 |
|
Prognosis |
Recrudesce |
35 |
70 |
No-recrudesce |
15 |
30 |
|
Immunohistochemistry (IHC)
IHC staining was performed with standard protocols [25]. Specific primary antibodies against TLR4, phospho-ERK (p-ERK), p65, α-SMA (Abcam, Cambridge, UK), and SDF-1α (Novus Biologicals, Littleton, CO, USA) were used. The results are expressed as the percentage of positively stained cells in five randomly selected microscopic fields counted by two independent researchers.
Cell culture
The human hepatic stellate cell line LX-2 and the human HCC cell lines HepG2 and SK-Hep-1 were obtained from Shanghai Advanced Research Institute, Chinese Academy of Sciences. Cells were maintained in DMEM supplemented with 10% fetal bovine serum and incubated in humidified air containing 5% CO2 at 37°C. Cultures were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions for the following experiments. To obtain conditioned media for the culture of LX-2 cells for specific experiments, we treated HepG2 and SK-Hep-1 cells with sorafenib/fluvastatin for 24 h and washed them three times with PBS. After routine culture of the cells for 48 h, the supernatants were collected.
Cell viability assay
HepG2 and SK-Hep-1 cells were seeded in 96-well plates (2000 cells/well) and were treated with fluvastatin or sorafenib for 72 h. The relative numbers of viable cells were detected by CCK8 reagents (DingGuo Bio, Beijing, China) [26].
TUNEL assay
Apoptotic cells in HCC tissues from rats were analyzed with a Click-iT™ TUNEL Colorimetric IHC Detection Kit (Thermo Fisher, Hudson, NH, USA) in accordance with the manufacturer’s instructions. TUNEL-stained apoptotic cells were counted in five randomly selected microscopic fields.
Apoptosis assay
Pretreated HCC cells were stained with an Annexin V-fluorescein isothiocyanate/PI apoptosis detection kit (BD Biosciences, La Jolla, CA, USA) according to the manufacturer’s instructions. Stained cells were analyzed by FACSCalibur.
Gene expression analysis
Total RNA was extracted and cDNA was synthesized according to standard protocols. The cDNA was further PCR-amplified by specific primers with an All-in-One qPCR Mix kit (GeneCopoeia, Rockville, MD, USA). Gapdh was used as a housekeeping gene. The primers used are listed in Supplementary Table 1.
Western blotting
Protein extracts were obtained through standard protocols [27]. Western blot analyses were performed as described previously [27]. Specific primary antibodies against COL1A1, COL3A1, Fibronectin, α-SMA, ERK, IκB, phospho-IκB, JNK, phospho-JNK (Cell Signaling Technology, Danvers, MA, USA), TLR4, p-ERK, p65 and SDF-1α (Abcam) were used. β-actin (Abcam) and Lamin A (Santa Cruz) were used as loading controls for total protein and nuclear protein, respectively.
ELISA
Rat serum AFP levels were determined by ELISA (Bio-Swamp, Shanghai, China) per the manufacturer’s instructions. The NF-κB nuclear concentrations and SDF-1α levels in the culture supernatants of treated HCC cells were also detected by ELISA (Abcam).
Luciferase reporter assay
Lipofectamine 2000 was used to transfect HepG2 cells with a pGL3 [luc/SV40] reporter plasmid containing the promoter sequence of hTLR4, or a pGL4.74 [hRluc/TK] control plasmid. Cells were subsequently incubated with different agents for 24 h. The firefly and Renilla luciferase activities of the cell extracts were measured with a Dual-Luciferase Reporter Assay System (Promega, Madison, WY, USA) according to the manufacturer’s instructions.
ChIP
P65 binding to the SDF-1α promoter was tested by ChIP analysis according to the manufacturer’s instructions. Promoter binding was examined by RT-PCR with the following primers: SDF-1α, F: TCTAGGGAAGTTCCAAACAGGAG; R: ACCAG GGAGCCATATTTTGGG.
Statistical analysis
All experimental data are expressed as the mean ± SEM from at least three experiments. Unpaired Student’s t-tests and analysis of variance (ANOVA) were used for statistical analysis. All statistical analyses were conducted with SPSS 13.0 software. A P-value less than 0.05 was considered statistically significant.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by the Postdoctoral Research Funding of Guangzhou Women and Children’s Medical Center [5001-2150172], the Guangdong Natural Science Foundation [2016A030310254], China Postdoctoral Science Foundation [2016M600648] and National Natural Science Foundation [81602646].
REFERENCES
1. Gao J, Xie L, Yang WS, Zhang W, Gao S, Wang J, Xiang YB. Risk factors of hepatocellular carcinoma—current status and perspectives. Asian Pac J Cancer Prev. 2012; 13:743–52.
2. Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004; 64:7099–109. doi: 10.1158/0008-5472.can-04-1443.
3. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008; 359:378–90. doi: 10.1056/NEJMoa0708857.
4. Almhanna K, Philip PA. Safety and efficacy of sorafenib in the treatment of hepatocellular carcinoma. Onco Targets Ther. 2009; 2:261–7.
5. Slawinska-Brych A, Zdzisinska B, Kandefer-Szerszen M. Fluvastatin inhibits growth and alters the malignant phenotype of the C6 glioma cell line. Pharmacol Rep. 2014; 66:121–9. doi: 10.1016/j.pharep.2014.01.002.
6. Kanugula AK, Dhople VM, Volker U, Ummanni R, Kotamraju S. Fluvastatin mediated breast cancer cell death: a proteomic approach to identify differentially regulated proteins in MDA-MB-231 cells. PLoS One. 2014; 9:e108890. doi: 10.1371/journal.pone.0108890.
7. Zhang W, Wu J, Zhou L, Xie HY, Zheng SS. Fluvastatin, a lipophilic statin, induces apoptosis in human hepatocellular carcinoma cells through mitochondria-operated pathway. Indian J Exp Biol. 2010; 48:1167–74.
8. Zhang S, Doudican NA, Quay E, Orlow SJ. Fluvastatin enhances sorafenib cytotoxicity in melanoma cells via modulation of AKT and JNK signaling pathways. Anticancer Res. 2011; 31:3259–65.
9. Wang Z, Yan J, Lin H, Hua F, Wang X, Liu H, Lv X, Yu J, Mi S, Wang J, Hu ZW. Toll-like receptor 4 activity protects against hepatocellular tumorigenesis and progression by regulating expression of DNA repair protein Ku70 in mice. Hepatology. 2013; 57:1869–81. doi: 10.1002/hep.26234.
10. Peng XX, Zhang SH, Wang XL, Ye TJ, Li H, Yan XF, Wei L, Wu ZP, Hu J, Zou CP, Wang YH, Hu XD. Panax Notoginseng flower saponins (PNFS) inhibit LPS-stimulated NO overproduction and iNOS gene overexpression via the suppression of TLR4-mediated MAPK/NF-kappa B signaling pathways in RAW264.7 macrophages. Chin Med. 2015; 10:15. doi: 10.1186/s13020-015-0045-x.
11. Chen Y, Huang Y, Reiberger T, Duyverman AM, Huang P, Samuel R, Hiddingh L, Roberge S, Koppel C, Lauwers GY, Zhu AX, Jain RK, Duda DG. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/C-X-C receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology. 2014; 59:1435–47. doi: 10.1002/hep.26790.
12. Hu S, Niu H, Inaba H, Orwick S, Rose C, Panetta JC, Yang S, Pounds S, Fan Y, Calabrese C, Rehg JE, Campana D, Rubnitz JE, et al. Activity of the multikinase inhibitor sorafenib in combination with cytarabine in acute myeloid leukemia. J Natl Cancer Inst. 2011; 103:893–905. doi: 10.1093/jnci/djr107.
13. Wang PP, Xie DY, Liang XJ, Peng L, Zhang GL, Ye YN, Xie C, Gao ZL. HGF and direct mesenchymal stem cells contact synergize to inhibit hepatic stellate cells activation through TLR4/NF-kB pathway. PLoS One. 2012; 7:e43408. doi: 10.1371/journal.pone.0043408.
14. Toffanin S, Cornella H, Harrington A, Llovet JM. HCC Is promoted by bacterial translocation and TLR-4 signaling: a new paradigm for chemoprevention and management. Hepatology. 2012; 56:1998–2000. doi: 10.1002/hep.26080.
15. Ci X, Ren R, Xu K, Li H, Yu Q, Song Y, Wang D, Li R, Deng X. Schisantherin A exhibits anti-inflammatory properties by down-regulating NF-kappaB and MAPK signaling pathways in lipopolysaccharide-treated RAW 264.7 cells. Inflammation. 2010; 33:126–36. doi: 10.1007/s10753-009-9166-7.
16. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007; 13:1324–32. doi: 10.1038/nm1663.
17. Jing YY, Han ZP, Sun K, Zhang SS, Hou J, Liu Y, Li R, Gao L, Zhao X, Zhao QD, Wu MC, Wei LX. Toll-like receptor 4 signaling promotes epithelial-mesenchymal transition in human hepatocellular carcinoma induced by lipopolysaccharide. BMC Med. 2012; 10:98. doi: 10.1186/1741-7015-10-98.
18. Wang L, Zhu R, Huang Z, Li H, Zhu H. Lipopolysaccharide-induced toll-like receptor 4 signaling in cancer cells promotes cell survival and proliferation in hepatocellular carcinoma. Dig Dis Sci. 2013; 58:2223–36. doi: 10.1007/s10620-013-2745-3.
19. Seifert L, Deutsch M, Alothman S, Alqunaibit D, Werba G, Pansari M, Pergamo M, Ochi A, Torres-Hernandez A, Levie E, Tippens D, Greco SH, Tiwari S, et al. Dectin-1 Regulates Hepatic Fibrosis and Hepatocarcinogenesis by Suppressing TLR4 Signaling Pathways. Cell Rep. 2015; 13:1909–21. doi: 10.1016/j.celrep.2015.10.058.
20. Bai T, Lian LH, Wu YL, Wan Y, Nan JX. Thymoquinone attenuates liver fibrosis via PI3K and TLR4 signaling pathways in activated hepatic stellate cells. Int Immunopharmacol. 2013; 15:275–81. doi: 10.1016/j.intimp.2012.12.020.
21. Kang N, Shah VH, Urrutia R. Membrane-to-Nucleus Signals and Epigenetic Mechanisms for Myofibroblastic Activation and Desmoplastic Stroma: Potential Therapeutic Targets for Liver Metastasis? Mol Cancer Res. 2015; 13:604–12. doi: 10.1158/1541-7786.mcr-14-0542.
22. Azzariti A, Mancarella S, Porcelli L, Quatrale AE, Caligiuri A, Lupo L, Dituri F, Giannelli G. Hepatic stellate cells induce hepatocellular carcinoma cell resistance to sorafenib through the laminin-332/alpha3 integrin axis recovery of focal adhesion kinase ubiquitination. Hepatology. 2016; 64:2103–17. doi: 10.1002/hep.28835.
23. Mazzocca A, Dituri F, Lupo L, Quaranta M, Antonaci S, Giannelli G. Tumor-secreted lysophostatidic acid accelerates hepatocellular carcinoma progression by promoting differentiation of peritumoral fibroblasts in myofibroblasts. Hepatology. 2011; 54:920–30. doi: 10.1002/hep.24485.
24. Chong LW, Hsu YC, Lee TF, Lin Y, Chiu YT, Yang KC, Wu JC, Huang YT. Fluvastatin attenuates hepatic steatosis-induced fibrogenesis in rats through inhibiting paracrine effect of hepatocyte on hepatic stellate cells. BMC Gastroenterol. 2015; 15:22. doi: 10.1186/s12876-015-0248-8.
25. Yang MH, Chen CL, Chau GY, Chiou SH, Su CW, Chou TY, Peng WL, Wu JC. Comprehensive analysis of the independent effect of twist and snail in promoting metastasis of hepatocellular carcinoma. Hepatology. 2009; 50:1464–74. doi: 10.1002/hep.23221.
26. Yao G, Ling L, Luan J, Ye D, Zhu P. Nonylphenol induces apoptosis of Jurkat cells by a caspase-8 dependent mechanism. Int Immunopharmacol. 2007; 7: 444–53. doi: 10.1016/j.intimp.2006.11.013.
27. Sun T, Zhao N, Zhao XL, Gu Q, Zhang SW, Che N, Wang XH, Du J, Liu YX, Sun BC. Expression and functional significance of Twist1 in hepatocellular carcinoma: its role in vasculogenic mimicry. Hepatology. 2010; 51:545–56. doi: 10.1002/hep.23311.