
INTRODUCTION
Cancer is an age related disease [1–5]. Consequently, exploration of the association between markers of ageing and cancer represents an obvious step to bring advances in both research areas.
Biomarkers that linearly change with chronological age are now available to the scientific community and span from anatomical (e.g. ocular biomarkers [6]) to molecular ones including micro-RNAs levels [7, 8], protein modifications [9] and telomeres’ length [10].
DNA methylation-based biomarkers have gained relevance in the last few years for many reasons. First, both genome-wide and high-throughput targeted approaches to measure DNA methylation are easily accessible and highly reproducible. Second, these markers show extremely high correlation with chronological age and with age-acceleration effects associated with pathological conditions, morbidity and mortality. Taken together, these results make it possible to hypothesize that a positive deviation from normal aging trajectories (i.e. higher biological than chronological age) could be predictive, if not causative, of the development of several diseases, including cancer [11].
To date a few studies have approached this idea, with still inconclusive results. Nan et al. found no association between the overall white blood cell (WBC) DNA methylation levels and colorectal cancer (CRC) risk among 358 females where blood samples had been collected prior to CRC diagnosis [12]. On the contrary, Pufulete et al. [13] and Lim et al. [14] reported significant association between hypomethylation in WBC DNA and an increased risk for colorectal adenomas. Finally, Walters et al. [15] described correlation between three DNA repetitive elements that present increased methylation levels in WBC from 539 cases diagnosed before 60 years of age and 242 healthy, cancer free, subjects.
Because of their ease of calculation and their prognostic potential, several methodologies have been developed to compute the epigenetic age.
Horvath’s epigenetic clock [16, 17], a multi-tissue predictor based on the methylation status of 353 CpG sites assessed by the Infinium HumanMethylation27 BeadChip (HM27) is among the most popular epigenetic age estimators. According to Horvath’s clock, age-acceleration was found in blood, brain and saliva from people affected by Down syndrome, a disease characterized by atypical aging patterns [18]. Similarly, the same clock successfully detected age acceleration in dorsolateral prefrontal cortex from patients with Alzheimer’s disease [19] and in whole blood from Parkinson’s disease patients [20]. Frailty [21], lifetime stress [22], HIV-1 infection [23], and menopause [24] were also found to accelerate epigenetic age of WBC. Finally it was demonstrated that epigenetic age estimated from whole blood DNA methylation is correlated to physical and cognitive fitness [25] and mortality [26–30] in large human cohorts. Importantly, Horvath’s clock is able to detect not only age-acceleration, but also age-deceleration effects in models of healthy aging and longevity [31].
Another epigenetic age-associated biomarker has been developed by Hannum et al. [29] and relies on the DNA methylation values of 71 CpG sites (only six being in common with Horvath’s) from the Infinium HumanMethylation450 BeadChip (HM450). Differently from Horvath’s clock, Hannum’s model was calibrated on whole blood only. Three studies demonstrated the association of this epigenetic clock with biological fitness and mortality [25, 26, 28], as well as its association with post-traumatic stress disorders [32].
The quantification of age acceleration (and deceleration) starting from easy-to-access blood samples has triggered efforts towards the simplification of Horvath’s and Hannum’s epigenetic clocks.
In this direction, a model based on 3 CpG sites (Weidner’s estimator) was found to significantly correlate with chronological age [33] but failed to predict mortality in the Lothian Birth Cohort 1921 study [34].
Finally, our group identified two HM450 CpG probes, cg16867657 in the CpG island of ELOVL2 and cg06639320 in the CpG island of FHL2 showing very high correlation with chronological age (Spearman correlation = 0.91) in whole blood DNA methylation data [35, 36]. These two loci were confirmed in several replicative tissues other than blood [36–40] and have been calibrated so far on teeth samples [41].
Age acceleration phenomena have been investigated also in cancer patients, owing to the peculiar observation that biomarkers of aging do not systematically show age acceleration in the tumour tissues, while they do in the blood of cancer-free people who develop cancer prospectively [42, 43]. To confirm an expand these promising findings we explored the reproducibility of this observation in an independent cohort collected by the Human Genetics Foundation (HuGeF, Turin, Italy) including prospective breast cancer and CRC data [44].
RESULTS AND DISCUSSION
Epigenetic age was estimated from DNA methylation blood data using 5 different methodologies: Horvath’s, Hannum’s, Weidner’s, ELOVL2 and FHL2 DNA methylation ages (DNAmAges) with and without adjustment for blood cell counts. We will use the term “Age Accel” to refer to non-adjusted age acceleration and IEAA otherwise. See Material and Methods for details.
Age Accel between females that developed breast cancer at follow-up and controls (cancer-free patients) was statistically significantly different only when using the ELOVL2 clock (Mann-Whitney-Wilcoxon test p-value = 0. 0432), with Age Accel values in tumor samples on average 0.9 years higher than in the control group (Supplementary Table 1 and Figure 1). Despite conservation of this trend (i.e. subjects that developed breast cancer still tend to have higher ELOVL2-based IEAA values than controls) statistical significance was lost when correction for blood cell counts was applied.
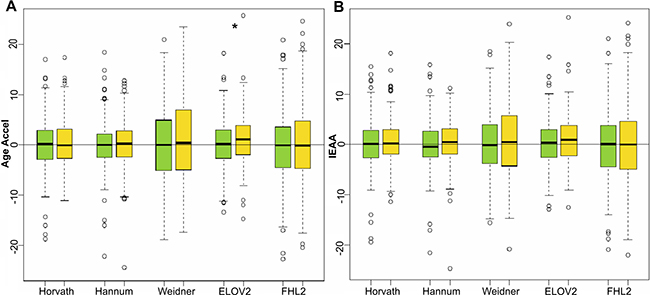
Figure 1: Age acceleration predictors in breast cancer samples. Boxplots of Age Accel (A) and IEAA (B) values for 233 female control subjects (green) and 233 female subjects that developed breast cancer at follow up (yellow), estimated by the 5 epigenetic predictors. Asterisks indicate significant differences according to Mann-Whitney-Wilcoxon test (p-value < 0.05), which was 0.0432 for ELOVL2 age acceleration estimators.
With respect to the male subjects that developed CRC, Horvath’s and FHL2 clocks returned a significant increase in Age Accel values (Mann-Whitney-Wilcoxon test p-value = 0.0421 and 0.0363 for Horvath’s and FHL2’s estimations respectively). Subjects that developed colon cancer were 1.6 and 2.5 years older using Horvath and FHL2 methods than their respective controls (Supplementary Table 1 and Figure 2). Although results by Hannum’s clock showed an evident trend towards higher Age Accel, this predictor did not give significant results, nor did Weidner’s nor ELOVL2 clocks. None of the 5 methods returned significant differences when IEAA values were compared, although a trend was visible with Horvath’s, Hannum’s and FHL2 clocks.
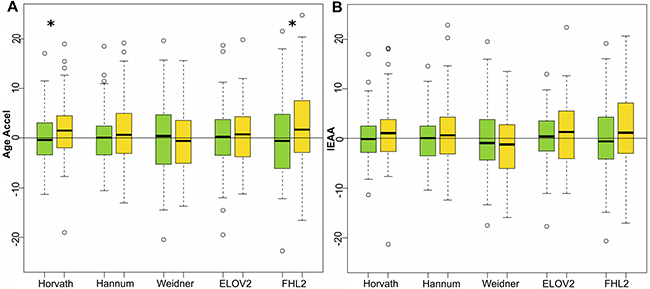
Figure 2: Age acceleration predictors in colorectal cancer male samples. Boxplots of Age Accel (A) and IEAA (B) values for 84 male control subjects (green) and 87 subjects that developed CRC at follow up (yellow), estimated by the 5 epigenetic predictors. Asterisks indicate significant differences according to Mann-Whitney-Wilcoxon test (p-value < 0.05), which were respectively 0.0421 and 0.0363 for Horvath and FHL2 age acceleration estimators.
For the CRC female counterpart, no significant differences were observed for Age Accel nor IEAA in any of the 5 predictors, despite a visible difference between the medians for Weidner, FHL2 and ELOV2 estimators (Supplementary Table 1 and Figure 3).
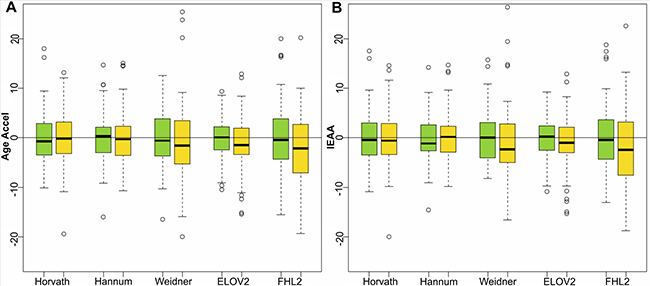
Figure 3: Age acceleration predictors in colorectal cancer female samples. Boxplots of Age Accel (A) and IEAA (B) values for 79 female control subjects (green) and 79 subjects that developed breast cancer at follow up (yellow), estimated by the 5 epigenetic predictors.
To explore these results further, we performed survival analysis using Kaplan-Meier method. For each of the five DNAmAge estimators we considered both Age Accel and IEAA values. Figure 4 shows the results for Age Accel and the corresponding IEAA obtained with Horvath, FHL2 and ELOV2, which are the estimators that were able to reveal significant differences in age acceleration between tumor and control samples. Results relative to all the other clocks and subgroups are reported in Supplementary Figures 1, 2 and 3. Log-rank test p-values are summarized in Table 1.
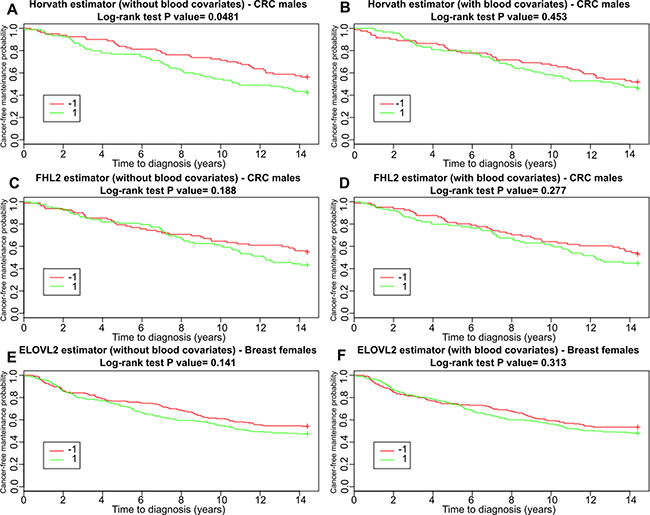
Figure 4: Survival functions for subjects belonging to the CRC males and breast cancer groups (including controls) incidence estimated with Kaplan-Meier method. Results are shown separately for accelerated (1) and decelerated (–1) age subjects, with age acceleration computed considering the estimators that showed significant differences between cases and controls: Horvath and FHL2 estimator for the CRC males dataset (A–D charts) and ELOVL2 for the breast dataset (E–F charts). In each chart title, we reported the Log-Rank test p-values comparing survival curves.
Table 1 : Survival analysis
Horvath |
Hannum |
Weidner |
ELOVL2 |
FHL2 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
Age Acc |
IEAA |
Age Acc |
IEAA |
Age Acc |
IEAA |
Age Acc |
IEAA |
Age Acc |
IEAA |
|
BRC females |
0.581 |
0.786 |
0.429 |
0.216 |
0.206 |
0.212 |
0.141 |
0.313 |
0.841 |
0.885 |
CRC males |
0.0481 |
0.453 |
0.346 |
0.68 |
0.313 |
0.527 |
0.767 |
0.231 |
0.188 |
0.277 |
CRC females |
0.732 |
0.67 |
0.541 |
0.202 |
0.479 |
0.165 |
0.0424 |
0.0395 |
0.423 |
0.479 |
Log-Rank test p-values for the three studied datasets and considering all five epigenetic age estimators, with and without correction for blood cell counts.
Overall, our analysis expands the results of Levine et al. focusing on lung cancer development using only Horvath’s epigenetic clock [43], and of Zheng et al. who applied both Horvath’s and Hannum’s predictors to a cohort of subjects that prospectively developed different types of cancer (mainly skin and prostate cancer) [42] and found that blood epigenetic age is related to cancer development and could be a potential biomarker for cancer early detection.
Here we observed that the two most used epigenetic clocks, Horvath’s and Hannum’s, are unable to detect age acceleration effects in blood of females that were later diagnosed with breast cancer, while significant differences were observed with ELOVL2 predictor. On the contrary, age acceleration computed with Horvath’s epigenetic clock, together with FHL2 clock, were associated with CRC development in males [42, 43].
The biological reasons behind the effectiveness of each clock is still to be unveiled, although the diverse epigenetic origin of each tumor type is bound to impact on the definition of CpG specific age acceleration.
In conclusion, we showed that different epigenetic estimators identify age acceleration effects in whole blood of subjects that prospectively developed cancer with a tumor type- and sex-specificity. These results reinforce the idea that a surrogate tissue can be used to evaluate the susceptibility to develop age-related diseases in other tissues and are encouraging for the fine tuning of more precise prognostic epigenetic biomarkers of age. In this sense, the observation that single CpG predictors, like ELOVL2 and FHL2, can detect epigenetic age deviations associated with future diagnosis of specific cancer types is of practical relevance.
On the cautious side, it is known that several variables like behavioral habits or previous health information (recently reviewed in [45]) may act as confounders of epigenetic age estimative. Therefore, the limited number of such variables made available (including batches) could be a limitation of this work. Given the potential of such results, a higher number of prospective studies of this type with freely accessible data is crucially needed to independently validate these findings.
MATERIAL AND METHODS
Blood dataset
We interrogated the Gene Expression Omnibus (GEO) repository using the search terms GPL13534 (GEO identifier for the HM450 platform), cancer and follow up. On February 2017, this search output 5 datasets, among those we selected the only one (GSE51032) whose sample size (hundreds of patients) is able to guarantee robustness of all findings and sufficient statistical power. The GSE51032 dataset contains DNA methylation measures on blood cells (buffy coats) from subjects that were prospectively followed by the Human Genetics Foundation (HuGeF) in Turin, Italy as part of the European Prospective Investigation into Cancer and Nutrition (EPIC). This study was conducted in ten European countries on populations that differ markedly in terms of dietary habits and cancer risk. The Italian EPIC cohort consists of 47,749 people recruited in the centers of Ragusa (6,404 subjects), Florence (13,597 subjects), Turin (10,604 subjects), Naples (5,062 subjects, women only) and Milan (12,079 subjects) [46, 47]. In Turin, the study recruitment began in 1993–1998 (people were aged 35-64 with no previous cancer) and was closed in 2010. The dataset includes DNA methylation data from 845 participants, selected as follows: 188 men and 657 women; at final follow-up 424 remained cancer-free (control samples), 235 had developed primary breast cancer, 166 had developed CRC and 20 had developed other primary cancers (5 bladder, 4 prostate gland, 4 skin, 2 bronchus and lung, 1 hemato reticuloendothelial, 1 corpus uteri, 1 kidney, 1 thyroid and endocrine glands, 1 unknown primary site lesion). In this work, we grouped colon, rectosigmoid and rectum data under the unifying label of CRC. To guarantee reproducibility and statistical power of our analyses, epigenetic age was calculated only for breast and CRC (>150 samples each).
All samples characteristics are reported in Table 2. Females and males were analyzed separately (stratification approach), for the two types of tumors, according to the recent report on sex-related differences in epigenetic age predictions [48]. Sex, in fact, is a confounding variable that affects both tumor incidence and age acceleration, and stratification has the advantage to take this into account, as well as to estimate the association between age acceleration and tumor incidence separately for females and males. Finally, to avoid unequal sample size issues, we randomly selected a subgroup of control samples with the same size and the same mean age of the group under study (Table 2).
Table 2: Sample characteristics
N |
Age at recruitment (mean years ± sd)/Median |
Time to diagnosis (mean years ± sd)/Median |
Wilcox test on Age at recruitment (p-value) |
|
|---|---|---|---|---|
All female control samples |
340 |
52.57 ± 7.4/(53.30) |
- |
|
All male control samples |
84 |
55.89 ± 5.6/(56.72) |
- |
|
Selected breast female controls |
233 |
52.57 ± 7.4/(53.27) |
- |
0.8678 |
Breast female cases |
233 |
52.37 ± 7.4/(53.70) |
3.84 ± 2.87/(2.69) |
|
CRC male controls |
84 |
55.89 ± 5.6/(56.72) |
- |
0.8821 |
CRC male cases |
87 |
55.97 ± 5.7/(56.53) |
- |
|
Selected CRC female controls |
79 |
53.71 ± 6.9/(53.71) |
- |
0.7306 |
CRC female cases |
79 |
54.09 ± 7.6/(54.25) |
5.11 ± 2.59/(4.99) |
Descriptive characteristics of the study samples. Mann-Whitney-Wilcoxon test was performed between each pair of cse and control samples to show that there were not differences between their chronological ages.
As reported more in details at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE51032, genomic DNA was extracted and purified from peripheral blood leukocytes and bisulfite converted before being amplified, fragmented and hybridized to Illumina Infinium HumanMethylation450 BeadChips finally imaged using standard protocols and settings.
Estimation of DNAmAge
We used five methodologies to estimate the epigenetic age (DNA methylation age, DNAmAge) in blood samples.
Horvath and Hannum DNAmAge were calculated using the online tool available at https://dnamage.genetics.ucla.edu/ [16]. The tool also provides counts estimates of naive CD8 T cells, exhausted CD8 T cells, plasma B cells (effector B cells), CD4 T cells, natural killer cells, monocytes, and granulocytes [43]. These estimates can be used to correct the DNAmAge taking into account possible variations due to the heterogeneity in blood cell counts between individuals (i.e. estimated cells abundance acting as covariates [31]). As mentioned above, we denote the non-adjusted epigenetic age acceleration as Age Accel, and we use the term IEAA (Intrinsic Epigenetic Age Acceleration of blood) when referring to regression residuals corrected by blood cell counts, in accordance with Horvath’s nomenclature [20].
Weidner’s epigenetic clock: Weidner et al. [33] generated a multivariate model based on the methylation values at 3 HM450 probes (α: cg02228185; β: cg25809905; γ: cg17861230). Weidner’s DNAmAge was calculated using the equation DNAmAge = 38.0–26.4 * α 23.7 * β + 164.7 * γ [33].
ELOVL2 and FHL2 [35]: linear regressions between beta values of each of the two probes and chronological age were computed on the Hannum’s dataset, resulting in the following models: ELOVL2 DNAmAge = 158.81 * (cg16867657 beta value) – 42.35; and FHL2 DNAm Age = 198.6 * (cg06639320 beta value) -30.12.
For Weidner, ELOVL2 and FHL2 clocks, we considered blood cell count adjusted and non-adjusted age acceleration (IEAA and Age Accel respectively), using the same cell counts estimates returned by Horvath’s online tool.
Statistical analysis
For each of the above-mentioned age predictors, we used regression analysis to calculate the relation between chronological age and DNAmAge in the control group. We fitted the model without including the tumor samples to obtain positive age acceleration for subjects whose epigenetic age is higher than the control group, chronological age being equal.
For each sample, the regression residuals provide an estimate of the epigenetic age acceleration (Age Accel and IEAA) in relation to the control group [16, 31].
Differences in age acceleration between controls and subjects who developed tumors were tested using the Wilcoxon-Mann-Whitney method to comply with the imperfect adherence to normality of the data.
The association between age acceleration and cancer incidence was evaluated through survival analysis, and performed considering all five epigenetic clocks. Survival functions for the accelerated (1) and decelerated (–1) age groups were fitted with Kaplan-Meier method.
Since the dataset does not provide the exact enrollment time for the control subjects, but specifies that they were recruited between 1993 and 1998 and that they were all followed up until 2010, we considered for the controls (censored data) a survival time of 14.5 years, that corresponds to an average recruitment time.
All statistical analyses and graphics were produced using the computing environment R.
ACKNOWLEDGMENTS AND FUNDING
We are grateful to all unknown individuals who participated in each study used in this work. This work was supported by the European Union’s Seventh Framework Programme (grant agreement 602757 “HUMAN”; grant agreement 305522 “COBRA”), by the European Union’s H2020 Programme (grant agreement 634821 “PROPAG-AGEING”), by the National Counsel of Technological and Scientific Development (CNPq) Science Technology and Innovation (MCTI) (n. 200891/2014-6) and by Roberto and Cornelia Pallotti Legacy for cancer research.
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
1. Adams PD, Jasper H, Rudolph KL. Aging-Induced Stem Cell Mutations as Drivers for Disease and Cancer. Cell Stem Cell. 2015; 16:601–12.
2. Lasry A, Ben-Neriah Y. Senescence-associated inflammatory responses: aging and cancer perspectives. Trends Immunol. 2015; 36:217–28.
3. Zane L, Sharma V, Misteli T. Common features of chromatin in aging and cancer: cause or coincidence? Trends Cell Biol. 2014; 24:686–94.
4. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013; 75:685–705.
5. Fraga MF, Agrelo R, Esteller M. Cross-talk between aging and cancer: the epigenetic language. Ann N Y Acad Sci. 2007; 1100:60–74.
6. Pathai S, Gilbert CE, Lawn SD, Weiss HA, Peto T, Cook C, Wong TY, Shiels PG. Assessment of candidate ocular biomarkers of ageing in a South African adult population: relationship with chronological age and systemic biomarkers. Mech Ageing Dev. 2013; 134:338–45.
7. Noren Hooten N, Fitzpatrick M, Wood WH, De S, Ejiogu N, Zhang Y, Mattison JA, Becker KG, Zonderman AB, Evans MK. Age-related changes in microRNA levels in serum. Aging (Albany NY). 2013; 5:725–40. doi: 10.18632/aging.100603.
8. Glass D, Viñuela A, Davies MN, Ramasamy A, Parts L, Knowles D, Brown AA, Hedman AK, Small KS, Buil A, Grundberg E, Nica AC, Di Meglio P, et al. Gene expression changes with age in skin, adipose tissue, blood and brain. Genome Biol. 2013; 14:R75. doi: 10.1186/gb-2013-14-7-r75.
9. Gorisse L, Pietrement C, Vuiblet V, Schmelzer CEH, Köhler M, Duca L, Debelle L, Fornès P, Jaisson S, Gillery P. Protein carbamylation is a hallmark of aging. Proc Natl Acad Sci U S A. 2016; 113:1191–6.
10. Zhang J, Rane G, Dai X, Shanmugam MK, Arfuso F, Samy RP, Lai MKP, Kappei D, Kumar AP, Sethi G. Ageing and the telomere connection: An intimate relationship with inflammation. Ageing Res Rev. Elsevier B.V.; 2016; 25:55–69.
11. Benayoun BA, Pollina EA, Brunet A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol. 2015; 16:593–610.
12. Nan H, Giovannucci EL, Wu K, Selhub J, Paul L, Rosner B, Fuchs CS, Cho E. Pre-Diagnostic Leukocyte Genomic DNA Methylation and the Risk of Colorectal Cancer in Women. PLoS One. 2013; 8. doi: 10.1371/journal.pone.0059455.
13. Pufulete M, Al-Ghnaniem R, Leather AJM, Appleby P, Gout S, Terry C, Emery PW, Sanders TAB. Folate status, genomic DNA hypomethylation, and risk of colorectal adenoma and cancer: A case control study. Gastroenterology. 2003; 124:1240–8.
14. Lim U, Flood A, Choi SW, Albanes D, Cross AJ, Schatzkin A, Sinha R, Katki HA, Cash B, Schoenfeld P, Stolzenberg-Solomon R. Genomic methylation of leukocyte DNA in relation to colorectal adenoma among asymptomatic women. Gastroenterology. 2008; 134:47–55.
15. Walters RJ, Williamson EJ, English DR, Young JP, Rosty C, Clendenning M, Walsh MD, Parry S, Ahnen DJ, Baron JA, Win AK, Giles GG, Hopper JL, et al. Association between hypermethylation of DNA repetitive elements in white blood cell DNA and early-onset colorectal cancer. Epigenetics. 2013; 8:748–55.
16. Horvath S. DNA methylation age of human tissues and cell types DNA methylation age of human tissues and cell types. Genome Biol. 2013; 14:1–19.
17. Horvath S. Erratum to : DNA methylation age of human tissues and cell types. Genome Biol. 2015; 16:1–5.
18. Horvath S, Garagnani P, Bacalini MG, Pirazzini C, Salvioli S, Gentilini D, Di Blasio AM, Giuliani C, Tung S, Vinters H V., Franceschi C. Accelerated epigenetic aging in Down syndrome. Aging Cell. 2015; 14:491–5.
19. Levine ME, Lu AT, Bennett DA, Horvath S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging (Albany NY). 2015; 7:1198–211. doi: 10.18632/aging.100864.
20. Horvath S, Ritz BR. Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging (Albany NY). 2015; 7:1130–42. doi: 10.18632/aging.100859.
21. Breitling LP, Saum K-U, Perna L, Schöttker B, Holleczek B, Brenner H. Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clin Epigenetics. Clinical Epigenetics; 2016; 8:21. doi: 10.1186/s13148-016-0186-5.
22. Zannas AS, Arloth J, Carrillo-Roa T, Iurato S, Röh S, Ressler KJ, Nemeroff CB, Smith AK, Bradley B, Heim C, Menke A, Lange JF, Brückl T, et al. Lifetime stress accelerates epigenetic aging in an urban, African American cohort: relevance of glucocorticoid signaling. Genome Biol. 2015; 16:266. doi: 10.1186/s13059-015-0828-5.
23. Horvath S, Levine AJ. HIV-1 infection accelerates age according to the epigenetic clock. J Infect Dis. 2015; 212:1563–73.
24. Levine ME, Lu AT, Chen BH, Hernandez DG, Singleton AB, Ferrucci L, Bandinelli S, Salfati E, Manson JE, Quach A, Kusters CDJ, Kuh D, Wong A, et al. Menopause accelerates biological aging. Proc Natl Acad Sci. 2016; 113:9327–32.
25. Marioni RE, Shah S, McRae AF, Ritchie SJ, Muniz-Terrera G, Harris SE, Gibson J, Redmond P, Cox SR, Pattie A, Corley J, Taylor A, Murphy L, et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int J Epidemiol. 2015; 44:1388–96.
26. Marioni RE, Shah S, McRae AF, Chen BH, Colicino E, Harris SE, Gibson J, Henders AK, Redmond P, Cox SR, Pattie A, Corley J, Murphy L, et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015; 16:25.
27. Perna L, Zhang Y, Mons U, Holleczek B, Saum K-U, Brenner H. Epigenetic age acceleration predicts cancer, cardiovascular, and all-cause mortality in a German case cohort. Clin Epigenetics. 2016; 8:64. doi: 10.1186/s13148-016-0228-z.
28. Christiansen L, Lenart A, Tan Q, Vaupel JW, Aviv A, Mcgue M, Christensen K. DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell. 2016; 15:149–54.
29. Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, Deconde R, Chen M, Rajapakse I, et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol Cell. 2013; 49:359–67.
30. Chen BH, Marioni RE, Colicino E, Peters MJ, Ward-Caviness CK, Tsai PC, Roetker NS, Just AC, Demerath EW, Guan W, Bressler J, Fornage M, Studenski S, et al. DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging (Albany NY). 2016; 8:1844–65. doi: 10.18632/aging.101020.
31. Horvath S, Pirazzini C, Bacalini MG, Gentilini D, Di Blasio AM, Delledonne M, Mari D, Arosio B, Monti D, Passarino G, De Rango F, D’Aquila P, Giuliani C, et al. Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging (Albany NY). 2015; 7:1159–70. doi: 10.18632/aging.100861.
32. Wolf EJ, Logue MW, Hayes JP, Sadeh N, Schichman SA, Stone A, Salat DH, Milberg W, McGlinchey R, Miller MW. Accelerated DNA methylation age: Associations with PTSD and neural integrity. Psychoneuroendocrinology. 2016; 63:155–62.
33. Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, Bauerschlag DO, Jöckel K-H, Erbel R, Mühleisen TW, Zenke M, Brümmendorf TH, Wagner W. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014; 15:R24. doi: 10.1186/gb-2014-15-2-r24.
34. Lin Q, Weidner CI, Costa IG, Marioni RE, Ferreira MRP, Deary IJ, Wagner W. DNA methylation levels at individual age-associated CpG sites can be indicative for life expectancy. Aging (Albany NY). 2016; 8:394–401. doi: 10.18632/aging.100908.
35. Garagnani P, Bacalini MG, Pirazzini C, Gori D, Giuliani C, Mari D, Di Blasio AM, Gentilini D, Vitale G, Collino S, Rezzi S, Castellani G, Capri M, et al. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell. 2012; 11:1132–4.
36. Florath I, Butterbach K, Müller H, Bewerunge-Hudler M, Brenner H. Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum Mol Genet. 2014; 23:1186–201.
37. Kananen L, Marttila S, Nevalainen T, Jylhävä J, Mononen N, Kähönen M, Raitakari OT, Lehtimäki T, Hurme M. Aging-associated DNA methylation changes in middle-aged individuals: the Young Finns study. BMC Genomics. 2016; 17:103. doi: 10.1186/s12864-016-2421-z.
38. Rönn T, Volkov P, Gillberg L, Kokosar M, Perfilyev A, Jacobsen AL, Jørgensen SW, Brøns C, Jansson PA, Eriksson KF, Pedersen O, Hansen T, Groop L, et al. Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum Mol Genet. 2015; 24:3792–813.
39. Steegenga WT, Boekschoten M V., Lute C, Hooiveld GJ, de Groot PJ, Morris TJ, Teschendorff AE, Butcher LM, Beck S, Müller M. Genome-wide age-related changes in DNA methylation and gene expression in human PBMCs. Age (Dordr). 2014; 36:9648. doi: 10.1007/s11357-014-9648-x.
40. Bacalini MG, Deelen J, Pirazzini C, De Cecco M, Giuliani C, Lanzarini C, Ravaioli F, Marasco E, van Heemst D, Suchiman HED, Slieker R, Giampieri E, Recchioni R, et al. Systemic Age-Associated DNA Hypermethylation of ELOVL2 Gene: In Vivo and In Vitro Evidences of a Cell Replication Process. J Gerontol A Biol Sci Med Sci. 2016. doi: 10.1093/gerona/glw185.
41. Giuliani C, Cilli E, Bacalini MG, Pirazzini C, Sazzini M, Gruppioni G, Franceschi C, Garagnani P, Luiselli D. Inferring chronological age from DNA methylation patterns of human teeth. Am J Phys Anthropol. 2016; 159:585–95.
42. Zheng Y, Joyce BT, Colicino E, Liu L, Zhang W, Dai Q, Shrubsole MJ, Kibbe WA, Gao T, Zhang Z, Jafari N, Vokonas P, Schwartz J, et al. Blood Epigenetic Age may Predict Cancer Incidence and Mortality. EBioMedicine. 2015; 5:68–73.
43. Levine ME, Hosgood HD, Chen B, Absher D, Assimes T, Horvath S. DNA methylation age of blood predicts future onset of lung cancer in the women’s health initiative. Aging (Albany NY). 2015; 7:690–700. doi: 10.18632/aging.100809.
44. Palli D, Berrino F, Vineis P, Tumino R, Panico S, Masala G, Saieva C, Salvini S, Ceroti M, Pala V, Sieri S, Frasca G, Giurdanella M, et al. A molecular epidemiology project on diet and cancer: the EPIC-Italy Prospective Study. Design and baseline characteristics of participants. Tumori. 2003; 89:586–93.
45. Bakulsky KM, Fallin MD. Epigenetic Epidemiology: Promises for PublicHealth Research. Environ Mol Mutagen. 2010; 51:229–35.
46. Riboli E, Hunt K, Slimani N, Ferrari P, Norat T, Fahey M, Charrondière U, Hémon B, Casagrande C, Vignat J, Overvad K, Tjønneland A, Clavel-Chapelon F, et al. European Prospective Investigation into Cancer and Nutrition (EPIC): study populations and data collection. Public Health Nutr. 2003; 5:1113–24.
47. Palli D, Berrino F, Vineis P, Tumino R, Panico S, Masala G, Saieva C, Salvini S, Ceroti M, Pala V, Sieri S, Frasca G, Giurdanella MC, et al. A molecular epidemiology project on diet and cancer: the EPIC-Italy Prospective Study. Design and baseline characteristics of participants. Tumori. 2003; 89:586–93.
48. Horvath S, Gurven M, Levine ME, Trumble BC, Kaplan H, Allayee H, Ritz BR, Chen B, Lu AT, Rickabaugh TM, Jamieson BD, Sun D, Li S, et al. An epigenetic clock analysis of race/ethnicity , sex , and coronary heart disease An epigenetic clock analysis of race/ethnicity , sex , and coronary heart disease. Genome. 2016; 17:1–23.