
Introduction
Neurofibromatosis type 2 (NF2) is a tumor suppressor gene on chromosome 22q12 that encodes a protein product named “merlin” (or schwannomin) affecting multiple cell signaling pathways (Figure 1)[1, 2]. Constitutional mutations in the NF2 gene cause an autosomal-dominant disorder (neurofibromatosis type 2) affecting about 1 in 33,000 people, and characterized by the development of tumors primarily affecting the nervous system[3]. Approximately half of the cases are due to de novo mutations not inherited from a family member [4]. NF2 mutations in neurofibromatosis 2 can either be germline (70%) or somatic mosaic (30%), the latter referring to mutations present in only a subset of cells[5, 6].
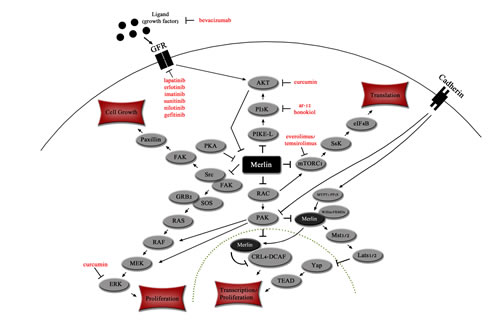
Figure 1: Merlin affects key signaling pathways in the cytoplasm and nucleus. Merlin inhibits PI3K signaling cascade at PIKE-L[19, 30] and mTORC1[32, 33]. The RAS and Src pathways are inhibited by merlin via the Src/FAK complex[34, 36]. Transcription is inhibited by merlin via Hippo kinase cascade[81, 82] and CRL4-DCAF[83]. Merlin can also down-regulates cell surface growth factor receptors. Dotted line indicates nuclear membrane. Targeted therapies are indicated in red.
Neurofibromatosis 2 follows the Knudson 2-hit hypothesis, with constitutional (germline) mutations occurring initially (first hit) and additional somatic mutations (second hit) being required for loss of heterozygosity and tumor suppressor inactivation followed by tumor formation[7-9]. The majority of tumors associated with neurofibromatosis type 2 are schwannomas, meningiomas, and ependymomas[7]. Other than these specific tumor types, there is no published evidence to support an increased incidence of cancer in individuals with neurofibromatosis 2[7]. In general, in neurofibromatosis 2, constitutional nonsense or frameshift NF2 mutations are associated with more severe disease, while missense mutations, large deletions, or somatic mosaicism results in milder disease (fewer tumors and older age of onset)
NF2 somatic mutations have also been found in multiple cancer types, including but not limited to mesothelioma, anaplastic thyroid cancer, breast cancers, endometrial and liver cancers, in patients not having constitutional NF2 mutations (Table 1)[10]. Missense mutations are more frequent in cancer than in neurofibromatosis 2; they occur in only a small subset of patients with latter condition. This may in part explain the lack of predisposition to developing cancer in patients with neurofibromatosis 2, though the explanation is not complete, since there remains considerable overlap between neurofibromatosis 2 and cancer-related NF2 aberrations (Figure 2)[11, 12].
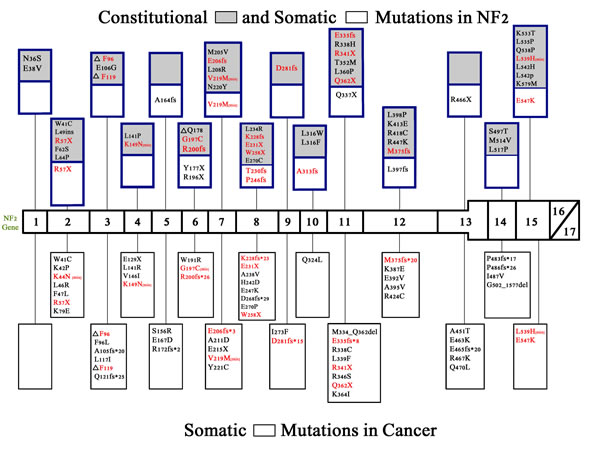
Figure 2: Constitutional and somatic NF2 mutations in neurofibromatosis 2 and somatic NF2 mutations in cancer. NF2 mutations have been found in neurofibromatosis 2 and also in numerous cancers[15]. Shown are the non-truncating constitutional and somatic mutations found in neurofibromatosis 2 [blue boxes] and non-truncating (missense) and truncating (nonsense or frameshift) mutations found in human cancer [black boxes][84]. Gray boxes indicate constitutional mutations while white boxes indicate somatic mutations. Because there are numerous truncating lesions found in neurofibromatosis 2[69] and in non-hereditary meningiomas, ependymomas, and schwannomas,[69, 85] these could not be depicted in figure 2. Truncating lesions that are found in both neurofibromatosis 2 and cancer are shown in red. Missense mutations that are found in both neurofibromatosis 2 and cancer are also shown in red followed by [mis]. Termination sites of translation for frameshift mutations is indicated with an (*) and for nonsense mutations with an (X).
Clinical Manifestations of Neurofibromatosis 2
The most common tumor associated with neurofibromatosis 2 is bilateral vestibular schwannoma (acoustic neuromas)[12, 13]. Initial symptoms of neurofibromatosis 2 include hearing loss, facial nerve impairment, visual disruptions, and skin tumors; with the majority of patients presenting before the age of 20[12]. Many patients with multiple tumor sites live only into their early thirties, and overall survival rate for patients 20 years after diagnosis is approximately 40%[12, 13]. Treatment plans for patients with NF2 revolve around monitoring, surgery, and radiation therapy[12]. Due to recent advancements in our understanding of the signaling pathways affected by merlin, several targeted therapies are now being tested[14]. However, there remains a paucity of clinical trials, and a pressing need to accelerate the pace of testing in the clinical setting.
Merlin
The NF2 gene contains 17 exons and its product—merlin—is a tumor suppressor that controls protean cell signaling pathways implicated in cell growth, proliferation, and morphology[15, 16]. Merlin acts as a membrane-cytoskeleton scaffolding (Ezrin–Radixin–Moesin (ERM)) protein that localizes underneath the plasma membrane at cell–cell junctions and other actin-rich sites[17, 18]. It links the cytoskeleton to the cell membrane. Two isoforms of merlin have been described that differ by the presence (type 2 merlin) or absence (type 1 merlin; includes exons 1-15 and 17) of exon 16 sequences inserted into the extreme carboxyl terminus of the protein.
Myosin phosphatase MYPT1–PP1δ directly activates merlin by dephosphorylating it. Inactivation (phosphorylation) of merlin contributes to malignant conversion in multiple cell types, as does loss of merlin expression because of mutation [19]. Importantly, phosphorylation not only causes the conformation of merlin to change to the open/inactive state, but also targets merlin for polyubiquitination and degradation by the proteasome [19-21]. The decreased ability of mutant merlin to effectively act as a tumor suppressor is at least in part a direct result of the decreased half-life of mutant compared to wild-type merlin[22, 23].
Functions of NF2/merlin
Several different signaling pathways crucial to cell proliferation are inhibited by merlin: PIKE-L/PI3K, mTORC1, Src/Fak, Mst1/2, Ras/Rac/PAK, ERK1/2, AKT and CRL4-DCAF (Figure 1)[1, 24, 25]. Merlin also acts as an unconventional cell cycle regulator, and it links receptors at the plasma membrane to their cytoplasmic kinases to facilitate contact inhibition [26, 27].
PI3K/PIKE-L:
The phosphatidylinositol 3-kinase (PI3K) pathway is activated in multiple human cancers, and contributes to increased cellular proliferation and metabolism, and decreased apoptosis when activated[28, 29]. Merlin suppresses the PI3K pathway by binding with the PI3K-enhancer-isoform-L (PIKE-L), hence preventing PIKE-L from binding PI3K[30, 31].
mTORC1:
Inhibition of mammalian-target-of-rapamycin-complex-1 (mTORC1) due to merlin activation leads to the inhibition of mRNA translation, which causes an increase in apoptosis limiting cell survival and blocking tumor initiation[32]. mTORC1 inhibition is accomplished through multiple pathways including AKT/ERK inhibition and integrin specific adhesion via p21-activated kinase (PAK)[32, 33]. mTORC1 has also been identified as an important pathway in meningioma and schwannoma cell growth[33].
ErbB2/Src/FAK/Paxillin:
Merlin binds the receptor ErbB2 and also to Src, inhibiting Src from binding to and being phosphorylated by ErbB2 in a competitive manner [34]. As a result, Src cannot activate focal adhesion kinase (FAK) and paxillin, which inhibits cellular proliferation and growth[34]. Interestingly, binding of merlin to ErbB2 is initiated by paxillin binding to merlin on exon 2 at residues 50-70, causing merlin to move to the plasma membrane where it binds ErbB2[35]. Merlin also inhibits FAK from binding to Src and PI3K [36].
ERK1/2, AKT, and PDGFR:
Schwannoma cell lines lacking the tumor suppressor activity of merlin have high basal levels of phosphorylated extracellular signal-regulated kinase (ERK1/2) and AKT, which are activated by the Src/FAK/Ras and PI3K/Raf signaling cascades and platelet-derived growth factor receptor beta (PDGFRβ) (Figure 1)[37]. Merlin inhibits the activity of ERK and MAPK via the upstream effector Raf-1[38, 39]. In normal cells, merlin promotes PDGFRβ degradation as well, thereby inhibiting cellular proliferation [37, 40]. In a positive feedback loop, activated AKT also binds and phosphorylates merlin on residues Thr230 and Ser315, which causes merlin to be ubiquitinated and targeted for degradation[19, 20, 41].
Rac/PAK:
Due to merlin’s sequence homology with ERM proteins, merlin binds the same proteins, but unlike ERM proteins merlin acts in an inhibitory manner by suppressing cell growth and proliferation [42, 43]. Merlin may suppress the oncogenic signaling of the small GTPases Ras and Rac by interfering with guanine nucleotide-exchange factor (GEF) activity, which is required for Ras and Rac activation[43, 44]. Merlin inhibits signaling of the Ras pathway downstream by inhibiting Rac activation, which results in the inhibition of phosphorylation/activation of the serine/threonine kinase PAK [43]. When PAK is not phosphorylated it is unable to phosphorylate RAF and MEK, which are both necessary to activate the Ras signaling pathway[43, 45].
NF2 mutations in cancer
Data collected from COSMIC and ICGC databases and published studies show that diverse cancers harbor somatic NF2 mutations (Table 1).
Table 1: Cancers associated with NF2 somatic mutations |
||
Cancer Type |
% Containing NF2 Somatic Mutations |
Source |
Acute myelogenous leukemia (AML) |
2% (1/45) |
Yoo et al, Pathology 2012 [9] COSMIC |
Aerodigestive tract (Squamous cell carcinoma) |
4% (1/23) |
COSMIC |
Bladder |
3% (1/14) |
Iyer G et al., Science 2012 [66] |
Bone (sarcoma) |
6% (2/34) |
COSMIC |
Breast Cancer |
1-2% (3/387) (1/42) |
COSMIC ICGC Data Portal (http://dcc.icgc.org)Bianchi et al, Nature Genetics 1994[13] Sjöblom T et al, Science 2006 [63] Ikediobi et al, Mol Cancer Ther 2006[64] |
Ependymoma* |
4% (18/433) |
COSMIC |
Colorectal Carcinoma |
5% (2/44) |
Arakawa H et al, HMG 1994 [87] |
Endometrium (mixed adenosquamous carcinoma) |
10% (1/10) |
COSMIC |
Glioma |
27% (37/135) |
Lau YK et al, Cancer Res 2008[25] |
Hepatocellular Carcinoma (HCC) |
2% (1/45) |
Yoo et al, Pathology 2012 [9] COSMIC |
Intestine (large) |
2% (8/335) |
COSMIC |
Liver Cancer |
23% (17/75) |
ICGC Data Portal (http://dcc.icgc.org) |
Lung (Adenocarcinoma) |
1% (1/163) |
ICGC Data Portal (http://dcc.icgc.org) |
Lung (Mixed) |
1% (3/586) |
COSMIC |
Lung (Squamous Cell Carcinoma) |
2% (1/45) |
Yoo et al, Pathology 2012 [9] COSMIC |
Melanoma |
5% (6/126) |
COSMIC Bianchi et al, Nature Genetics 1994 [13] |
Meningioma * |
31% (363/1164) |
COSMIC |
Mesothelioma |
30-50% (8/27) |
COSMIC Carbone and Yang, CCR 2012 [88] Sekido Y, et al, Cancer Res 1995 [89] |
Ovary (serous carcinoma) |
1% (2/149) |
COSMIC |
Pituitary |
100% (1/1) |
COSMIC Szijan et al, Neuromolecular Med 2003 [91] |
Renal cell carcinoma |
1-2% (5/412) (7/428) |
Staller P, Future Oncology 2010 [92] COSMIC |
Schwannoma * |
42% (271/647) |
COSMIC |
Stomach |
3% (2/66) |
COSMIC |
Thyroid (anaplastic carcinoma) |
18% (2/11) |
COSMIC |
Urinary tract |
11% (2/18) |
COSMIC |
*Sporacic tumors, patients do not have neurofibromatosis 2 COSMIC (http://www.sanger.ac.uk/cosmic) |
||
Thoracic tumors (mesothelioma and lung cancer):
The most common cancer linked to NF2 aberrations is mesothelioma, with approximately 30-50% of tumors having mutations in the NF2 coding regio[22, 46-48]. The NF2 mutations found in neurofibromatosis 2 differ from the mutations found in mesotheliomas in that mutations in hereditary NF2 are not usually missense mutations. Only ~5% of neurofibromatosis 2 patients have constitutional missense mutations and these types of mutations are typically associated with a milder version of NF2 [49, 50]. This dichotomy may partially explain why patients with neurofibromatosis 2 do not develop mesothelioma[51]. A variety of lung cancers have also been found to harbor NF2 mutations, but at a much lower rate (1-2%)[11].
Sporadic schwannomas, meningiomas, and ependymomas:
Sporadic schwannomas, meningiomas, and ependymomas fund in patients who do not suffer from constitutional neurofibromatosis 2 have somatic NF2 mutations at rates of 42%, 27%, and 4%, respectively[46]. These are also the most prevalent tumor types found in individuals with constitutional NF2 mutations[7]. Mutations found in these sporadic tumors tend to include more frameshift mutations, while those found in neurofibromatosis 2 are often nonsense mutations[52, 53].
Thyroid cancer:
NF2 mutations have also been discerned in 18% (two of 11) anaplastic thyroid cancers. These are uncommon (accounting for only 2% of all thyroid cancers) but very aggressive cancers, with a median survival of only a few months[54]. Of interest, other pathways (PIK3/AKT/MTOR, RAS/RAK and ERK) activated via mutation in this cancer type are negatively regulated by active merlin [54-59].
Breast cancer:
One to two percent of breast cancers have NF2 mutations [15, 60-62]. A decrease in merlin expression correlates with increase in tumor grade [47]. When merlin expression is reestablished in breast xenograft models, tumorigenesis is reduced [47]. Since NF2 mutations activate mTor, we treated a patient with NF2-mutant metaplastic breast cancer (a highly refractory form of breast cancer) with a temsirolimus-based regimen, which resulted in a complete remission [63].
Glioblastoma multiforme:
Decreased expression of merlin RNA and protein levels have been observed in several human glioblastoma multiforme tumors; 27% of grade 4 gliomas have loss of merlin expression [46]. Merlin decreases proliferation and invasiveness while increasing apoptosis-induced cell death when its tumor suppressor capabilities are reestablished in the corresponding glioma cell lines [46].
Endometrial cancer:
Ten percent of endometrial carcinomas harbor NF2 mutations. This observation is of interest since NF2 mutations can activate mTor, the downstream effector of PIK3CA, and a high percentage of this cancer type (>80%) have PI3K pathway aberrations [30, 64].
Hepatocellular tumors:
Twenty-three percent of liver cancers have mutations in NF2. Mice with heterozygous NF2 mutations develop hepatocellular carcinoma and cholangiocarcinoma [65, 66].
Comparing NF2 aberrations in hereditary neurofibromatosis and in cancer
Overlap in truncating and non-truncating mutations found in cancers and hereditary neurofibromatosis 2 are shown in Figure 2 (red lettering). There have not been any mutations found in exons 16 and 17, which are the two exons affected by alternative splicing, and no known proteins have been found to bind to exon 16[33, 67]. Tumor suppressor activity can be attenuated by truncating mutations in any of the other exons 1-15[68]. In neurofibromatosis 2, truncating mutations (frameshift and nonsense) are associated with an increase in disease severity when compared with missense mutations[7]. Nonsense mutations are more frequent in hereditary neurofibromatosis 2 than in sporadic schwannomas, ependymomas, and meningiomas where frameshift mutations are more common [53, 69]. Missense mutations are more frequent in cancer, but occur in only about 5% of hereditary neurofibromatosis 2.
There are several binding sites in the merlin protein that are crucial for merlin’s function, being critical to interactions with other proteins or for correct merlin folding and activation. Merlin must associate with itself in order to change conformation and become active. This is achieved first by the folding of the N-terminal domain, which involves the interaction of amino acids 8-121 with amino acids 200-320[67, 70]. After the conformational change of the N-terminus is complete, merlin can transition to the active, closed conformation by the association of the N-terminal amino acids 302-308 with the C-terminal amino acids 580-595[67, 70]. When examining Figure 2, it is remarkable that no mutations are found in the region where the C-terminus and N-terminus interact, but multiple mutations are found in the regions where the N-terminus interacts with itself.
The phosphatase (MYPT1-PP1δ) responsible for the dephosphorylation and activation of merlin (at residue S518) interacts with merlin via its MYPT1 subunit at residues 312-341 of merlin (exons 10 and 11)[67]. As shown in figure 2, this region has multiple missense mutations in both cancer and NF2.
P21-activating kinases (PAK) are regulated by merlin and also regulate the activity of merlin by phosphorylating S518 and causing merlin to change to the open, inactive state[67]. PAKs interact with merlin at the N-terminus between amino acids 1-313, and there are many missense mutations in both cancer and neurofibromatosis 2 found within this region (Figure 2)[67].
PIKE-L binds to merlin in the region containing amino acids 1-332, whereas mutant merlin harboring the L64P missense mutation does not bind PIKE-L[30, 67]. Importantly, merlin cannot suppress tumorigenesis, including cellular proliferation, via inhibition of the PI3K pathway without binding to PIKE-L[30, 67]. Merlin’s interaction with PIKE-L could also regulate downstream effectors of PI3K such as AKT and mTOR[67]. The L64P mutation has not been found in any cancers but is reported in neurofibromatosis 2.
Clinical Trials
Novel targeted therapies are now being used in a small number of cases to treat hereditary neurofibromatosis 2 and cancers harboring NF2 mutations (Table 2). The vast majority of these trials are ongoing and results are not yet published. A search of clinicaltrials.gov was undertaken in order to find clinical trials treating NF2 patients.
Table 2: Ongoing Clinical Trials for Neurofibromatosis 2 Patients |
|||
Drug Name |
Mechanism |
Phase of Study |
Trial Reference Number |
Lapatinib |
Dual tyrosine kinase inhibitor (Her2/EGFR) |
Phase 0 |
NCT00863122 |
PD-0332991 |
CDK inhibitor |
Phase 1 |
NCT01602887 |
Bevacizumab |
Anti-VEGF monoclonal antibody |
Phase 2 |
NCT01207687, NCT01125046 |
Everolimus (RAD-001) |
mTOR inhibitor |
Phase 2 |
NCT01490476, NCT01345136, NCT01419639 |
Lapatinib |
ErbB2/EGFR inhibitor |
Phase 2 |
NCT00973739 |
Nilotinib |
Receptor tyrosine kinase inhibitor (Bcr-Abl, PDGFR, c-KIT) |
Phase 2 |
NCT01201538 |
PTC299 |
Decrease production of VEGF |
Phase 2 |
NCT00911248 |
Sunitinib |
Receptor tyrosine kinase inhibitor (PDGFR, VEGFR, c-KIT) |
Phase 2 |
NCT00561665, NCT00589784 |
Receptor tyrosine kinase (RTK) Inhibitors:
Lapatinib, which targets EGFR and ErbB-2, is being examined in both a phase 0 study treating vestibular schwannomas (NCT00863122) and a phase II (NCT00973739) trial for its effectiveness in all NF2 related tumor types. Clinical analysis has shown that vestibular schwannomas overexpress ErbB2/3 and that EGFR and its ligand are up-regulated in the majority of NF2-related vestibular schwannomas[71, 72].
Sporadic and neurofibromatosis 2-related vestibular schwannomas overexpress c-kit and PDGFR, which are targets of the receptor tyrosine kinase (RTK) inhibitor imatinib. In vitro studies using the vestibular schwannoma cell line HEI-193 show a decrease in proliferation and an increase in apoptosis in response to treatment with imatinib. Currently sunitinib and nilotinib, are being tested in patients with neurofibromatosis 2[13]. Sunitinib is an RTK inhibitor that targets multiple receptors such as PDGFR, VEGFR, and c-KIT. Two phase II clinical trials are currently evaluating the effectiveness of sunitinib in unresectable meningiomas (NCT00561665) and (NCT00589784). Nilotinib is a RTK inhibitor that targets Bcr-Abl, PDGFR, and c-Kit, and is also being used in a phase II clinical trial (NCT01201538) focusing on progressing vestibular schwannomas.
Vascular Endothelial Growth Factor (VEGF) Inhibitors:
Bevacizumab is an anti-VEGF monoclonal antibody that inhibits angiogenesis, slowing tumor growth and formation. Schwannomas produce high levels of VEGF. In a study of ten NF2 patients treated with bevacizumab, nine had tumor shrinkage, and seven experienced hearing improvement. Two phase II clinical trials (NCT01207687, NCT01125046) are assessing the effectiveness of bevacizumab in treating neurofibromatosis 2 patients with symptomatic vestibular schwannomas and recurrent or progressive meningiomas. Another drug PTC299 targets VEGF by inhibiting its synthesis upstream and interfering with post-transcriptional processing [73]. PTC299 has been studied in neurofibromatosis 2 patients via a phase II clinical trial [73].
mTOR Inhibitors:
Everolimus (RAD-001) is an mTOR inhibitor. Merlin is a negative regulator of mTOR. Therefore, in patients with deactivating NF2 mutations, one could hypothesize that an mTOR inhibitor would restore merlin’s inhibition of mTOR and arrest tumor formation [32]. There are three phase II clinical trials using everolimus to treat NF2-related tumors: NCT01490476, NCT01345136 and NCT01419639. A fourth phase II clinical trial (NCT01024946) is available for treatment of malignant pleural mesotheliomas using NF2/merlin loss as a biomarker to predict everolimus sensitivity. The mTOR inhibitor temsirolimus was tried in combination with the anti-VEGF antibody bezacizumab in a clinical case series focusing on patients with neurofibromatosis 2; of the two patients on this regimen, one achieved a 33% reduction in tumor size[74]. Of interest, one patient with metaplastic breast cancer who harbored an NF2 mutation achieved a complete remission on a temsirolimus-containing regimen [63].
RAS and CDK inhibitors:
Merlin blocks RAS activation. When merlin activity is lost, RAS can then move into the active state and promote cell growth and proliferation [13]. S-trans, trans-farnesyl-thiosalicylic-acid (FTS) is a RAS inhibitor that was administered to two patients with hereditary neurofibromatosis 2, one of whom achieved stable disease for over 4.5 years[74, 75].
Preclinical Studies
Treatment of human schwannoma cells in vitro with curcumin (diferuloylmethane), caused dephosphorylation of AKT and ERK1/2 and activation of the merlin phosphatase MYPT1-pp1δ, which is responsible for the dephosphorylation of S518 [76]. Interestingly, hsp70 was up-regulated following curcumin treatment, which could serve as a resistance mechanism. Hence, a heat shock protein inhibitor (KNK437) was used in combination with curcumin to block this potential resistance mechanism [76]. Merlin loss also causes constitutive activation of receptor tyrosine kinases (EGFR, ErbB2, and ErbB3), which can be targeted by EGFR and ErbB2 inhibitors such as erlotinib or lapatinib[13]. Finally, sorafinib, a PDGFR and c-RAF inhibitor has been shown to decrease proliferation in human schwannoma cell line [37].
Conclusions
NF2 is a complex gene with somatic mutations being associated with various cancers (e.g. 30 to 50 percent of mesotheliomas harbor NF2 mutations (Table 1)). Germline mutations cause the autosomal dominant disorder neurofibromatosis 2. Although the somatic mutations sometimes overlap with those in hereditary NF2 (Figure 2), there are no published papers documenting an increased risk of cancer in neurofibromatosis patients; individuals with hereditary NF2 do develop schwannomas, ependymomas, and meningiomas. Though there is overlap in the types of NF2 mutations/aberrations between NF2-related conditions (Figure 2), nonsense mutations are more frequent in neurofibromatosis 2, frameshift mutations in sporadic schwannomas, meningiomas, and ependymomas, and missense mutations in cancer[69].
Merlin inhibits PIK3CA and mTOR function along with several other pathways including Src/Fak, Mst1/2 (hippo), Ras/Rac/PAK, ERK1/2, AKT and CRL4-DCAF (Figure 1). A small series of mostly anecdotal reports describe activity for anti-angiogenesis agents (bevacizumab), a RAS inhibitor, and the mTor inhibitor temsirolimus in patients with hereditary neurofibromatosis 2[74, 77]. In patients with malignancy, an increasing number of studies have established that matching targeted therapy to even a single aberration in patients whose tumors harbor multiple genomic abnormalities can at times result in remarkable responses[78-80]. Since neurofibromatosis 2, unlike cancer, is a single gene disorder, it seems conceivable that proper targeting would result in tumor regressions. In order to explore this possibility, a variety of relevant agents should be explored in the clinical setting. This is especially important because of the morbidity and mortality associated with neurofibromatosis 2, and because many agents that impact NF2-related pathways are already available. Rare conditions are an accrual challenge for larger trials. Therefore, pilot trials whose aim is tumor response may be a mechanism to initially establish activity of an agent, and more of these trials are urgently warranted.
References
1. Okada T, You L and Giancotti FG. Shedding light on Merlin’s wizardry. Trends in cell biology. 2007; 17(5):222-229.
2. Evans DG, Trueman L, Wallace A, Collins S and Strachan T. Genotype/phenotype correlations in type 2 neurofibromatosis (NF2): evidence for more severe disease associated with truncating mutations. Journal of medical genetics. 1998; 35(6):450-455.
3. Evans DG, Wallace AJ, Wu CL, Trueman L, Ramsden RT and Strachan T. Somatic mosaicism: a common cause of classic disease in tumor-prone syndromes? Lessons from type 2 neurofibromatosis. American journal of human genetics. 1998; 63(3):727-736.
4. Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V, Strachan T and Harris R. A genetic study of type 2 neurofibromatosis in the United Kingdom. II. Guidelines for genetic counselling. Journal of medical genetics. 1992; 29(12):847-852.
5. Baser ME, Kuramoto L, Woods R, Joe H, Friedman JM, Wallace AJ, Ramsden RT, Olschwang S, Bijlsma E, Kalamarides M, Papi L, Kato R, Carroll J, Lazaro C, Joncourt F, Parry DM, et al. The location of constitutional neurofibromatosis 2 (NF2) splice site mutations is associated with the severity of NF2. Journal of medical genetics. 2005; 42(7):540-546.
6. Moyhuddin A, Baser ME, Watson C, Purcell S, Ramsden RT, Heiberg A, Wallace AJ and Evans DG. Somatic mosaicism in neurofibromatosis 2: prevalence and risk of disease transmission to offspring. Journal of medical genetics. 2003; 40(6):459-463.
7. Ahronowitz I, Xin W, Kiely R, Sims K, MacCollin M and Nunes FP. Mutational spectrum of the NF2 gene: a meta-analysis of 12 years of research and diagnostic laboratory findings. Human mutation. 2007; 28(1):1-12.
8. Zirn B, Arning L, Bartels I, Shoukier M, Hoffjan S, Neubauer B and Hahn A. Ring chromosome 22 and neurofibromatosis type II: proof of two-hit model for the loss of the NF2 gene in the development of meningioma. Clinical genetics. 2012; 81(1):82-87.
9. Knudson AG, Jr. Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Sciences of the United States of America. 1971; 68(4):820-823.
10. Sekido Y, Pass HI, Bader S, Mew DJ, Christman MF, Gazdar AF and Minna JD. Neurofibromatosis type 2 (NF2) gene is somatically mutated in mesothelioma but not in lung cancer. Cancer research. 1995; 55(6):1227-1231.
11. Yoo NJ, Park SW and Lee SH. Mutational analysis of tumour suppressor gene NF2 in common solid cancers and acute leukaemias. Pathology. 2012; 44(1):29-32.
12. Evans DG. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet journal of rare diseases. 2009; 4:16.
13. Blakeley JO, Evans DG, Adler J, Brackmann D, Chen R, Ferner RE, Hanemann CO, Harris G, Huson SM, Jacob A, Kalamarides M, Karajannis MA, Korf BR, Mautner VF, McClatchey AI, Miao H, et al. Consensus recommendations for current treatments and accelerating clinical trials for patients with neurofibromatosis type 2. American journal of medical genetics Part A. 2012; 158A(1):24-41.
14. Evans DG, Kalamarides M, Hunter-Schaedle K, Blakeley J, Allen J, Babovic-Vuskanovic D, Belzberg A, Bollag G, Chen R, DiTomaso E, Golfinos J, Harris G, Jacob A, Kalpana G, Karajannis M, Korf B, et al. Consensus recommendations to accelerate clinical trials for neurofibromatosis type 2. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009; 15(16):5032-5039.
15. Bianchi AB, Hara T, Ramesh V, Gao J, Klein-Szanto AJ, Morin F, Menon AG, Trofatter JA, Gusella JF, Seizinger BR and et al. Mutations in transcript isoforms of the neurofibromatosis 2 gene in multiple human tumour types. Nature genetics. 1994; 6(2):185-192.
16. Laulajainen M, Melikova M, Muranen T, Carpen O and Gronholm M. Distinct overlapping sequences at the carboxy-terminus of merlin regulate its tumour suppressor and morphogenic activity. Journal of cellular and molecular medicine. 2012; 16(9):2161-2175.
17. Pearson MA, Reczek D, Bretscher A and Karplus PA. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell. 2000; 101(3):259-270.
18. Shimizu T, Seto A, Maita N, Hamada K, Tsukita S and Hakoshima T. Structural basis for neurofibromatosis type 2. Crystal structure of the merlin FERM domain. The Journal of biological chemistry. 2002; 277(12):10332-10336.
19. Ye K. Phosphorylation of merlin regulates its stability and tumor suppressive activity. Cell adhesion & migration. 2007; 1(4):196-198.
20. Laulajainen M, Muranen T, Nyman TA, Carpen O and Gronholm M. Multistep phosphorylation by oncogenic kinases enhances the degradation of the NF2 tumor suppressor merlin. Neoplasia. 2011; 13(7):643-652.
21. Ammoun S and Hanemann CO. Emerging therapeutic targets in schwannomas and other merlin-deficient tumors. Nature reviews Neurology. 2011; 7(7):392-399.
22. Yang C, Asthagiri AR, Iyer RR, Lu J, Xu DS, Ksendzovsky A, Brady RO, Zhuang Z and Lonser RR. Missense mutations in the NF2 gene result in the quantitative loss of merlin protein and minimally affect protein intrinsic function. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108(12):4980-4985.
23. Sherman L, Xu HM, Geist RT, Saporito-Irwin S, Howells N, Ponta H, Herrlich P and Gutmann DH. Interdomain binding mediates tumor growth suppression by the NF2 gene product. Oncogene. 1997; 15(20):2505-2509.
24. Li W, Cooper J, Karajannis MA and Giancotti FG. Merlin: a tumour suppressor with functions at the cell cortex and in the nucleus. EMBO reports. 2012; 13(3):204-215.
25. Zhou L and Hanemann CO. Merlin, a multi-suppressor from cell membrane to the nucleus. FEBS letters. 2012; 586(10):1403-1408.
26. Beltrami S, Kim R and Gordon J. Neurofibromatosis type 2 protein, NF2: an uncoventional cell cycle regulator. Anticancer research. 2013; 33(1):1-11.
27. Stamenkovic I and Yu Q. Merlin, a “magic” linker between extracellular cues and intracellular signaling pathways that regulate cell motility, proliferation, and survival. Current protein & peptide science. 2010; 11(6):471-484.
28. Wen PY, Lee EQ, Reardon DA, Ligon KL and Alfred Yung WK. Current clinical development of PI3K pathway inhibitors in glioblastoma. Neuro-oncology. 2012; 14(7):819-829.
29. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nature reviews Cancer. 2009; 9(8):550-562.
30. Rong R, Tang X, Gutmann DH and Ye K. Neurofibromatosis 2 (NF2) tumor suppressor merlin inhibits phosphatidylinositol 3-kinase through binding to PIKE-L. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101(52):18200-18205.
31. Schulze KM, Hanemann CO, Muller HW and Hanenberg H. Transduction of wild-type merlin into human schwannoma cells decreases schwannoma cell growth and induces apoptosis. Human molecular genetics. 2002; 11(1):69-76.
32. Lopez-Lago MA, Okada T, Murillo MM, Socci N and Giancotti FG. Loss of the tumor suppressor gene NF2, encoding merlin, constitutively activates integrin-dependent mTORC1 signaling. Molecular and cellular biology. 2009; 29(15):4235-4249.
33. James MF, Han S, Polizzano C, Plotkin SR, Manning BD, Stemmer-Rachamimov AO, Gusella JF and Ramesh V. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Molecular and cellular biology. 2009; 29(15):4250-4261.
34. Houshmandi SS, Emnett RJ, Giovannini M and Gutmann DH. The neurofibromatosis 2 protein, merlin, regulates glial cell growth in an ErbB2- and Src-dependent manner. Molecular and cellular biology. 2009; 29(6):1472-1486.
35. Fernandez-Valle C, Tang Y, Ricard J, Rodenas-Ruano A, Taylor A, Hackler E, Biggerstaff J and Iacovelli J. Paxillin binds schwannomin and regulates its density-dependent localization and effect on cell morphology. Nature genetics. 2002; 31(4):354-362.
36. Poulikakos PI, Xiao GH, Gallagher R, Jablonski S, Jhanwar SC and Testa JR. Re-expression of the tumor suppressor NF2/merlin inhibits invasiveness in mesothelioma cells and negatively regulates FAK. Oncogene. 2006; 25(44):5960-5968.
37. Ammoun S, Flaiz C, Ristic N, Schuldt J and Hanemann CO. Dissecting and targeting the growth factor-dependent and growth factor-independent extracellular signal-regulated kinase pathway in human schwannoma. Cancer research. 2008; 68(13):5236-5245.
38. Lim JY, Kim H, Jeun SS, Kang SG and Lee KJ. Merlin inhibits growth hormone-regulated Raf-ERKs pathways by binding to Grb2 protein. Biochemical and biophysical research communications. 2006; 340(4):1151-1157.
39. Chadee DN, Xu D, Hung G, Andalibi A, Lim DJ, Luo Z, Gutmann DH and Kyriakis JM. Mixed-lineage kinase 3 regulates B-Raf through maintenance of the B-Raf/Raf-1 complex and inhibition by the NF2 tumor suppressor protein. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103(12):4463-4468.
40. Fraenzer JT, Pan H, Minimo L, Jr., Smith GM, Knauer D and Hung G. Overexpression of the NF2 gene inhibits schwannoma cell proliferation through promoting PDGFR degradation. International journal of oncology. 2003; 23(6):1493-1500.
41. Sekido Y. Inactivation of Merlin in malignant mesothelioma cells and the Hippo signaling cascade dysregulation. Pathology international. 2011; 61(6):331-344.
42. Legg JW and Isacke CM. Identification and functional analysis of the ezrin-binding site in the hyaluronan receptor, CD44. Current biology : CB. 1998; 8(12):705-708.
43. Morrison H, Sperka T, Manent J, Giovannini M, Ponta H and Herrlich P. Merlin/neurofibromatosis type 2 suppresses growth by inhibiting the activation of Ras and Rac. Cancer research. 2007; 67(2):520-527.
44. Ryu CH, Kim SW, Lee KH, Lee JY, Kim H, Lee WK, Choi BH, Lim Y, Kim YH, Hwang TK, Jun TY and Rha HK. The merlin tumor suppressor interacts with Ral guanine nucleotide dissociation stimulator and inhibits its activity. Oncogene. 2005; 24(34):5355-5364.
45. Slack-Davis JK, Eblen ST, Zecevic M, Boerner SA, Tarcsafalvi A, Diaz HB, Marshall MS, Weber MJ, Parsons JT and Catling AD. PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. The Journal of cell biology. 2003; 162(2):281-291.
46. Lau YK, Murray LB, Houshmandi SS, Xu Y, Gutmann DH and Yu Q. Merlin is a potent inhibitor of glioma growth. Cancer research. 2008; 68(14):5733-5742.
47. Morrow KA, Das S, Metge BJ, Ye K, Mulekar MS, Tucker JA, Samant RS and Shevde LA. Loss of tumor suppressor Merlin in advanced breast cancer is due to post-translational regulation. The Journal of biological chemistry. 2011; 286(46):40376-40385.
48. Carbone M and Yang H. Molecular pathways: targeting mechanisms of asbestos and erionite carcinogenesis in mesothelioma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012; 18(3):598-604.
49. Baser ME, Rai H, Wallace AJ and Evans DG. Neurofibromatosis 2 (NF2) and malignant mesothelioma in a man with a constitutional NF2 missense mutation. Familial cancer. 2005; 4(4):321-322.
50. Bianchi AB, Mitsunaga SI, Cheng JQ, Klein WM, Jhanwar SC, Seizinger B, Kley N, Klein-Szanto AJ and Testa JR. High frequency of inactivating mutations in the neurofibromatosis type 2 gene (NF2) in primary malignant mesotheliomas. Proceedings of the National Academy of Sciences of the United States of America. 1995; 92(24):10854-10858.
51. Baser ME, De Rienzo A, Altomare D, Balsara BR, Hedrick NM, Gutmann DH, Pitts LH, Jackler RK and Testa JR. Neurofibromatosis 2 and malignant mesothelioma. Neurology. 2002; 59(2):290-291.
52. Evans DG, Maher ER and Baser ME. Age related shift in the mutation spectra of germline and somatic NF2 mutations: hypothetical role of DNA repair mechanisms. Journal of medical genetics. 2005; 42(8):630-632.
53. Lee JD, Kwon TJ, Kim UK and Lee WS. Genetic and epigenetic alterations of the NF2 gene in sporadic vestibular schwannomas. PLoS ONE. 2012; 7(1):e30418.
54. Smallridge RC, Marlow LA and Copland JA. Anaplastic thyroid cancer: molecular pathogenesis and emerging therapies. Endocrine-related cancer. 2009; 16(1):17-44.
55. Garcia-Rostan G, Costa AM, Pereira-Castro I, Salvatore G, Hernandez R, Hermsem MJ, Herrero A, Fusco A, Cameselle-Teijeiro J and Santoro M. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer research. 2005; 65(22):10199-10207.
56. Hou P, Liu D, Shan Y, Hu S, Studeman K, Condouris S, Wang Y, Trink A, El-Naggar AK, Tallini G, Vasko V and Xing M. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007; 13(4):1161-1170.
57. Liu Z, Hou P, Ji M, Guan H, Studeman K, Jensen K, Vasko V, El-Naggar AK and Xing M. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. The Journal of clinical endocrinology and metabolism. 2008; 93(8):3106-3116.
58. Santarpia L, El-Naggar AK, Cote GJ, Myers JN and Sherman SI. Phosphatidylinositol 3-kinase/akt and ras/raf-mitogen-activated protein kinase pathway mutations in anaplastic thyroid cancer. The Journal of clinical endocrinology and metabolism. 2008; 93(1):278-284.
59. Garcia-Rostan G, Zhao H, Camp RL, Pollan M, Herrero A, Pardo J, Wu R, Carcangiu ML, Costa J and Tallini G. ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2003; 21(17):3226-3235.
60. Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006; 314(5797):268-274.
61. Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O’Meara S, Santarius T, Avis T, Barthorpe S, Brackenbury L, Buck G, Butler A, Clements J, Cole J, Dicks E, Forbes S, et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Molecular cancer therapeutics. 2006; 5(11):2606-2612.
62. Arakawa H, Hayashi N, Nagase H, Ogawa M and Nakamura Y. Alternative splicing of the NF2 gene and its mutation analysis of breast and colorectal cancers. Human molecular genetics. 1994; 3(4):565-568.
63. Moulder S, Moroney J, Helgason T, Wheler J, Booser D, Albarracin C, Morrow PK, Koenig K and Kurzrock R. Responses to liposomal Doxorubicin, bevacizumab, and temsirolimus in metaplastic carcinoma of the breast: biologic rationale and implications for stem-cell research in breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011; 29(19):e572-575.
64. Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, Lu Y, Stemke-Hale K, Dyer MD, Zhang F, Ju Z, Cantley LC, Scherer SE, Liang H, Lu KH, Broaddus RR, et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer discovery. 2011; 1(2):170-185.
65. McClatchey AI, Saotome I, Mercer K, Crowley D, Gusella JF, Bronson RT and Jacks T. Mice heterozygous for a mutation at the Nf2 tumor suppressor locus develop a range of highly metastatic tumors. Genes & development. 1998; 12(8):1121-1133.
66. Kalamarides M, Niwa-Kawakita M, Leblois H, Abramowski V, Perricaudet M, Janin A, Thomas G, Gutmann DH and Giovannini M. Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes & development. 2002; 16(9):1060-1065.
67. Scoles DR. The merlin interacting proteins reveal multiple targets for NF2 therapy. Biochimica et biophysica acta. 2008; 1785(1):32-54.
68. Jacoby LB, MacCollin M, Barone R, Ramesh V and Gusella JF. Frequency and distribution of NF2 mutations in schwannomas. Genes, chromosomes & cancer. 1996; 17(1):45-55.
69. Baser ME. The distribution of constitutional and somatic mutations in the neurofibromatosis 2 gene. Human mutation. 2006; 27(4):297-306.
70. Gutmann DH, Sherman L, Seftor L, Haipek C, Hoang Lu K and Hendrix M. Increased expression of the NF2 tumor suppressor gene product, merlin, impairs cell motility, adhesionand spreading. Human molecular genetics. 1999; 8(2):267-275.
71. Doherty JK, Ongkeko W, Crawley B, Andalibi A and Ryan AF. ErbB and Nrg: potential molecular targets for vestibular schwannoma pharmacotherapy. Otology & neurotology : official publication of the American Otological Society, American Neurotology Society [and] European Academy of Otology and Neurotology. 2008; 29(1):50-57.
72. Karajannis MA, Legault G, Hagiwara M, Ballas MS, Brown K, Nusbaum AO, Hochman T, Goldberg JD, Koch KM, Golfinos JG, Roland JT and Allen JC. Phase II trial of lapatinib in adult and pediatric patients with neurofibromatosis type 2 and progressive vestibular schwannomas. Neuro-oncology. 2012; 14(9):1163-1170.
73. Kissil JL, Blakeley JO, Ferner RE, Huson SM, Kalamarides M, Mautner VF, McCormick F, Morrison H, Packer R, Ramesh V, Ratner N, Rauen KA, Stevenson DA, Hunter-Schaedle K and North K. What’s new in neurofibromatosis? Proceedings from the 2009 NF Conference: new frontiers. American journal of medical genetics Part A. 2010; 152A(2):269-283.
74. Subbiah V, Slopis J, Hong DS, Ketonen LM, Hamilton J, McCutcheon IE and Kurzrock R. Treatment of patients with advanced neurofibromatosis type 2 with novel molecularly targeted therapies: from bench to bedside. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012; 30(5):e64-68.
75. Halaschek-Wiener J, Wacheck V, Schlagbauer-Wadl H, Wolff K, Kloog Y and Jansen B. A novel Ras antagonist regulates both oncogenic Ras and the tumor suppressor p53 in colon cancer cells. Mol Med. 2000; 6(8):693-704.
76. Angelo LS, Wu JY, Meng F, Sun M, Kopetz S, McCutcheon IE, Slopis JM and Kurzrock R. Combining curcumin (diferuloylmethane) and heat shock protein inhibition for neurofibromatosis 2 treatment: analysis of response and resistance pathways. Molecular cancer therapeutics. 2011; 10(11):2094-2103.
77. Plotkin SR, Stemmer-Rachamimov AO, Barker FG, 2nd, Halpin C, Padera TP, Tyrrell A, Sorensen AG, Jain RK and di Tomaso E. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. The New England journal of medicine. 2009; 361(4):358-367.
78. Tsimberidou AM, Iskander NG, Hong DS, Wheler JJ, Falchook GS, Fu S, Piha-Paul S, Naing A, Janku F, Luthra R, Ye Y, Wen S, Berry D and Kurzrock R. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012; 18(22):6373-6383.
79. Janku F, Wheler JJ, Naing A, Stepanek VM, Falchook GS, Fu S, Garrido-Laguna I, Tsimberidou AM, Piha-Paul SA, Moulder SL, Lee JJ, Luthra R, Hong DS and Kurzrock R. PIK3CA mutations in advanced cancers: characteristics and outcomes. Oncotarget. 2012; 3(12):1566-1575.
80. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Janne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, Fidias P, Stubbs H, Engelman JA, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. The New England journal of medicine. 2010; 363(18):1693-1703.
81. Angus L, Moleirinho S, Herron L, Sinha A, Zhang X, Niestrata M, Dholakia K, Prystowsky MB, Harvey KF, Reynolds PA and Gunn-Moore FJ. Willin/FRMD6 expression activates the Hippo signaling pathway kinases in mammals and antagonizes oncogenic YAP. Oncogene. 2012; 31(2):238-250.
82. Yu J, Zheng Y, Dong J, Klusza S, Deng WM and Pan D. Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Developmental cell. 2010; 18(2):288-299.
83. Li W, You L, Cooper J, Schiavon G, Pepe-Caprio A, Zhou L, Ishii R, Giovannini M, Hanemann CO, Long SB, Erdjument-Bromage H, Zhou P, Tempst P and Giancotti FG. Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell. 2010; 140(4):477-490.
84. Bourn D, Carter SA, Mason S, Gareth D, Evans R and Strachan T. Germline mutations in the neurofibromatosis type 2 tumour suppressor gene. Human molecular genetics. 1994; 3(5):813-816.
85. Fong B, Barkhoudarian G, Pezeshkian P, Parsa AT, Gopen Q and Yang I. The molecular biology and novel treatments of vestibular schwannomas. Journal of neurosurgery. 2011; 115(5):906-914.