
INTRODUCTION
Chronic kidney disease (CKD), regardless of the initial cause of the renal disease, is characterized by the progressive deterioration of kidney function, the relentless accumulation and deposition of extracellular matrix, and progressive tissue fibrosis. The incidence and prevalence of CKD has been estimated to be 8–16% worldwide and is increasing [1]. Renal fibrosis is a common feature of CKD, contributes prominently to the progressive loss of organ structure and function and leads to end-stage renal failure [2]. To inhibit the CKD process, it is important to prevent renal fibrosis, though CKD remains incurable. Macrophages (MΦ), which have diverse functions and phenotypic plasticity, have been recognized as key factors in renal fibrosis. MΦ have been classified into classically activated M1 and alternatively activated M2 subgroups. M1 macrophages play a pro-inflammatory role, enhancing renal inflammation by secreting pathogenic mediators, resulting in renal fibrosis. M2 macrophages play an anti-inflammatory role, suppressing renal inflammation by releasing anti-inflammatory mediators, such as interleukin (IL)-10, resulting in reduced renal fibrosis [3, 4, 5].
Relaxin is an ovarian and cardiovascular hormone that has emerged as a rapid-acting but safe antifibrotic that inhibits renal fibrosis in several experimental models, including the unilateral ureteral obstruction (UUO) and renal ischemia/reperfusion injury models [6, 7, 8]. The clinical assessment of the vasodilatory benefits in acute heart failure has been well developed, but the mechanism of relaxin for the antifibrotic potential is not clear [9].
After UUO or renal ischemia/reperfusion injury, changing the macrophage polarization conditions plays an important role in renal fibrosis, but whether relaxin ameliorates renal fibrosis by changing the macrophage polarization is still unknown. However, relaxin has the potential to shift macrophage polarization toward the M2 phenotype. Figueiredo et al. found that in response to inflammatory stimuli, relaxin can suppress expression of the typical M1-cytokine IL-1β in macrophages, thus promoting the acquisition of an immunosuppressive M2 phenotype in macrophages [10].
Toll-like receptors (TLRs) are an innate family of receptors that can sense tissue damage and orchestrate a cascade of inflammation. Recent evidence indicates that TLR4-mediated inflammation is a critical pathogenic link between innate immunity and renal fibrosis. TLR4, which is upregulated in mouse kidneys after UUO, can also promote fibrosis after UUO [11]. TLR4 also plays an important role in the transduction of polarization phenotype response and the release of pro-inflammatory cytokines in macrophages. HMGB1, the ligand of TLR4, and TLR4 signaling pathway expression are essential for the M1 macrophage transition [12, 13]. Otherwise, global TLR4 deficiency shifts macrophage polarization toward the M2 phenotype and consequently reduces adipose tissue inflammation [14]. However, it is unclear whether relaxin ameliorates renal fibrosis by inducing the macrophage phenotype transition. We hypothesized that relaxin induces M2 macrophage polarization by inhibiting the TLR4-NF-κB signaling pathway and alleviates the inflammation and renal fibrosis at the early stages of UUO. Our results showed that relaxin can downregulate the TLR4-NF-κB signaling pathway, shift macrophage polarization toward the M2 phenotype and ameliorate renal fibrosis at the early stages of UUO. In vitro experiments confirmed that relaxin can downregulate the TLR4-NF-κB signaling pathway and induce the M2 macrophage transition. Furthermore, the macrophage phenotype transition actions of relaxin were significantly blocked by TAK-242, a TLR4 antagonist, in vitro.
RESULTS
Relaxin alleviates fibrosis following UUO
To determine the functional significance of relaxin in renal fibrosis, we analyzed fibrotic kidneys following hematoxylin and eosin (H&E), Sirius red and protein fibronectin. Mice were pretreated with relaxin (0.5 mg/kg per day) or deionized water as control. Their kidneys were assessed at day 5 (in the early stage of UUO and when renal fibrosis is well established). H&E and Sirius red staining showed that marked interstitial inflammation and fibrosis occurred 5 days after UUO and that relaxin can reduce cell infiltration and interstitial fibrosis (Figure 1A–1C). Compared with the kidneys of mice pretreated with relaxin, the kidneys of day 5 UUO-injured mice had higher fibronectin levels (Figure 1D, 1E), suggesting that relaxin can alleviate renal fibrosis after UUO.
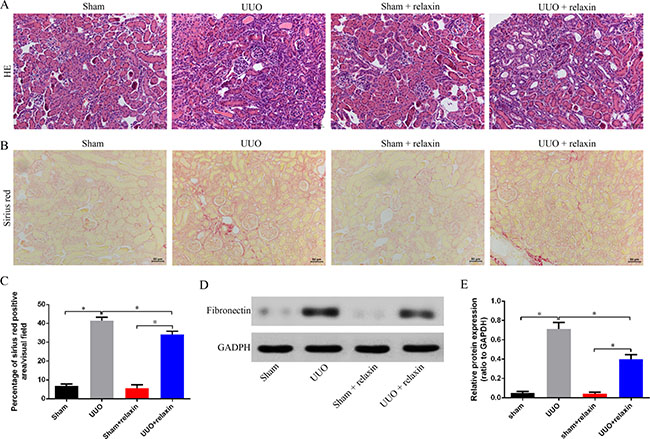
Figure 1: Relaxin alleviates fibrosis following UUO. (A–B) Representative images (eight visual fields for each tissue analyzed) of H&E (A), Sirus red (B) staining of kindrys frong the indicated experimental groups. Scale bars, 50 μm. (C) Interstitial fibrosis on basis of Sirius red staining. n = 8 per group;*p < 0.05. (D, E) Whole kidney lysates from kidneys of the mice pretreated with deionized water or relaxin following UUO (sham, UUO, sham + relaxin and UUO + relaxin) were analyzed for changes in fibrosis (fibronectin) by western blot analysis. Expression of the indicated proteins in the kidneys was analyzed by densitometry normalized to glyceraldehyde 3-phosphate dehydrogenase (GADPH) and expressed as mean ± sd. n = 8 per group; *p < 0.05.
Relaxin shifts macrophage polarization toward the M2 phenotype in vivo following UUO
To characterize the differential phenotypes of macrophages 5 days following UUO, macrophages were isolated from the obstructed and contralateral kidneys and analyzed for the expression of M1 and M2 markers by real-time PCR. Five days following UUO, gene expression analysis of macrophages from the kidneys revealed increased both M1 and M2 markers (Figure 2A–2J). Relaxin pretreatment caused significant increases in the gene expression levels of most renal macrophage M2 markers (MRC (mannose receptor), IL-4, arginase, IL-10, and Ym1); only the CX3CR1 (M2) gene was not differentially expressed in the macrophages from the two groups (Figure 2E–2J). However, the macrophage M1 markers (iNOS (inducible nitric oxide synthase), TNF-α (tumor necrosis factor-α), CCL-3 (chemokine (C-C motif) ligand 3), IL-23) were significantly decreased after relaxin pretreatment (Figure 2A–2D).
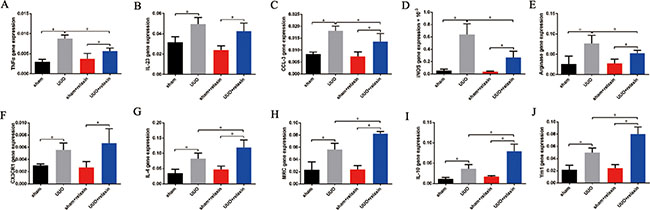
Figure 2: Relaxin shifts macrophage polarization toward the M2 phenotype in vivo following UUO. (A–J) Gene expression analysis of M1 markers, including tumor necrosis factor-α (TNF-α), interleukin (IL)-23, chemokine (C-C motif) ligand 3 (CCL3), inducible nitric oxide synthase (iNOS), and M2 markers, including arginase, CX3CR1, IL-4, mannose receptor (MRC), IL-10, Ym1 on macrophages isolated from kidneys of the mice pretreated with deionized water or relaxin following UUO. Experiments were performed at least three times and gene expression data were normalized to GADPH, analyzed, and represented as mean ± sd; n = 8 per group; *p < 0.05.
Relaxin shifts macrophage polarization toward the M2 phenotype in vitro
To further investigate the correlation between relaxin treatment and macrophage polarization, we polarized Raw264.7 cells to M1 or M2 macrophages in vitro using interferon-γ (IFNγ) or IL-4 and treated them with relaxin or deionized water as vehicle control, respectively. After treatment with relaxin, iNOS protein expression was decreased and Arg expression was increased in M0, M1 and M2 macrophages (Figure 3A–3C). The expression levels of M1 marker genes (iNOS, TNF-α, CCL-3, IL-23) were upregulated and M2 marker genes (arginase, CX3CR1, IL-4, Arg-1, IL-10, Ym1) were downregulated after treatment with relaxin in Raw264.7 cells, corroborating our in vivo data showing that relaxin treatment can increase the propensity to polarize M0 macrophages to the M2 phenotype (Figure 3D). Similarly, when M1 macrophages or M2 macrophages were treated with relaxin, the expression levels of most of the M1 marker genes decreased while M2 markers increased (Figure 3E, 3F), indicating that relaxin can shift M1 macrophage polarization toward the M2 phenotype.
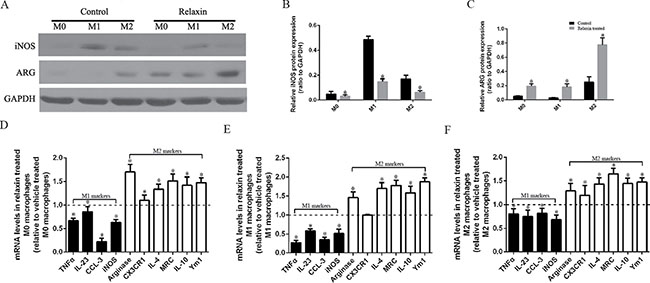
Figure 3: Relaxin shifts macrophage polarization toward the M2 phenotype in vitro. Raw 264.7 cells were treated with deionized water (M0), interferonγ(IFNγ) (M1) or IL-4(M2) and (A–C) analyzed for the expression of M1 marker iNOS and M2 marker arginase (Arg) after treated with vehicle (deionized water) or relaxin by western blot analysis. Protein expression data were normalized to GADPH, analyzed, and represented as mean ± sd; *p < 0.05 vs. vehicle treated groups(control groups). (D–F) M0, M1 and M2 macrophages were analyzed for the expression of M1 (TNF-α, IL-23, CCL3, iNOS) and M2 (arginase, CX3CR1, IL-4, MRC, IL-10, Ym-1) genes after treated with vehicle (deionized water) or relaxin by real-time PCR. Experiments were performed at least three independent times and gene expression data were normalized to GAPDH, analyzed, and represented as mean ± sd; *p < 0.05 vs. vehicle treated groups (control groups).
Relaxin downregulates the TLR4-NF-κB signaling pathway in vivo following UUO
Given the significance of the TLR4 signaling pathway for M1 macrophage transition, we hypothesized that relaxin induces M2 macrophage polarization by inhibiting the TLR4-NF-κB signaling pathway. The mice were pretreated with relaxin or deionized water as control, and the kidneys were collected at day 5 following UUO. TLR4, Myd88, NF-κB (p65) and phospho-NF-κB (p-p65) were measured by western blotting and real-time PCR. TLR4, Myd88, p65 and pp65 expression levels were upregulated at day 5 following UUO, while relaxin significantly decreased their mRNA and protein expression levels (Figure 4A–4E).
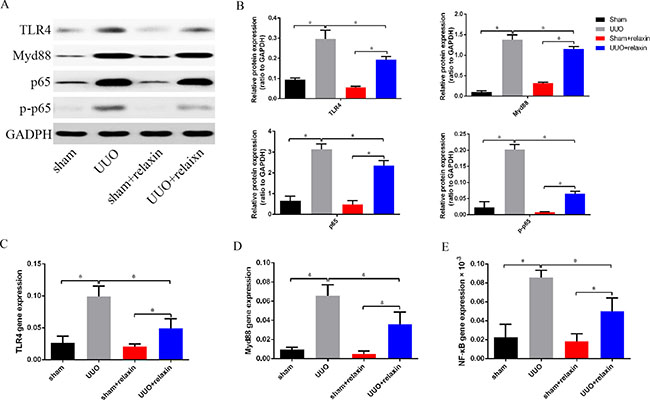
Figure 4: Relaxin downregulates the TLR4-NF-κB signaling pathway in vivo following UUO. (A, B) Whole kidney lysates from kidneys of the mice pretreated with deionized water or relaxin following UUO (sham, UUO, sham + relaxin and UUO + relaxin) were analyzed for changes in TLR4, Myd88, p65, p-p65 by western blot analysis. Expression of the indicated proteins in the kidneys was analyzed by densitometry normalized to GADPH and expressed as mean ± sd; n = 8 per group; *p < 0.05. (C–E) Macrophages isolated from kidneys of the mice pretreated with deionized water or relaxin following UUO were analyzed for the expression of TLR4, Myd88 and p65 genes. Experiments were performed at least three independent times and gene expression data were normalized to GAPDH, analyzed, and represented as mean ± sd; n = 8 per group;*p < 0.05.
Relaxin downregulates the TLR4-NF-κB signaling pathway in vitro
To elucidate the significance of relaxin in the regulation of the TLR4 signaling pathway of the different macrophage phenotypes in vitro, and as described earlier in this study, we polarized Raw264.7 cells to M1 or M2 macrophages and analyzed the mRNA and protein expression profiles of the TLR4 signaling pathway of the different macrophage phenotypes after treatment with relaxin or vehicle. Compared with those treated with vehicle, the mice treated with relaxin downregulated TLR4, Myd88, p65 and pp65 protein expression in both M0, M1 and M2 macrophages (Figure 5A–5L). Consistent with the protein expression data, real-time PCR results also indicated that relaxin could inhibit the TLR4 signaling pathway. TLR4, Myd88, p65 and pp65 genes were expressed at low levels after relaxin treatment in the different macrophage phenotypes compared with those treated with vehicle (Figure 5M–5O).
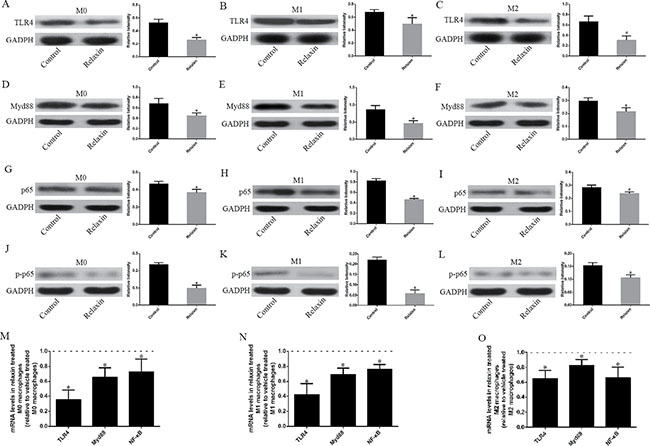
Figure 5: Relaxin downregulates the TLR4-NF-κB signaling pathway in vitro. Raw 264.7 cells were treated with deionized water (M0), interferonγ (IFNγ) (M1) or IL-4(M2) and (A–L) analyzed for the expression of TLR4, Myd88, p65 after treated with vehicle (deionized water) or relaxin by western blot analysis. Protein expression data were normalized to GADPH, analyzed, and represented as mean ± sd; *p < 0.05 vs. vehicle treated groups (control groups). (M, N, O) M0, M1 and M2 macrophages were analyzed for the expression of TLR4, Myd88, p65, p-p65 genes after treated with vehicle (deionized water) or relaxin by real-time PCR. Experiments were performed at least three independent times and gene expression data were normalized to GAPDH, analyzed, and represented as mean ± sd; *p < 0.05 vs. vehicle treated groups (control groups).
The effects of relaxin on macrophage polarization regulation are blocked by the TLR4 antagonist TAK-242 in vitro
After inducing Raw264.7 cells into M1 or M2 macrophages, the cells were pre-treated with TAK-242 (1 μM) or deionized water as vehicle control for 1 h and were continuously treated with relaxin (100 ng/ml; 16.8 nmol/l) for 72 h. Untreated cells and cells treated with TAK-242 alone (1 μM) for 72 h were used as appropriate controls. Treatment of different phenotypes of macrophages with relaxin can shift macrophage polarization toward the M2 phenotype in vitro and in vivo (Figures 2A–2J, 3A–3F). However, compared with their expression in the cells treated with TAK-242 only, the M1 markers (TNF-α, IL-23, CCL-3, iNOS,) and most of the M2 markers (arginase, CX3CR1, IL-4,MRC, Ym1) were not differentially expressed in the M0, M1 and M2 macrophages treated with TAK-242 and relaxin; only the IL-10 (M2) genes were increased in the M2 macrophages after treated with both TAK-242 and relaxin (Figure 6C–6E). Consistent with the gene expression data, the protein expression profiles of iNOS (M1) in M1 macrophage and Arg (M2) in M2 macrophage were not different between those treated with TAK-242 and those treated with TAK-242 and relaxin (Figure 6A, 6B). The results indicated that the effects of relaxin on regulating macrophage polarization are blocked by the TLR4 antagonist TAK-242 in vitro.
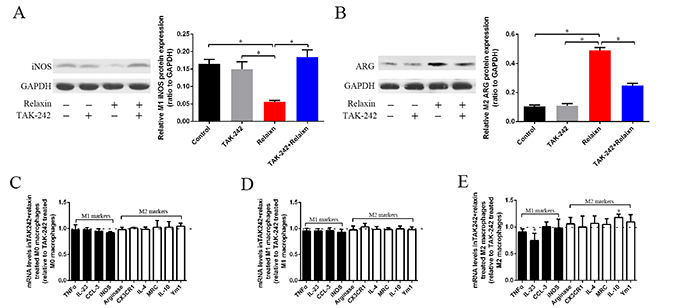
Figure 6: The effects of relaxin on macrophages polarization regulation are blocked by the TLR4 antagonist TAK-242 in vitro. (A, B) Raw 264.7 cells were treated with IFNγ (M1) to induce the macrophage to M1 macrophages, the cells were pre-treated with TAK-242 or vehicle (deionized water) for 1 h and were continuously treated with vehicle (deionized water) or relaxin for over 72 h and analyzed for the expression of M1 marker iNOS (A) and M2 marker arginase (Arg) (B) by western blot analysis. Protein expression data were normalized to GADPH, analyzed, and represented as mean ± sd; *p < 0.05. (C–E) M0, M1 and M2 macrophages were analyzed for the expression of M1 (TNF-α, IL-23, CCL3, iNOS) and M2 (arginase, CX3CR1, IL-4, MRC, IL-10, Ym-1) genes after treated with relaxin and TAK-242 or TAK-242 only by real-time PCR. Experiments were performed at least three independent times and gene expression data were normalized to GAPDH, analyzed, and represented as mean ± sd; *p < 0.05 vs. treated with TAK-242 only.
DISCUSSION
This study explored whether the H2 relaxin ameliorated renal fibrosis at the early stage of UUO by shifting macrophage polarization toward the M2 phenotype. Consistent with previous studies, relaxin can promote the acquisition of an immunosuppressive M2 phenotype in macrophages; M2 macrophages suppress kidney inflammation by releasing anti-inflammatory mediators, such as IL-10, thus inhibiting renal fibrosis [3, 4, 5, 10]. We also found that relaxin can downregulate the TLR4-NF-ΚB signaling pathway at all levels. Surprisingly, however, the macrophage phenotype transition function of relaxin was significantly blocked by the TLR4 antagonist TAK-242 in vitro. Therefore, we demonstrated for the first time that H2 relaxin can shift macrophage polarization toward the M2 phenotype by downregulating the TLR4-NF-ΚB signaling pathway, thus alleviating renal fibrosis.
Relaxin is a fast-acting but safe antifibrotic that inhibits renal fibrosis in UUO [6, 7, 8]. Recent studies have demonstrated that relaxin abrogates the TGF-β1/pSmad2 axis through an RXFP1-pERK1/2-nNOSNO-cGMP-dependent pathway and regulates the collagen-degrading matrix-degrading matrix metalloproteinases (MMPs) (MMP-1/-13, MMP-2 and MMP-9) [15, 16, 17]. However, our results show that relaxin can shift macrophage polarization toward the anti-inflammatory macrophage phenotype (M2), thus ameliorating renal fibrosis at the early stage of UUO. Our results indicate that relaxin can suppress fibrosis indirectly through anti-inflammatory actions. Consistent with our result, recent studies have shown that relaxin has the potential to shift macrophage polarization toward the M2 phenotype. Figueiredo et al. found that in response to inflammatory stimuli, relaxin can suppress expression of the typical M1-cytokine IL-1β in macrophages, thus promoting acquisition of an immunosuppressive M2 phenotype in macrophages [10]. Binder et al. investigated the mechanism of relaxin-enhanced breast tumor growth and found that tumor growth was enhanced because relaxin can switch macrophages toward the M2 phenotype [18].
MΦ show a phase-dependent balance of pro-inflammatory and anti-inflammatory effects in UUO. In the early stages of UUO, the pro-inflammatory macrophage (M1) is the predominant macrophage phenotype, directly inducing renal fibrosis by releasing pathogenic mediators such as TNF-α, IL-1β, CCL2 and reactive oxygen species (ROS). In the late stage of disease, MΦ undergo a switch to an anti-inflammatory macrophage (M2), which can suppress renal inflammation by secreting IL-10, leading to reduced renal fibrosis [4]. Consistent with previous studies, our findings indicate that relaxin could switch M1 macrophages to an M2 phenotype at the early stage of obstructive injury, resulting in reduced renal fibrosis.
TLRs, activated by pathogen-associated motifs and the release of endogenous stress ligands, can orchestrate an inflammatory response during tissue injury. TLR4, expressed in a wide range of cell types, including macrophage and renal epithelial cells, provides an important response after UUO [19]. TLR4 expression is significantly increased after UUO and induces renal fibrosis [20]. One day after unilateral ureteral obstruction, TLR4-deficient mice had more tubular damage and fewer proliferating tubular epithelial cells than WT mice but developed considerably less renal fibrosis [11]. Their studies suggest that UUO can induce a dramatic increase in TLR4 expression, therefore promoting renal fibrosis. In our study, we found that relaxin can downregulate the TLR4-NF-ΚB signaling pathway, indicating that relaxin can indirectly suppress fibrosis through anti-inflammatory activity.
Considering our finding that relaxin could switch M1 macrophages to an M2 phenotype and that the mechanism by which relaxin modulates macrophage polarization and function is unknown, we investigated whether relaxin shift macrophage polarization toward the M2 phenotype via inhibition of TLR4 signaling. We found that the macrophage phenotype transition action of relaxin was significantly blocked by the TLR4 antagonist TAK-242 in vitro. This finding indicates that relaxin shift macrophage polarization toward the M2 phenotype via inhibition of TLR4 signaling. Consistent with our results, previous studies have suggested that TLR4 also plays an important role in the transduction of polarization phenotype signals and the release of pro-inflammatory cytokines in macrophages. Delta-like 4 (DLL4) can increase M1 macrophage markers, such as IL-12 and iNOS expression, and can reprogram macrophages to the M1 phenotype via the TLR4/NF-κB-dependent pathway [21]. HMGB1, the ligand of TLR4, can promote renal fibrosis by facilitating the M1 macrophage phenotype [13]. Zhou et al. also found that curcumin can inhibit M1 macrophage polarization via the inhibition of expression along the TLR4 signaling pathway [12]. The mechanisms of macrophage heterogeneity remain unknown; however, it is reasonable to confirm that the TLR4 signaling pathway is a candidate switch between M1 and M2 phenotypes.
This study has several inherent limitations. First, we used Raw264.7 cells rather than other macrophages to investigate the effect of relaxin on macrophage polarization via TLR4 signaling pathways in vitro. Further studies should investigate relaxin-mediated macrophage polarization in macrophages isolated from mice following UUO and other macrophages. Additionally, it is necessary to use in vivo transgenic mouse experiments to further validate the effect of relaxin-mediated macrophage polarization and its mechanism.
Taken together, these results have demonstrated a novel mechanism by which relaxin ameliorates renal fibrosis by regulating macrophage polarization. The TLR4 signaling pathway is critically required for relaxin to shift macrophage polarization toward the M2 phenotype in vitro. This essential requirement is achieved through downregulation of the TLR4-NF-ΚB signaling pathway. However, the observation that TLR4 expression is low under normal physiological conditions and that it is upregulated in models of UUO may explain why the antifibrotic effects of relaxin are only observed under pathological conditions.
MATERIALS AND METHODS
Animal model
C57BL/6 male mice were supplied by the laboratory animal center of our institute. Ethical approval for all animal experiments was obtained from the Institutional Animal Care and Use Committee at Shanghai First People’s Hospital of Shanghai Jiao Tong University.
Mice were preventatively treated with recombinant H2 relaxin (PeproTech, Rocky Hill, NJ, USA) (0.5 mg/kg per day) or deionized water (PeproTech, Rocky Hill, NJ, USA) as control via subcutaneously implanted osmotic minipumps (model 1007D; Alzet, Cupertino, CA, USA) from 2 days before UUO until 5 days after injury. In various models of renal disease, regardless of etiology, this dose of relaxin had previously been used to successfully prevent or reverse fibrosis progression [7, 8].
As described in a previous study, a mouse model of UUO was used as an experimental model of primary tubulointerstitial fibrosis [22]. After general anesthesia, mice underwent ligation of the right ureter, with the left kidney remaining intact. The right ureter was exposed after a midline abdominal incision. The ligated animals were completely obstructed 1 cm below the renal pelvis with 5.0 silk ligature; sham operated animals were manipulated similarly but not ligated. Five days after surgery, the kidneys were collected, rinsed with isotonic saline, dissected and stored in liquid nitrogen or fixed in 4% formaldehyde for further analysis.
Cell culture and differentiation
Raw264.7 macrophages were obtained from the Shanghai cell bank of the Chinese Academy of Sciences. Raw264.7 macrophages were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Life Technologies, Waltham, MA, USA). The cells, induced to differentiate as previously described, were stimulated with 100 U/ml murine recombinant IFNγ (PeproTech, Rocky Hill, NJ, USA) or 20 ng/ml murine recombinant IL-4 (PeproTech, Rocky Hill, NJ, USA) for 24 h to induce M1 or M2 polarization, respectively [23].
After Raw264.7 cells were induced into M1 or M2 macrophages, the cells were pre-treated with TAK-242 (1 μM; MedChemExpress, Monmouth Junction, NJ, USA) or deionized water (PeproTech, Rocky Hill, NJ, USA) as vehicle control for 1 h and were continuously treated with relaxin (100 ng/ml; 16.8 nmol/l) for 72 h. Cells treated with deionized water or TAK-242 alone (1 μM) for 72 h were used as appropriate controls.
Histological analysis
Kidney tissues were embedded in paraffin after fixation in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 24 h. A paraffin microtome with stainless steel knives was used to cut the paraffin blocks into 8-μm sections. Before immunostaining, the renal tissue sections were mounted on glass slides, dewaxed in xylene, rehydrated with decreasing concentrations of ethanol, washed in PBS, and then stained with H&E. After staining, the sections were dehydrated with increasing concentrations of ethanol and xylene.
To evaluate collagen deposition, the renal tissue sections were stained with Sirius red as previously described [23]. The renal tissue sections were deparaffinized with xylene and then rehydrated in water with graded ethanol washes. After two washes in acidified water, the sections were incubated in Picrosirius red for 1 h. The sections were then dehydrated, cleared, and mounted in a resinous medium. The area of collagen deposition was measured using color image analysis software (Image-Pro Plus, Media Cybernetics, Houston, TX, USA).
Isolation and enrichment of renal macrophages
CD11b-expressing cells in the kidney single-cell suspensions were enriched using mouse CD11b Microbeads and MACS columns (Milteni Biotec Auburn, CA, USA) following the manufacturer’s protocol.
Quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from kidney tissue or cells using Trizol (Invitrogen, Carlsbad, CA, USA). RNA reverse transcription was performed using a PrimeScript RT reagent kit 127 (Takara, Otsu, Japan) according to the manufacturer’s instructions. Gene transcripts were quantified using a SYBR Premix Ex Taq TM II kit (Takara, Otsu, Japan). GAPDH was used as an internal control. The expression levels relative to β-actin were calculated using the 2−ΔΔCT method. Real-time primers used in this study are described in Table 1.
Table 1: Primers for real-time PCR analysis
Gene |
Primer sequence (5′-3′) |
|---|---|
GAPDH Fwd |
ATCATCCCTGCATCCACT |
GAPDH Rew |
ATCCACGACGGACACATT |
TNFα Fwd |
ACGGCATGGATCTCAAAGAC |
TNFα Rew |
AGATAGCAAATCGGCTGACG |
IL-23 Fwd |
CAGCAGCTCTCTCGGAATCT |
IL-23 Rew |
TGGATACGGGGCACATTATT |
CCL-3 Fwd |
GCCCTTGCTGTTCTTCTCTG |
CCL-3 Rew |
GATGAATTGGCGTGGAATCT |
iNOS Fwd |
GGAATCTTGGAGCGAGTTGT |
iNOS Rew |
GCAGCCTCTTGTCTTTGACC |
Ariginase Fwd |
GCAGAGGTCCAGAAGAATGG |
Ariginase Rew |
AGCATCCACCCAAATGACAC |
CX3CR1 Fwd |
TCCCTTCCCATCTGCTCAG |
CX3CR1 Rew |
ACAATGTCGCCCAAATAACAGG |
IL-4 Fwd |
TCTGTAGGGCTTCCAAGGTG |
IL-4 Rew |
ATCGAAAAGCCCGAAAGAGT |
MRC Fwd |
GGAGGCTGATTACGAGCAGT |
MRC Rew |
CATAGGAAACGGGAGAACCA |
IL-10 Fwd |
CCAAGCCTTATCGGAAATGA |
IL-10 Rew |
TCCTGAGGGTCTTCAGCTTC |
Ym1 Fwd |
TTCTTGTCACAGGTCTGG |
Ym1 Rew |
TCCTTAGCCCAACTGGTATAG |
TLR4 Fwd |
CCTGATGACATTCCTTCT |
TLR4 Rew |
AGCCACCAGATTCTCTAA |
Myd88 Fwd |
GCCAGAGTGGAAAGCAGTGT |
Myd88 Rew |
TATCGTTGGGGCAGTAGCAG |
P65 Fwd |
TAACAGCAGGACCCAAGGAC |
P65 Rew |
AGCCCCTAATACACGCCTCT |
Western blotting analysis
Harvested cells or collected tissues were lysed in radioimmunoprecipitation assay (RIPA) buffer (Beyotime Biotechnology, Shanghai, China) with protease inhibitor (Sigma-Aldrich, Darmstadt, Germany). The protein was quantified using the bicinchoninic acid (BCA) protein assay (Thermo Scientific, Waltham, MA, USA). Total proteins were resolved by 12% Tris-glycine sodium dodecyl sulfate polyacrylamide gel electrophoresis and were transferred to a polyvinylidene fluoride membrane (Millipore, Darmstadt, Germany). Membranes were blocked with 5% nonfat dry milk in PBST for 1 h and then incubated with primary antibodies against mouse Fibronectin (ab45688, Abcam), TLR4 (ab13867, Abcam), NF-κB (#8242, CST), phospho-NF-κB p65 (#3031, CST), MyD88 (#4283, CST) or GAPDH (#5174, CST) followed by incubation with a peroxidase-conjugated goat anti-rabbit (or mouse) IgG antibody. Densitometry analysis was performed, and the results were normalized to GAPDH expression and expressed as the fold changes over controls.
Data analysis
All statistical analyses were performed using SPSS (version 19.0.0, SPSS Inc., Chicago, IL, USA). The results are expressed as the means ± standard deviations. Differences between groups were analyzed using two-sided Student’s t-test or one-way ANOVA. Statistical significance was set at P < 0.05.
ACKNOWLEDGMENTS AND FUNDING
This work was in part supported by National Natural Science Foundation of China (NO. 81200504), by Project of Biomedical Engineering Research Foundation of Shanghai Jiao Tong University.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
Authors’ contributions
Y.Shao, S-J.Xia and L.Chen designed the study; L.Chen, D.Li, Y.-P.Zhu, X.-J.Wang and C.-Y.Jiang conducted experiments; L.Chen analyzed data and wrote the manuscript.
REFERENCES
1. Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, Saran R, Wang AY, Yang CW. Chronic kidney disease: global dimension and perspectives. Lancet. 2013; 382: 260–272.
2. Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011; 7:684–696.
3. Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest. 2008; 118: 3522–3530.
4. Cao Q, Wang Y, Harris DC. Pathogenic and protective role of macrophages in kidney disease. Am J Physiol Renal Physiol. 2013; 305:F3–11.
5. Li MO, Flavell RA. Contextual regulation of inflammation: a duet by transforming growth factor-beta and interleukin-10. Immunity. 2008; 28:468–476.
6. Bathgate RAD, Hsueh AJW, Sherwood OD. CHAPTER 16 - Physiology and Molecular Biology of the Relaxin Peptide Family A2 - Neill, Jimmy D. Knobil and Neill's Physiology of Reproduction (Third Edition). (St Louis: Academic Press), 2006; 679–768.
7. Samuel CS, Hewitson TD, Unemori EN, Tang ML. Drugs of the future: the hormone relaxin. Cell Mol Life Sci. 2007; 64:1539–1557.
8. Samuel CS, Hewitson TD. Relaxin and the progression of kidney disease. Curr Opin Nephrol Hypertens. 2009; 18:9–14.
9. Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, Ponikowski P, Unemori E, Voors AA, Adams KF Jr., Dorobantu MI, Grinfeld LR, Jondeau G, et al. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet. 2013; 381:29–39.
10. Figueiredo KA, Rossi G, Cox ME. Relaxin promotes clustering, migration, and activation states of mononuclear myelocytic cells. Ann N Y Acad Sci. 2009; 1160:353–360.
11. Pulskens WP, Rampanelli E, Teske GJ, Butter LM, Claessen N, Luirink IK, van der Poll T, Florquin S, Leemans JC. TLR4 promotes fibrosis but attenuates tubular damage in progressive renal injury. J Am Soc Nephrol. 2010; 21:1299–1308.
12. Zhou Y, Zhang T, Wang X, Wei X, Chen Y, Guo L, Zhang J, Wang C. Curcumin Modulates Macrophage Polarization Through the Inhibition of the Toll-Like Receptor 4 Expression and its Signaling Pathways. Cell Physiol Biochem. 2015; 36:631–641.
13. Tian S, Zhang L, Tang J, Guo X, Dong K, Chen SY. HMGB1 exacerbates renal tubulointerstitial fibrosis through facilitating M1 macrophage phenotype at the early stage of obstructive injury. Am J Physiol Renal Physiol. 2015; 308:F69–75.
14. Orr JS, Puglisi MJ, Ellacott KL, Lumeng CN, Wasserman DH, Hasty AH. Toll-like receptor 4 deficiency promotes the alternative activation of adipose tissue macrophages. Diabetes. 2012; 61:2718–2727.
15. Heeg MH, Koziolek MJ, Vasko R, Schaefer L, Sharma K, Muller GA, Strutz F. The antifibrotic effects of relaxin in human renal fibroblasts are mediated in part by inhibition of the Smad2 pathway. Kid Int. 2005; 68:96–109.
16. Mookerjee I, Hewitson TD, Halls ML, Summers RJ, Mathai ML, Bathgate RA, Tregear GW, Samuel CS. Relaxin inhibits renal myofibroblast differentiation via RXFP1, the nitric oxide pathway, and Smad2. Faseb J. 2009; 23: 1219–1229.
17. Chow BS, Chew EG, Zhao C, Bathgate RA, Hewitson TD, Samuel CS. Relaxin signals through a RXFP1-pERK-nNOS-NO-cGMP-dependent pathway to up-regulate matrix metalloproteinases: the additional involvement of iNOS. PloS One. 2012; 7:e42714.
18. Binder C, Chuang E, Habla C, Bleckmann A, Schulz M, Bathgate R, Einspanier A. Relaxins enhance growth of spontaneous murine breast cancers as well as metastatic colonization of the brain. Clin Exp Metastasis. 2014; 31: 57–65.
19. Rosetto M, Engstrom Y, Baldari CT, Telford JL, Hultmark D. Signals from the IL-1 receptor homolog, Toll, can activate an immune response in a Drosophila hemocyte cell line. Biochem Biophys Res Commun. 1995; 209: 111–116.
20. Campbell MT, Hile KL, Zhang H, Asanuma H, Vanderbrink BA, Rink RR, Meldrum KK. Toll-like receptor 4: a novel signaling pathway during renal fibrogenesis. J Surg Res. 2011; 168:e61–69.
21. Fung E, Tang SM, Canner JP, Morishige K, Arboleda-Velasquez JF, Cardoso AA, Carlesso N, Aster JC, Aikawa M. Delta-like 4 induces notch signaling in macrophages: implications for inflammation. Circulation. 2007; 115:2948–2956.
22. Saliba Y, Karam R, Smayra V, Aftimos G, Abramowitz J, Birnbaumer L, Fares N. Evidence of a Role for Fibroblast Transient Receptor Potential Canonical 3 Ca2+ Channel in Renal Fibrosis. J Am Soc Nephrol. 2015; 26:1855–1876.
23. Bolisetty S, Zarjou A, Hull TD, Traylor AM, Perianayagam A, Joseph R, Kamal AI, Arosio P, Soares MP, Jeney V, Balla J, George JF, Agarwal A. Macrophage and epithelial cell H-ferritin expression regulates renal inflammation. Kidney Int. 2015; 88:95–108.