
Introduction
Classically, Akt has been viewed as the main upstream activator of mammalian target of rapamycin (mTOR). Indeed, activated Akt phosphorylates and inhibits tuberous sclerosis 2 (TSC2), allowing Ras homolog enriched in brain (Rheb) to accumulate in the GTP-bound state and trigger activation of the mTOR complex1 (mTORC1) pathway. mTORC1 is composed by mTOR, regulatory associated protein of mTOR (Raptor), mLST8/G-protein β-subunit like protein (GβL), RAS40 and Deptor. The activation of mTOR in mTORC1 leads to phosphorylation of ribosomal S6 protein kinase 1 (S6K1) and eIF4E-binding protein 1 (4E-BP1), mediators of protein translation and cell growth [1]. mTOR response to a wide range of intracellular (energy and stress) and extracellular (nutrients, growth factors, hormones) signals is mediated through these effectors. In response to nutrient and growth factor availability, mTORC1 suppresses autophagy, a process by which metabolically stressed cells recycle cytoplasmic components including organelles, to recover energy necessary for their survival. mTORC1 has also been recently identified as orchestrating anabolic cell growth by stimulating nucleotide synthesis through the pyrimidine synthesis pathway [2].
Different from mTORC1, mTORC2 is composed of mTOR, rapamycin-insensitive companion of mTOR (Rictor), mLST8/GβL, stress-activated-protein-kinase-interacting protein 1 (Sin1), proline-rich repeat protein-5 (PRR-5)/protein observed with Rictor-1 (Protor-1), and Deptor. The upstream regulation of mTORC2 is not well defined, although ribosome association appears to be a major, if not the sole, mechanism of mTORC2 activation [3]. mTORC2 plays an important role in cell survival, metabolism, proliferation and cytoskeleton organization, as it phosphorylates Protein Kinase Cα (PKCα), Serum/glucocorticoid-regulated kinase 1 (SGK1), as well as Akt, allowing for complete activation of Akt [4-7]. Akt is therefore both an upstream activator of mTORC1 and downstream effector of mTORC2 (Figure 1).
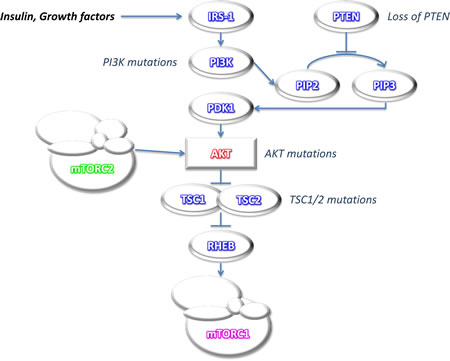
Figure 1: Simplified scheme of mTOR pathway activation. Akt activation releases Rheb from the inhibitory effects of TSC1/2 thus allowing mTOR activation. Signaling defects upstream of mTOR in the PI3K/Akt/mTOR pathway (mutations in PTEN, PI3K, Akt and TSCs) lead to mTOR deregulation. Akt can also be a downstream effector of mTOR, due to mTOR association with different protein partners to form two functionally distinct signaling complexes, mTORC1 and mTORC2.
Because it plays such a crucial role in cancer biology, mTOR has emerged as a potential target for drug development. Several mTOR inhibitors have already gone through clinical trials for treating various cancers without great success. Nevertheless, the role of mTOR inhibitors in cancer therapy continues to evolve, as new compounds are synthetized.
In the present review we have focused on the role for mTOR in orchestrating key physiological and pathological processes, with a particular emphasis on colorectal cancer (CRC), which remains the second leading cause of cancer death in the United States [8].
mTOR as a proto-oncogene
Although mTOR is frequently activated in human cancers, mutation of the mTOR gene has been found only occasionally [9-11]. This means that over-activation of the mTOR pathway is mostly due to signaling defects upstream of mTOR in the phosphatidylinositol-3-kinase (PI3K)/Akt/mTOR pathway. Mutations in PI3K alpha catalytic subunit kinase domain (PIK3CA) generally arise late in tumorigenesis, and can be identified in 32% of CRC tumors [12]. Loss of heterozygosity (LOH) and mutations in Phosphatase and tensin homolog (PTEN), a negative regulator of PI3K activity, have also been reported in CRC [13]. Both PIK3CA mutations and PTEN loss lead to mTOR over-activation. Although mutations in Akt genes are rarely found in CRC [14], a somatic missense mutation of Akt1 (E17K) in the pleckstrin homology (PH) domain resulting in constitutive association of Akt1 with the plasma membrane and Akt1 prolonged activation has been reported in CRC, which can lead to mTOR deregulation [15]. Similarly, although rare, germline TSC gene mutations, which have been associated with colonic hamartomatous polyps, account for 1% CRC, possibly through mTOR pathway triggering [16, 17] (Figure 1).
Several studies have attempted to characterize activating mutations in the mTOR gene [18-24]. These studies, mostly conducted using yeast, have demonstrated the impact of mutations on the function of mTOR and in vitro oncogenicity in some cases. However, the tumorigenic potential of the mTOR gene has only recently been established. By introducing mutations into evolutionarily conserved amino acid residues in major functional domains of human mTOR, Murugan et al. demonstrated that the mTOR gene is a proto-oncogene that possesses strong tumorigenicity when genetically activated [25]. Specifically, eight mutants in the HEAT repeats (M938T) and the FAT (W1456R and G1479N) and kinase (P2273S, V2284M, V2291I, T2294I, and E2288K) domains of mTOR were generated. These mutants showed increased protein kinase activities and activated the mTOR/p70S6K and Akt signaling pathways in human embryonic kidney cells (HEK293T). The kinase domain mutants which exhibited the greatest gains in activity, P2273S and E2288K were subsequently selected to explore the oncogenic potential of the mTOR gene in the mouse embryonic fibroblast cell line NIH3T3. mTOR mutant-expressing cells showed morphologic transformation and anchorage-independent growth, possessed invasive ability and in vivo tumorigenicity. This discovery of oncogenic mTOR mutations may facilitate the design of drugs targeting mTOR, as well as help predict their efficacy. For example, in yeast, resistance to Rapalogs has been associated with mutations in FK506 binding protein 12 (FKBP12) or the FKBP-rapamycin-binding (FRB) domain of TOR [26].
mTOR’s role in proliferation, differentiation and senescence
While emerging evidence supports a central role of the mTOR pathway in cell growth and cancer progression, increased mTOR activity can also play a role mediating the depletion of the epithelial stem cell compartment. Indeed, the aberrant activation of the mTOR pathway can paradoxically cause cells to undergo differentiation or senescence, thereby exiting the proliferative cell pool [27]. This concept is well demonstrated by the fact that persistent activation of mTOR by wingless-related MMTV integration site 1 (Wnt1) leads to accelerated epithelial stem cell senescence and premature aging in mice [28, 29]. Accordingly, inhibition of mTOR prevents the loss of proliferative epithelial progenitor stem cells upon radiation and enhances their tissue repopulating capacity [30]. Similarly, mTOR inhibition by Rapamycin enriches CD133+ subpopulations in liver tumor cells [31]. This enrichment is most likely achieved through blocking differentiation of the CD133+ subpopulations, enhancing apoptosis in the CD133- subsets, and triggering the conversion of CD133- to CD133+ cells. Thus, the maintenance of CD133+ cells in vivo by Rapamycin leads to high continuous tumorigenic potential in the context of liver cancer. These data suggest that mTOR signaling is involved in regulating the balance of proliferation and differentiation of cancer stem cells (CSCs) and that transient inhibition of mTOR can promote tumor re-emergence in certain tumor types via enrichment of CSCs.
The molecular mechanism(s) underlying these paradoxical effects of mTOR are not fully understood. It has been suggested that strong oncogenic signals (RAS, PI3K) concomitantly induce cell cycle arrest and activation of growth-promoting (i.e., anabolic) pathways such mTOR. Cell cycle arrest by itself is not yet senescence [32]. Nevertheless, in the presence of growth-stimulation, cell cycle blockage eventually leads to senescence. This mechanism by which arrested cells are converted to senescent cells has been named gerogenic conversion or geroconversion [33]. To avoid geroconversion, cancer cells must lose expression of cell cycle inhibitors, such as p53. Thus, cross-talk between p53 and the mTOR signaling pathways can determine whether stressed cells undergo apoptosis, reversible quiescence or irreversible senescence [34]. Inhibitors of mTOR can suppress geroconversion, protecting adult stem cells from undergoing premature cell senescence while simultaneously preventing their oncogenic transformation [35]. Amongst mTOR inhibitors, Rapamycin has been defined as a “longevity enhancer and cancer preventative agent” in the context of p53 deficiency [36]. Indeed, continuous treatment with Rapamycin or a novel Rapamycin formulation (Rapatar) delayed carcinogenesis in tumor-prone p53+/-and p53-/-mice respectively, most likely by slowing down the process of aging [37, 38]. Similarly, chronic treatment of mice with an enterically released formulation of Rapamycin (eRapa) delayed the onset and/or progression of neuroendocrine tumors in Rb1+/- mice [39]. Likewise, hypoxia can decelerate geroconversion and extend lifespan. Indeed, not only does hypoxia arrests cell cycle, but also inhibits the mTOR pathway, thus preventing irreversible cellular senescence [40]. It turns out that in stem cell niches, stem cells might be protected from senescence and maintained in a quiescent status instead, thanks to the low oxygen levels which characterize stem cell niches [41]. Overall, these studies point out molecular differences in normal and cancer cells that can be exploited to prevent tumor growth without disrupting the function of normal tissues and cells.
Development of mTOR inhibitors: progress and challenges
Rapamycin, a macrolide antibiotic produced by Streptomyces hygroscopicus, was the first mTOR inhibitor discovered. More precisely, by exploiting Rapamycin’s antifungal properties researchers were able to identify mTOR [42]. Guba et al. described Rapamycin antitumor effects for the first time in 2002, in their Nature Medicine paper [43]. Traditionally used as an immunosuppressant for organ transplants, Rapamycin was found to suppress tumor growth by inhibiting angiogenesis. Since then, additional inhibitors of mTOR function were synthesized with similar characteristics to Rapamycin, constituting the family of Rapalogs. Rapalogs, which also include Everolimus, Temsirolimus, and Ridaforolimus, bind FKBP12 and interfere with the FRB domain of mTOR. These compounds have gone through clinical trials as single agents for treating various cancers without great success. However, Temsirolimus and Everolimus were both effective against advanced renal cell carcinoma (RCC) [44]. While resistance to Rapalogs has been associated with mutations in FKBP12 or the FKB domain of TOR in yeast [26], in human, it has been mostly associated with KRAS and BRAF mutations [45]. Additional mechanisms of resistance include up-regulation of the Proviral integration site for Moloney murine leukemia virus (PIM) family of oncogenic serine/threonine kinases [46], oxidative stress [47], and over-expression of anti-apoptotic proteins [48]. Nevertheless, suppression of several negative feedback loops following mTORC1 targeting is mostly responsible for failure of mTOR inhibitors. Biopsies taken from patients affected by malignancies other than RCC, like colon and breast tumors, showed activated Akt following treatment with Rapalogs [49, 50]. Pro-survival rather than anticancer effects of Rapalogs likely results from disruption of the mTORC1-dependent negative feedback loop to mTORC2 and IRS-1/PI3K. Particularly, mTORC1-activated S6K1 phosphorylates Rictor and/or Insulin receptor substrate (IRS)-1, thus inhibiting mTORC2 and PI3K/Akt signaling, respectively [51, 52]. More recent findings indicate that mTORC1 also phosphorylates Growth Factor Receptor Bound Protein 10 (Grb10), leading to accumulation of Grb10 and negative feedback inhibition of PI3K and the Microtubule-associated protein kinase/Extracellular-signal regulated kinase (MAPK/ERK) pathway [53]. Thus, over-activation of upstream pathways following suppression of the above-mentioned feedback loops potentially counterbalances the antiproliferative effects of mTOR inhibitors. To overcome the detrimental consequences of feedback loop activation, a second generation of inhibitors, which compete with ATP in the catalytic site of mTOR, and inhibit both complexes, was developed [54]. Figure 2 depicts activators and effectors of both mTOR complexes, together with the effects of mTOR inhibition using different mTOR inhibitors. Table 1 and Table 2 provide a list of different ATP-competitive mTOR-selective or PI3K/mTOR dual inhibitors that have been validated in preclinical cancer studies. These studies showed inhibition of tumor growth in a number of xenograft models [55-147]. As shown in Table 1, ATP-competitive mTOR inhibitors have often been used in combination with ionizing radiation (IR) as well as many chemicals. Clinical trials have been completed for AZD8055, INK-128, OSI-027, NVP-BEZ23, and XL765. Additional clinical trials utilizing mTOR-inhibiting agents are ongoing.
Table 1: In vivo pre-clinical studies using ATP-competitive inhibitors of mTOR.
Name |
Xenograft models |
Combinatorial Therapy |
References |
AZD2014 |
Patient-derived ER+ breast cancer |
/ |
[55] |
AZD8055 |
U87MG, BT474c, A549, Calu-3, LoVo, SW620, PC3, MES-SA |
/ |
[56] |
PTEN(+/-)LKB1(+/hypo) |
/ |
[85] |
|
Patient-derived primary HCC |
SAHA |
[57] |
|
MTT |
/ |
[86] |
|
RD |
AZD6244 |
[94] |
|
INK-128 |
MDA-MB361 |
Lapatinib |
[62] |
KU-0063794 |
786-O |
/ |
[130] |
OSI-027 |
COLO 205, GEO |
/ |
[65] |
GEO, H292, Ovcar-5 |
Sunitinib (H292, Ovcar-5) |
[128] |
|
Jeko |
/ |
[146] |
|
HNSCC |
Cetuximab |
[126] |
|
OXA-01 |
GEO |
/ |
[128] |
PP242 |
p190BCR-ABL, SUP-B15 |
Imatinib (p190BCR-AB), Dasatinib (SUP-B15) |
[135] |
8226 MM |
/ |
[134] |
|
LS174T |
UO126 |
[105] |
|
MDA-MB-231 |
/ |
[71] |
|
LS174T, SW480 |
/ |
[61] |
|
HS Sultan cells stably transfected with exogenous VEGF |
/ |
[133] |
|
Patient-derived colon cancer |
/ |
[132] |
|
U251 |
IR |
[131] |
|
DLD-1 |
Erlotinib |
[129] |
|
Torin-1 |
U87MG |
/ |
[72] |
Patient-derived colon CSCs |
/ |
[171] |
|
WYE-125132 (WYE-132) |
MDA361, U87MG, |
Bevacizumab |
[76] |
WYE-354 |
PC3MM2, U87MG |
/ |
[75] |
A panel of second-generation mTOR inhibitors is listed. Information regarding xenograft models as well as combinatorial drugs used in preclinical cancer studies for each mTOR inhibitor is provided. For information about combinatorial drugs, please consult legend of Table 2.
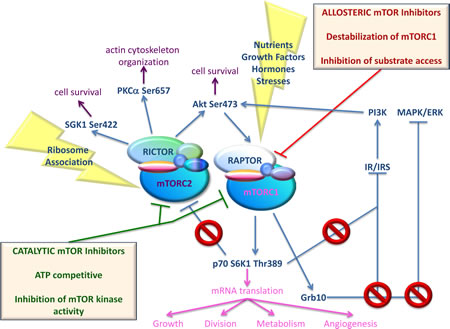
Figure 2: Simplified scheme of activators and effectors of both mTOR complexes, together with effects of mTOR inhibition using different mTOR inhibitors. mTOR can be shared by two different complexes, mTORC1 and mTORC2. Phosphorylation of mTOR at Ser2448 and Ser2481 are indicative of mTORC1 and mTORC2 activation, respectively. mTORC1 can be activated by nutrients (amino acids, glucose), growth factors (Insulin, Insulin-like Growth Factor-1), hormones (leptin), and stresses (starvation, hypoxia, and DNA damage). Through mTORC1, these signals accelerate the synthesis of key proteins, involved in growth, division, metabolism and angiogenesis. mTORC1-activated p70 S6K1 and Grb10 mediate IRS-1 degradation, thus inhibiting PI3K/Akt activation. Grb10 also leads to negative feedback inhibition of MAPK/ERK pathway. Finally, activated p70 S6K1 inhibits mTORC2 signaling by phosphorylating Rictor on Thr1135. Allosteric mTOR inhibitors exert an incomplete inhibition of mTORC1 and are inactive against mTORC2 under short-term conditions. Moreover, they disrupt the mTORC1-dependent negative feedback loop to IRS-1/PI3K, MAPK/ERK and mTORC2. As a consequence, treatment with mTOR allosteric inhibitors often results in increased mTORC2 activity. mTORC2 functions upstream of Akt providing the critical second phosphorylation of Akt at Ser473, which is necessary for Akt full activation. mTORC2 also regulates cytoskeletal dynamics by activating PKCα, and regulates growth via SGK1 phosphorylation. The upstream regulation of mTORC2 is not well defined although ribosome association appears to be a major, if not the sole, mechanism of mTORC2 activation. Catalytic mTOR inhibitors are able to suppress activity of both mTORC1 and mTORC2 complexes, avoiding oncogenic signaling pathway activation
Table 2: In vivo pre-clinical studies using PI3K and mTOR dual inhibitors.
Name |
Xenograft models |
Combinatorial Therapy |
References |
BEZ235 (NVP-BEZ235) |
PC3M, U87MG |
Temozolomide (U87MG) |
[89] |
BN472 |
/ |
[95] |
|
HER2+ BT474 |
/ |
[96] |
|
Tet-op-PIK3CA H1047R–CCSP-rtTA, LSL Kras G12D |
ARRY-142886 (LSL Kras G12D) |
[83] |
|
Patient-derived primary pancreatic cancer |
/ |
[81] |
|
B16BL6 |
/ |
[147] |
|
ENU-treated Tsc2+- |
/ |
[93] |
|
MM.1S |
/ |
[91] |
|
U87 |
/ |
[88] |
|
CCSP-rtTA/Tet-op-K-Ras (FVB/N), H460 |
IR (H460) |
[87] |
|
EGFR T790M-L858R |
AZD6244 |
[84] |
|
RD/18 |
/ |
[92] |
|
TC-71, RD/18 |
Vincristine |
[90] |
|
BC-1 PEL |
/ |
[80] |
|
786-0, A498 |
/ |
[82] |
|
FL-18 |
/ |
[98] |
|
Patient-derived glioblastoma (SJ28P3) |
SL327 |
[99] |
|
A172, patient-derived glioblastoma (SJ28P3) |
/ |
[100] |
|
Ptch+/−Hic+/− |
NVP-LDE225 |
[101] |
|
GS2 |
Chloroquine |
[102] |
|
DU145 |
Taxotere |
[103] |
|
LSL-K-ras(G12D/+)Pten(loxP/loxP) |
/ |
[104] |
|
LS174T |
UO126 |
[105] |
|
A549 |
RAD001 |
[106] |
|
N87, MKN28, MKN45 |
/ |
[107] |
|
JHH-7 |
/ |
[108] |
|
A549 |
Chloroquine |
[109] |
|
786-0 |
Sorafenib |
[110] |
|
CAL62, TT |
RAF265 |
[111] |
|
GEM |
/ |
[112] |
|
AsPC-1 |
EMAP and/or Gemcitabine |
[113] |
|
Met-1, MCNeuA in the MKR mouse |
/ |
[114] |
|
LS174T, SW480 |
/ |
[61] |
|
HEC-59, AN3CA |
/ |
[60] |
|
Tyr-HRas(G12V) Ink4a/Arf−/−, T11 OST, C3-TAg GEMM, MMTV-Neu GEMM |
AZD6244 |
[121] |
|
A549 |
Ganetespib |
[115] |
|
8505C |
Paclitaxel |
[58] |
|
H295R |
/ |
[59] |
|
U-87 MG, 786-O |
RAD001 |
[118] |
|
Patient-derived PDAC |
Panobinostat |
[123] |
|
U937 cells expressing doxycycline-inducible shRNAs against both Bcl-2 and Bcl-xL |
ABT-737 |
[120] |
|
PC-9/HGF |
/ |
[122] |
|
PC3 |
IR |
[119] |
|
4T1, 67NR |
Dovitinib |
[116] |
|
LoVo, HCT116 |
5-FU or irinotecan |
[117] |
|
CNE2, HONE1 |
Cisplatin |
[124] |
|
RD |
AZD6244 |
[94] |
|
C4-2AT6 |
Docetaxel |
[125] |
|
NVP-BBD130 |
B16BL6 |
/ |
[147] |
PI-103 |
U87:ΔEGFR |
/ |
[141] |
U87MG, HCT116, PC3, MDA-MB-435, MDA-MB-468, SKOV3, IGROV-1 |
/ |
[66] |
|
U251 MG |
IR |
[139] |
|
p190 L-CFCs (hCD4+) |
Imatinib |
[143] |
|
EC-vGPCR |
/ |
[138] |
|
U87MG |
Temozolomide |
[142] |
|
Patient-derived neuroblastoma |
Doxorubicin |
[137] |
|
518A2 |
Rapamycin |
[144] |
|
Gli36-EvIII-FmC, mNSC-S-TRAIL |
/ |
[136] |
|
PC-9 remixed with HGF high producing MRC-5 cells |
Gefitinib |
[140] |
|
SH-EP |
TRAIL |
[145] |
|
SK-N-BE(2) |
/ |
[67] |
|
HGC27 |
5-FU |
[69] |
|
Huh7 |
Sorafenib |
[68] |
|
XL765 |
Patient-derived glioblastoma |
Temozolomide |
[77] |
BxPC-3 |
Chloroquine |
[78] |
|
STS26T |
Chloroquine |
[79] |
|
GH3 |
Temozolomide |
[127] |
A panel of second-generation mTOR inhibitors is listed. Information regarding xenograft models as well as combinatorial drugs used in preclinical cancer studies for each mTOR inhibitor is provided. SAHA and Panobinostat are histone deacetylase (HDAC) inhibitors; AZD6244, UO126, ARRY-142886, and SL327 are MEK inhibitors; Lapatinib, Cetuximab, Erlotinib, and Gefitinib are EGFR inhibitors; Sunitinib and Sorafenib are multitargeted kinases inhibitors; Imatinib and Dasatinib are tyrosine kinases inhibitors; Bevacizumab is an anti-vascular endothelial growth factor (VEGF) monoclonal antibody; Taxotere is a microtubule inhibitor; RAF265 is a BRAF inhibitor; Ganetespib is a heat shock protein 90 (Hsp90) inhibitor; ABT-737 is a Bcl-2 inhibitor; Dovitinib is an inhibitor of fibroblast growth factor (FGF), VEGF, and platelet-derived growth factor (PDGF) receptors; Vincristine and Paclitaxel are mitosis inhibitors; Irinotecan is a topoisomerase-I inhibitor; Temozolomide is an alkylating agent; NVP-LDE225 is a Smoothened (Smo) antagonist; Chloroquine is a lysosomotropic agent; Endothelial-monocyte activating polypeptide (EMAP) is a proinflammatory cytokine and a mediator of programmed endothelial cell death; 5-Fluorouracil, Cisplatin, and Doxorubicin are chemotherapy drugs; Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a cytotoxic protein and a mediator of programmed cell death.
mTOR kinase inhibitors (mTorKIs) do not cause Akt feedback activation observed with Rapalogs. However, Rodrik-Outmezguine et al. showed a biphasic effect of these drugs on Akt [148]. Inhibition of mTORC2 led to Akt Ser473 de-phosphorylation and a rapid but transient inhibition of Akt Thr308 phosphorylation and Akt signaling. Nevertheless, inhibition of mTOR kinase also relieves feedback inhibition of Receptor tyrosine kinases (RTKs) leading to subsequent PI3K activation and re-phosphorylation of Akt Thr308 sufficient to reactivate Akt activity and signaling. Importantly, similar mechanisms might also exist in tumor-associated stromal cells such as endothelial cells. Phosphorylation of Akt Thr308, Akt substrates [PRAS40, FoxO1 (forkhead box protein O1), and GSK-3 (glycogen synthase kinase2013;3)], and SGK substrate [NDRG1 (N-myc downstream regulated 1)] rebounds as early as 4 hours after adding mTorKIs in human umbilical vein endothelial cells (HUVECs), implying that PI3K is activated in response to mTOR inhibition in these cells [149]. Particularly, mTorKIs induce the expression of multiple RTK-encoding genes and signaling by promoting the de-phosphorylation of FoxO1, which eventually result in increased activation of PI3K. Thus mTorKIs may render endothelial cells more prone to PI3K inhibition, suggesting that combinations of PI3K and mTOR inhibitors might have synergistic effects and fully inhibit endothelial cell growth [149].
An additional limitation to the use of catalytic mTOR inhibitors is that they inhibit mTOR within the mTOR complexes and do not block intrinsic activity of mTOR-binding partners, which, although deprived of mTOR kinase activity, could continue conveying growth and survival signals in response to death stimuli. As opposed to Rapalogs and mTorKIs, P529 is a first-in-class allosteric inhibitor of mTORC1/mTORC2 that can dissociate both mTORC1 and mTORC2 complexes, thus overcoming possible activity of mTOR-binding partners. However, it is not clear whether P529 directly interacts with the mTOR complexes causing their dissociation. It may inhibit a chaperone, which assembles the mTOR complexes. Nevertheless, P529 was reported to inhibit tumor growth, angiogenesis, and vascular permeability [150], and to be effective in the treatment of prostate cancer [151, 152]. Moreover, P529 was explored as a therapeutic option for the treatment of keloid disease [153]. Aside from its association with cancer, deregulation of the mTOR pathway is in fact linked to several other diseases, including ocular, fibrotic, viral, skin and central nervous system diseases.
mTOR signaling in colon: past and present evidence
mTORC1 is a major sensor of the organismal nutritional state. Indeed, caloric restriction lowers mTORC1 signaling in Paneth cells, a key constituent of the mammalian intestinal stem-cell (ISC) niche. Paneth cells, in turn, stimulate small-intestinal stem cells to proliferate [154]. Thus, mTOR inhibition can improve intestinal regeneration in patients affected by intestinal atrophy. While mTORC1 inhibition can increase the number and regenerative capacity of ISCs, excessive mTORC1 stimulation can lead to the onset of cancer. Indeed, the importance of mTORC1 pathway in intestinal polyp formation has been well described in several papers using the Apc∆716 mice, a mouse model of familial adenomatous polyposis (FAP). Through these studies, mTORC1 was found to stimulate chromosomal instability (CIN) through anaphase bridge formation, enhancing, as a consequence, both tumor initiation and progression [155]. Similarly, ex vivo immunohistochemical studies on human colorectal adenomas and cancers confirmed that mTORC1 signaling occurs as an early event in the process of tumorigenesis, and participates in the progression of normal cells to a neoplastic phenotype [156], sustaining the bases of mTORC1-targeted drug development for therapy and prevention of colon polyps and cancers. Accordingly, Everolimus-mediated mTORC1 inhibition suppressed polyp formation and reduced mortality in Apc∆716 mice [157]. However, blocking of a specific pathway may disrupt the balance between signaling pathways and enhance oncogenic signals. In that regard, in parallel with its cytostatic effect, mTORC1 inhibition by Rapamycin strongly increased MAPK kinase (MEK)/ERK activity, resulting in the appearance of a spindle morphology and higher invasiveness of KRAS-transformed intestinal epithelial cells (IECs) [158]. In this system, Rapamycin treatment also increased Bcl-2 levels, further indicating the need to develop new therapeutic drugs capable of overcoming the relief of feedback inhibition of pro-survival, pro-invasive and pro-metastatic pathways. Besides mTORC1, mTORC2 has been shown to be overexpressed in CRC and to play an important role in cancer biology. Down-regulation of mTORC2 reduced proliferation of colon cancer cell lines and inhibited the formation of tumor xenografts in vivo [159, 160]. Moreover both mTORC1 and mTORC2 complexes were shown to regulate epithelial-mesenchymal transition (EMT), motility, and metastasis of CRCs via RhoA and Rac1 GTPases, providing the rationale for including ATP-competitive mTOR-selective or PI3K/mTOR dual inhibitors for therapy of CRC patients [161]. Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 was shown to induce tumor regression in a genetically engineered mouse (GEM) model for sporadic CRC [112]. Consistent with this finding, a more recent study also demonstrated the efficacy of NVP-BEZ235 and of an additional catalytic mTOR inhibitor, pp242, in human colon cancer cell line xenografts [61]. Nevertheless, a remarkable intrinsic drug resistance of a large proportion of CRC cell lines to new-generation mTOR inhibitors, including both NVP-BEZ235 and pp242 compounds, was also described, warranting further studies [162]. By screening a panel of over 600 human cancer cell lines to identify markers of resistance and sensitivity to pp242, Ducker et al. found that KRAS mutations are responsible for conferring resistance to pp242 [132]. This resistance was specifically linked to changes in the level of phosphorylation of 4E-BP1, and was not evident in either a tumor with wild type KRAS or a tumor with a PIK3CA mutation in addition to KRAS. This and other studies [163] highlight the importance of monitoring the phosphorylation status of 4E-BP1 to assess responses to mTorKIs. Additional experimental evidence has revealed that pp242 treatment of a panel of CRC cell lines transiently inhibits Akt Ser473 phosphorylation while increasing the phosphorylation of epidermal growth factor receptor (EGFR) at Tyr1068 [129]. A parallel increase of Akt Ser473 and EGFR Tyr1068 in cells following pp242 treatment raises the possibility that, apart from KRAS mutations, EGFR phosphorylation, might also contribute to the incomplete inhibition of mTORC2 by pp242. Accordingly, the combination treatment of pp242 and erlotinib, an EGFR inhibitor, completely blocked both mTORC1 and mTORC2 activity, inhibited cell growth and suppressed the progression of CRC xenografts [129].
An additional and previously undescribed mechanism leading to Rapamycin resistance, based on 3-Phosphoinositide–dependent protein kinase-1 (PDK1)/Polo-like kinase 1 (PLK1)/Myc signaling, has also been reported in the context of CRC. Specifically, epigenetic loss of Protein phosphatase 2, regulatory subunit B, beta (PPP2R2B), occurring in >90% colorectal tumor samples, was indicated as a molecular event affecting the sensitivity of CRC to mTOR inhibitors [164]. On loss of PPP2R2B, Rapamycin triggers a compensatory Myc phosphorylation in PDK1-dependent, but PI3K and AKT-independent manner, resulting in resistance. Re-expression of PPP2R2B, genetic ablation of PDK1 or pharmacologic inhibition of PDK1 (using the small molecule BX912) abrogates Rapamycin-induced Myc phosphorylation, leading to Rapamycin sensitization. Additional studies revealed that PDK1 directly induces phosphorylation of PLK1, which in turn induces Myc phosphorylation and protein accumulation [165]. Importantly, the PLK1 inhibitor BI2536 worked in synergy with NVP-BEZ235 to induce robust apoptosis and tumor growth inhibition in CRC [165]. This study emphasizes the importance of epigenetic mechanisms in regulating oncogenic signaling and therapeutic response. Moreover, it indicates PPP2R2B may serve as a predictive marker for patient selection, whereas Myc phosphorylation can serve as a surrogate marker to evaluate the drug response.
mTOR inhibitors for the ablation of colon cancer stem-like cells: future hopes
Over the last decade, proposed theories in cancer biology have drastically changed. Contrary to the long-standing clonal evolution model described by Nowell in the late 70’s [166], the CSC hypothesis proposed that not every cell of the body could be the target of malignant transformation. The limited lifespan of a committed cell is likely shorter than the time necessary to accumulate tumor-inducing genetic changes. Therefore, cancer-initiating capability could be a unique feature of long-lived, self renewing stem cells [167]. Indeed, much experimental evidence indicates that stem cell transformation might be an early event in carcinogenesis. Transformation of normal stem cells through loss of APC is an extremely efficient route towards initiating intestinal adenomas [168]. The CSC hypothesis is neither a universal model for all cancers nor for all patients with the same disease. While some cancers have been hypothesized to initiate as a stem cell disease, they may then progress by clonal evolution of their CSCs, as CRC has been suggested to do through CIN [169].
To our knowledge, only one study investigated the effects of mTOR inhibitors in cancer stem-like cells so far. mTOR signaling was shown to be activated in colorectal cell line-derived spheres in serum-free medium [170]. Treatment with Rapamycin and pp242 diminished sphere-forming capacity as well as aldehyde dehydrogenase isoform 1 (ALDH1) activity. However, only pp242 suppressed the enrichment of ALDH+ cells induced by chemotherapy, thus highlighting an essential role of mTORC2 signaling in the maintenance of the CRC stem-like phenotype. Although this study further confirmed the importance of mTOR signaling in CRC, the authors did not perform sufficient functional experiments to assess the effect of mTOR inhibition on biological properties and tumorigenic potentials of CRC stem-like cells. Moreover, a controversy exists in the literature regarding the use of ALDH+ cells isolated from cancer cell lines as in vitro models for CSC study, further indicating the need to study the effect of mTOR inhibition using alternative methods to identify and characterize CSCs. Multiple cell-surface proteins have been proposed as potential candidate markers for colon CSCs, and our in vitro system, based on a feeder layer derived from rat mammary adenocarcinoma, efficiently enriches for these cells [169]. Recently, we analyzed colon CSCs for expression of major mTORC1/2 pathway components [171]. Colon CSCs exhibited unexpectedly low Akt signaling. Nevertheless, they showed mTORC2 activation. We indicated SGK1 as the possible main mTORC2 effector in colon CSCs. Akt hypo-phosphorylation and dependence of SGK family members for viability are known to occur most frequently in the context of wild-type PTEN, and helical PIK3CA mutations [172]. Future studies are needed to confirm whether this genetic signature can predict resistance or sensitivity of colon CSCs to different mTorKIs. Unfortunately, the prognostic and predictive value of common mutations in patients with CRC is controversial, due to a bias in research settings [173]. In our opinion, mTorKI resistance might also occur through less well-studied but equally important epigenetic mechanisms [174].
mTOR inhibitors affected colon CSCs differently, resulting in proliferation, induction of autophagy or apoptosis. The apoptosis-inducing mTOR inhibitor Torin-1 hindered growth, motility, invasion, and survival of CD326+/CD24+/CD49f+/CD29+ and CD326+/CD44+/CD166+ CRC subpopulations in vitro, and suppressed tumor growth in vivo with a concomitant reduction in vessel formation. Torin-1 also affected the expression of markers for cell proliferation, angio-/lympho-genesis, and stemness in vivo. Our study also indicated that although Torin-1 resistant clones can emerge, they are poorly tumorigenic, thus encouraging its potential use for CRC therapy. Because normal stem cells and CSCs share many traits, it seems reasonable to think that any therapy targeting CSCs may also destroy healthy tissues [175]. Through an innovative system based on the use of the mouse lymph node as an in vivo bioreactor [176], we showed that Torin-1 does not affect the survival of normal colon stem cells in vivo, suggesting its selectivity towards cancer cells. All these data support further development of Torin-1 for the clinical treatment of CRC.
Concluding remarks: towards personalized medicine
mTOR is frequently activated in human cancers and is a commonly sought anticancer therapeutic target. Although direct substrates and downstream effectors of mTOR are well studied, the broad physiological role of mTOR implies that there are still downstream pathways to be identified. A more comprehensive understanding of the dynamics of mTOR signaling networks is therefore required for the design of safe and effective drug molecules targeting the mTOR pathway.
ATP competitive mTOR inhibitors have both advantages and disadvantages. Undoubtedly, they have a well-defined target, their biological effects are easy to be observed, and they transit quickly to clinical trials. On the other hand, they often have a high level of cross-reactivity with other kinases, are not pharmacologically tolerated, and easily lead to the development of drug resistance. Identifying biological factors that may predict efficacy or resistance to mTOR inhibitors is still challenging, since mTOR inhibitors may exert antitumor effects through multiple mechanisms of action. Moreover, crosstalk of the mTOR pathway with other pathways gives rise to compensatory loops for tumor cells to escape anti-tumor stimuli. This reveals the adaptive capabilities of oncogenic signaling networks and the limitations of monotherapy for inhibiting feedback-regulated pathways. Drug combinations targeting multiple pathways have been exploited to overcome this resistance but it is not clear which strategy will yield the greater therapeutic benefit. Additionally, the use of multiple drugs increases side effects and adverse reactions. In our study, a single mTOR inhibitor, Torin-1 was able to counteract CRC progression, supporting the rationale for its clinical testing [171]. Nevertheless, the precise mechanism by which Torin-1 acts remains to be elucidated. It must be noted that not all CRC patients could benefit from this therapy due to large individual variability in drug efficacy and safety. The observed discrepancy in outcome using different mTOR inhibitors in a population of colon CSCs is not only related to the difference of mTOR inhibitors in terms of pharmacokinetic and pharmacologic properties, but also to intrinsic variations in colon CSCs. A major goal of clinical pharmacology and pharmacogenomics is to establish phenotype-genotype associations that reveal drug response and toxicity. Unfortunately, although the impact of variations of the human genome sequence on the response to drug therapy has been increasingly considered in recent years, for complex diseases like CRC, it could be very difficult to determine unequivocally an exact phenotype or genotype that would predict therapy outcome. Genetic variations can result from single-nucleotide polymorphism (SNP), insertion, deletion, or duplication of DNA sequences. SNP of drug targets, drug-metabolizing enzymes, drug transporters, DNA repair enzymes, are likely to affect drug response [177]. In addition, non-genetic factors could contribute to variability in drug effects. Even if causal relations between variations and individual drug responses would be fully unraveled, the rapid development of resistance to targeted anticancer agents could represent a major challenge in targeted cancer therapy. Such resistance often results from secondary mutations of drug targets in cancer cells. Acquired resistance to imatinib in chronic myeloid leukemia (CML) and gastrointestinal stromal tumor (GISTs) through a secondary gene mutation in BCR-ABL and KIT genes, respectively, are examples of the adaptive capability of cancer cells to kinase inhibitors [178, 179]. Similarly, a dominant drug-resistant allele of mTOR (S2035T) eliminates the cellular actions of Rapamycin in the yeast Saccharomyces cerevisae [180]. It is not excluded that although primary mTOR mutations are barely observed in human cancers, the mTOR kinase could be hit by a secondary or even a de novo gene mutation in response to therapy as a self-defense mechanism.
In conclusion, although targeted therapy agents are increasingly available for clinical applications, many of these promising drugs have produced disappointing results when tested in clinical trials. Because most tumors are heterogeneous, a single drug regimen for patients with the same tumor type/histology is not always appropriate. Gene and protein signatures have been identified in several diseases, but this information struggles to be translated into clinically meaningful improvements. Nevertheless, these observations can help defeat the challenge of achieving individualized drug therapy, hopefully in the near future.
Acknowledgements
This work was supported by Ri.MED foundation (MGF). The authors thank Lynda Guzik for English editing.
References
1. Fingar DC, Salama S, Tsou C, Harlow E and Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes & development. 2002; 16(12):1472-1487.
2. Ben-Sahra I, Howell JJ, Asara JM and Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013; 339(6125):1323-1328.
3. Zinzalla V, Stracka D, Oppliger W and Hall MN. Activation of mTORC2 by association with the ribosome. Cell. 2011; 144(5):757-768.
4. Sarbassov DD, Guertin DA, Ali SM and Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005; 307(5712):1098-1101.
5. Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A and Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nature cell biology. 2004; 6(11):1122-1128.
6. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P and Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Current biology : CB. 2004; 14(14):1296-1302.
7. Garcia-Martinez JM and Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). The Biochemical journal. 2008; 416(3):375-385.
8. Jemal A, Siegel R, Xu J and Ward E. Cancer statistics, 2010. CA: a cancer journal for clinicians. 2010; 60(5):277-300.
9. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455(7216):1061-1068.
10. Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C, Teague J, Andrews J, Barthorpe S, Beare D, Buck G, Campbell PJ, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010; 463(7279):360-363.
11. Robbins CM, Tembe WA, Baker A, Sinari S, Moses TY, Beckstrom-Sternberg S, Beckstrom-Sternberg J, Barrett M, Long J, Chinnaiyan A, Lowey J, Suh E, Pearson JV, Craig DW, Agus DB, Pienta KJ, et al. Copy number and targeted mutational analysis reveals novel somatic events in metastatic prostate tumors. Genome research. 2011; 21(1):47-55.
12. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B and Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004; 304(5670):554.
13. Zhou XP, Loukola A, Salovaara R, Nystrom-Lahti M, Peltomaki P, de la Chapelle A, Aaltonen LA and Eng C. PTEN mutational spectra, expression levels, and subcellular localization in microsatellite stable and unstable colorectal cancers. The American journal of pathology. 2002; 161(2):439-447.
14. Soung YH, Lee JW, Nam SW, Lee JY, Yoo NJ and Lee SH. Mutational analysis of AKT1, AKT2 and AKT3 genes in common human carcinomas. Oncology. 2006; 70(4):285-289.
15. Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007; 448(7152):439-444.
16. Devroede G, Lemieux B, Masse S, Lamarche J and Herman PS. Colonic hamartomas in tuberous sclerosis. Gastroenterology. 1988; 94(1):182-188.
17. Zbuk KM and Eng C. Hamartomatous polyposis syndromes. Nature clinical practice Gastroenterology & hepatology. 2007; 4(9):492-502.
18. Reinke A, Chen JC, Aronova S and Powers T. Caffeine targets TOR complex I and provides evidence for a regulatory link between the FRB and kinase domains of Tor1p. The Journal of biological chemistry. 2006; 281(42):31616-31626.
19. Urano J, Sato T, Matsuo T, Otsubo Y, Yamamoto M and Tamanoi F. Point mutations in TOR confer Rheb-independent growth in fission yeast and nutrient-independent mammalian TOR signaling in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104(9):3514-3519.
20. Ohne Y, Takahara T, Hatakeyama R, Matsuzaki T, Noda M, Mizushima N and Maeda T. Isolation of hyperactive mutants of mammalian target of rapamycin. The Journal of biological chemistry. 2008; 283(46):31861-31870.
21. Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM and Abraham RT. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer research. 2000; 60(13):3504-3513.
22. Edinger AL and Thompson CB. An activated mTOR mutant supports growth factor-independent, nutrient-dependent cell survival. Oncogene. 2004; 23(33):5654-5663.
23. Sato T, Nakashima A, Guo L, Coffman K and Tamanoi F. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene. 2010; 29(18):2746-2752.
24. Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ and Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature. 2013; 497(7448):217-223.
25. Murugan AK, Alzahrani A and Xing M. Mutations in critical domains confer the human mTOR gene strong tumorigenicity. The Journal of biological chemistry. 2013; 288(9):6511-6521.
26. Lorenz MC and Heitman J. TOR mutations confer rapamycin resistance by preventing interaction with FKBP12-rapamycin. The Journal of biological chemistry. 1995; 270(46):27531-27537.
27. Iglesias-Bartolome R and Gutkind JS. Signaling circuitries controlling stem cell fate: to be or not to be. Current opinion in cell biology. 2011; 23(6):716-723.
28. Castilho RM, Squarize CH, Chodosh LA, Williams BO and Gutkind JS. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell stem cell. 2009; 5(3):279-289.
29. Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, Malide D, Rovira, II, Schimel D, Kuo CJ, Gutkind JS, Hwang PM and Finkel T. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007; 317(5839):803-806.
30. Iglesias-Bartolome R, Patel V, Cotrim A, Leelahavanichkul K, Molinolo AA, Mitchell JB and Gutkind JS. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell stem cell. 2012; 11(3):401-414.
31. Yang Z, Zhang L, Ma A, Liu L, Li J, Gu J and Liu Y. Transient mTOR inhibition facilitates continuous growth of liver tumors by modulating the maintenance of CD133+ cell populations. PloS one. 2011; 6(12):e28405.
32. Blagosklonny MV. Cell cycle arrest is not senescence. Aging. 2011; 3(2):94-101.
33. Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging. 2012; 4(3):159-165.
34. Galluzzi L, Kepp O and Kroemer G. TP53 and MTOR crosstalk to regulate cellular senescence. Aging. 2010; 2(9):535-537.
35. Iglesias-Bartolome R and Gutkind JS. Exploiting the mTOR paradox for disease prevention. Oncotarget. 2012.
36. Donehower LA. Rapamycin as longevity enhancer and cancer preventative agent in the context of p53 deficiency. Aging. 2012; 4(10):660-661.
37. Comas M, Toshkov I, Kuropatwinski KK, Chernova OB, Polinsky A, Blagosklonny MV, Gudkov AV and Antoch MP. New nanoformulation of rapamycin Rapatar extends lifespan in homozygous p53-/- mice by delaying carcinogenesis. Aging. 2012; 4(10):715-722.
38. Komarova EA, Antoch MP, Novototskaya LR, Chernova OB, Paszkiewicz G, Leontieva OV, Blagosklonny MV and Gudkov AV. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53+/- mice. Aging. 2012; 4(10):709-714.
39. Livi CB, Hardman RL, Christy BA, Dodds SG, Jones D, Williams C, Strong R, Bokov A, Javors MA, Ikeno Y, Hubbard G, Hasty P and Sharp ZD. Rapamycin extends life span of Rb1+/- mice by inhibiting neuroendocrine tumors. Aging. 2013; 5(2):100-110.
40. Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV and Blagosklonny MV. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109(33):13314-13318.
41. Mohyeldin A, Garzon-Muvdi T and Quinones-Hinojosa A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell stem cell. 2010; 7(2):150-161.
42. Heitman J, Movva NR and Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991; 253(5022):905-909.
43. Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, Bruns CJ, Zuelke C, Farkas S, Anthuber M, Jauch KW and Geissler EK. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nature medicine. 2002; 8(2):128-135.
44. Battelli C and Cho DC. mTOR inhibitors in renal cell carcinoma. Therapy. 2011; 8(4):359-367.
45. Mohseni M and Park BH. PIK3CA and KRAS mutations predict for response to everolimus therapy: now that’s RAD001. The Journal of clinical investigation. 2010; 120(8):2655-2658.
46. Fox CJ, Hammerman PS and Thompson CB. The Pim kinases control rapamycin-resistant T cell survival and activation. The Journal of experimental medicine. 2005; 201(2):259-266.
47. Neklesa TK and Davis RW. Superoxide anions regulate TORC1 and its ability to bind Fpr1:rapamycin complex. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105(39):15166-15171.
48. Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, Loda M, Lane HA and Sellers WR. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nature medicine. 2004; 10(6):594-601.
49. O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J and Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer research. 2006; 66(3):1500-1508.
50. Tabernero J, Rojo F, Calvo E, Burris H, Judson I, Hazell K, Martinelli E, Ramon y Cajal S, Jones S, Vidal L, Shand N, Macarulla T, Ramos FJ, Dimitrijevic S, Zoellner U, Tang P, et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008; 26(10):1603-1610.
51. Julien LA, Carriere A, Moreau J and Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Molecular and cellular biology. 2010; 30(4):908-921.
52. Carlson CJ, White MF and Rondinone CM. Mammalian target of rapamycin regulates IRS-1 serine 307 phosphorylation. Biochemical and biophysical research communications. 2004; 316(2):533-539.
53. Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, Kubica N, Hoffman GR, Cantley LC, Gygi SP and Blenis J. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011; 332(6035):1322-1326.
54. Zhou H, Luo Y and Huang S. Updates of mTOR inhibitors. Anti-cancer agents in medicinal chemistry. 2010; 10(7):571-581.
55. Guichard S. AZD2014, a dual mTORC1 and mTORC2 inhibitor is differentiated from allosteric inhibitors of mTORC1 in ER+ breast cancer. Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research. 2012; Cancer Res 2012;72(8 Suppl):Abstract nr 917.
56. Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini P, James D, Howard Z, Dudley P, Hughes G, Smith L, Maguire S, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer research. 2010; 70(1):288-298.
57. Shao H, Gao C, Tang H, Zhang H, Roberts LR, Hylander BL, Repasky EA, Ma WW, Qiu J, Adjei AA, Dy GK and Yu C. Dual targeting of mTORC1/C2 complexes enhances histone deacetylase inhibitor-mediated anti-tumor efficacy in primary HCC cancer in vitro and in vivo. Journal of hepatology. 2012; 56(1):176-183.
58. Lin SF, Huang YY, Lin JD, Chou TC, Hsueh C and Wong RJ. Utility of a PI3K/mTOR Inhibitor (NVP-BEZ235) for Thyroid Cancer Therapy. PloS one. 2012; 7(10):e46726.
59. Doghman M and Lalli E. Efficacy of the novel dual PI3-kinase/mTOR inhibitor NVP-BEZ235 in a preclinical model of adrenocortical carcinoma. Molecular and cellular endocrinology. 2012; 364(1-2):101-104.
60. Shoji K, Oda K, Kashiyama T, Ikeda Y, Nakagawa S, Sone K, Miyamoto Y, Hiraike H, Tanikawa M, Miyasaka A, Koso T, Matsumoto Y, Wada-Hiraike O, Kawana K, Kuramoto H, McCormick F, et al. Genotype-dependent efficacy of a dual PI3K/mTOR inhibitor, NVP-BEZ235, and an mTOR inhibitor, RAD001, in endometrial carcinomas. PloS one. 2012; 7(5):e37431.
61. Blaser B, Waselle L, Dormond-Meuwly A, Dufour M, Roulin D, Demartines N and Dormond O. Antitumor activities of ATP-competitive inhibitors of mTOR in colon cancer cells. BMC cancer. 2012; 12:86.
62. Garcia-Garcia C, Ibrahim YH, Serra V, Calvo MT, Guzman M, Grueso J, Aura C, Perez J, Jessen K, Liu Y, Rommel C, Tabernero J, Baselga J and Scaltriti M. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012; 18(9):2603-2612.
63. Garcia-Martinez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM and Alessi DR. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). The Biochemical journal. 2009; 421(1):29-42.
64. Liu Q, Thoreen C, Wang J, Sabatini D and Gray NS. mTOR Mediated Anti-Cancer Drug Discovery. Drug discovery today Therapeutic strategies. 2009; 6(2):47-55.
65. Bhagwat SV, Gokhale PC, Crew AP, Cooke A, Yao Y, Mantis C, Kahler J, Workman J, Bittner M, Dudkin L, Epstein DM, Gibson NW, Wild R, Arnold LD, Houghton PJ and Pachter JA. Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: distinct from rapamycin. Molecular cancer therapeutics. 2011; 10(8):1394-1406.
66. Raynaud FI, Eccles S, Clarke PA, Hayes A, Nutley B, Alix S, Henley A, Di-Stefano F, Ahmad Z, Guillard S, Bjerke LM, Kelland L, Valenti M, Patterson L, Gowan S, de Haven Brandon A, et al. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer research. 2007; 67(12):5840-5850.
67. Segerstrom L, Baryawno N, Sveinbjornsson B, Wickstrom M, Elfman L, Kogner P and Johnsen JI. Effects of small molecule inhibitors of PI3K/Akt/mTOR signaling on neuroblastoma growth in vitro and in vivo. International journal of cancer Journal international du cancer. 2011; 129(12):2958-2965.
68. Gedaly R, Angulo P, Chen C, Creasy KT, Spear BT, Hundley J, Daily MF, Shah M and Evers BM. The role of PI3K/mTOR inhibition in combination with sorafenib in hepatocellular carcinoma treatment. Anticancer research. 2012; 32(7):2531-2536.
69. Bhattacharya B, Akram M, Balasubramanian I, Tam KK, Koh KX, Yee MQ and Soong R. Pharmacologic synergy between dual phosphoinositide-3-kinase and mammalian target of rapamycin inhibition and 5-fluorouracil in PIK3CA mutant gastric cancer cells. Cancer biology & therapy. 2012; 13(1):34-42.
70. Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, Hoffman R, Williams RL, Shokat KM and Knight ZA. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nature chemical biology. 2008; 4(11):691-699.
71. Li H, Lin J, Wang X, Yao G, Wang L, Zheng H, Yang C, Jia C, Liu A and Bai X. Targeting of mTORC2 prevents cell migration and promotes apoptosis in breast cancer. Breast cancer research and treatment. 2012; 134(3):1057-1066.
72. Liu Q, Chang JW, Wang J, Kang SA, Thoreen CC, Markhard A, Hur W, Zhang J, Sim T, Sabatini DM and Gray NS. Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benz o[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. Journal of medicinal chemistry. 2010; 53(19):7146-7155.
73. Liu Q, Wang J, Kang SA, Thoreen CC, Hur W, Ahmed T, Sabatini DM and Gray NS. Discovery of 9-(6-aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h][1,6]naphthyridin-2( 1H)-one (Torin2) as a potent, selective, and orally available mammalian target of rapamycin (mTOR) inhibitor for treatment of cancer. Journal of medicinal chemistry. 2011; 54(5):1473-1480.
74. Benjamin D, Colombi M, Moroni C and Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nature reviews Drug discovery. 2011; 10(11):868-880.
75. Yu K, Toral-Barza L, Shi C, Zhang WG, Lucas J, Shor B, Kim J, Verheijen J, Curran K, Malwitz DJ, Cole DC, Ellingboe J, Ayral-Kaloustian S, Mansour TS, Gibbons JJ, Abraham RT, et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer research. 2009; 69(15):6232-6240.
76. Yu K, Shi C, Toral-Barza L, Lucas J, Shor B, Kim JE, Zhang WG, Mahoney R, Gaydos C, Tardio L, Kim SK, Conant R, Curran K, Kaplan J, Verheijen J, Ayral-Kaloustian S, et al. Beyond rapalog therapy: preclinical pharmacology and antitumor activity of WYE-125132, an ATP-competitive and specific inhibitor of mTORC1 and mTORC2. Cancer research. 2010; 70(2):621-631.
77. Prasad G, Sottero T, Yang X, Mueller S, James CD, Weiss WA, Polley MY, Ozawa T, Berger MS, Aftab DT, Prados MD and Haas-Kogan DA. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro-oncology. 2011; 13(4):384-392.
78. Mirzoeva OK, Hann B, Hom YK, Debnath J, Aftab D, Shokat K and Korn WM. Autophagy suppression promotes apoptotic cell death in response to inhibition of the PI3K-mTOR pathway in pancreatic adenocarcinoma. J Mol Med (Berl). 2011; 89(9):877-889.
79. Ghadimi MP, Lopez G, Torres KE, Belousov R, Young ED, Liu J, Brewer KJ, Hoffman A, Lusby K, Lazar AJ, Pollock RE and Lev D. Targeting the PI3K/mTOR axis, alone and in combination with autophagy blockade, for the treatment of malignant peripheral nerve sheath tumors. Molecular cancer therapeutics. 2012; 11(8):1758-1769.
80. Bhatt AP, Bhende PM, Sin SH, Roy D, Dittmer DP and Damania B. Dual inhibition of PI3K and mTOR inhibits autocrine and paracrine proliferative loops in PI3K/Akt/mTOR-addicted lymphomas. Blood. 2010; 115(22):4455-4463.
81. Cao P, Maira SM, Garcia-Echeverria C and Hedley DW. Activity of a novel, dual PI3-kinase/mTor inhibitor NVP-BEZ235 against primary human pancreatic cancers grown as orthotopic xenografts. British journal of cancer. 2009; 100(8):1267-1276.
82. Cho DC, Cohen MB, Panka DJ, Collins M, Ghebremichael M, Atkins MB, Signoretti S and Mier JW. The efficacy of the novel dual PI3-kinase/mTOR inhibitor NVP-BEZ235 compared with rapamycin in renal cell carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010; 16(14):3628-3638.
83. Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nature medicine. 2008; 14(12):1351-1356.
84. Faber AC, Li D, Song Y, Liang MC, Yeap BY, Bronson RT, Lifshits E, Chen Z, Maira SM, Garcia-Echeverria C, Wong KK and Engelman JA. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106(46):19503-19508.
85. Garcia-Martinez JM, Wullschleger S, Preston G, Guichard S, Fleming S, Alessi DR and Duce SL. Effect of PI3K- and mTOR-specific inhibitors on spontaneous B-cell follicular lymphomas in PTEN/LKB1-deficient mice. British journal of cancer. 2011; 104(7):1116-1125.
86. Giubellino A, Bullova P, Nolting S, Turkova H, Powers JF, Liu Q, Guichard S, Tischler AS, Grossman AB and Pacak K. Combined inhibition of mTORC1 and mTORC2 signaling pathways is a promising therapeutic option in inhibiting pheochromocytoma tumor growth: in vitro and in vivo studies in female athymic nude mice. Endocrinology. 2013; 154(2):646-655.
87. Konstantinidou G, Bey EA, Rabellino A, Schuster K, Maira MS, Gazdar AF, Amici A, Boothman DA and Scaglioni PP. Dual phosphoinositide 3-kinase/mammalian target of rapamycin blockade is an effective radiosensitizing strategy for the treatment of non-small cell lung cancer harboring K-RAS mutations. Cancer research. 2009; 69(19):7644-7652.
88. Liu TJ, Koul D, LaFortune T, Tiao N, Shen RJ, Maira SM, Garcia-Echevrria C and Yung WK. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Molecular cancer therapeutics. 2009; 8(8):2204-2210.
89. Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Molecular cancer therapeutics. 2008; 7(7):1851-1863.
90. Manara MC, Nicoletti G, Zambelli D, Ventura S, Guerzoni C, Landuzzi L, Lollini PL, Maira SM, Garcia-Echeverria C, Mercuri M, Picci P and Scotlandi K. NVP-BEZ235 as a new therapeutic option for sarcomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010; 16(2):530-540.
91. McMillin DW, Ooi M, Delmore J, Negri J, Hayden P, Mitsiades N, Jakubikova J, Maira SM, Garcia-Echeverria C, Schlossman R, Munshi NC, Richardson PG, Anderson KC and Mitsiades CS. Antimyeloma activity of the orally bioavailable dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Cancer research. 2009; 69(14):5835-5842.
92. Nanni P, Nicoletti G, Landuzzi L, Croci S, Murgo A, Palladini A, Antognoli A, Ianzano ML, Stivani V, Grosso V, Maira SM, Garcia-Echeverria C, Scotlandi K, De Giovanni C and Lollini PL. High metastatic efficiency of human sarcoma cells in Rag2/gammac double knockout mice provides a powerful test system for antimetastatic targeted therapy. Eur J Cancer. 2010; 46(3):659-668.
93. Pollizzi K, Malinowska-Kolodziej I, Stumm M, Lane H and Kwiatkowski D. Equivalent benefit of mTORC1 blockade and combined PI3K-mTOR blockade in a mouse model of tuberous sclerosis. Molecular cancer. 2009; 8:38.
94. Renshaw J, Taylor KR, Bishop R, Valenti M, De Haven Brandon A, Gowan S, Eccles SA, Ruddle R, Johnson LD, Raynaud FI, Selfe J, Thway K, Pietsch T, Pearson AD and Shipley J. Dual blockade of the PI3K/AKT/mTOR (AZD8055) and RAS/MEK/ERK (AZD6244) pathways synergistically inhibits rhabdomyosarcoma cell growth in vitro and in vivo. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013.
95. Schnell CR, Stauffer F, Allegrini PR, O’Reilly T, McSheehy PM, Dartois C, Stumm M, Cozens R, Littlewood-Evans A, Garcia-Echeverria C and Maira SM. Effects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 on the tumor vasculature: implications for clinical imaging. Cancer research. 2008; 68(16):6598-6607.
96. Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, Maira M, Garcia-Echeverria C, Parra JL, Arribas J and Baselga J. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer research. 2008; 68(19):8022-8030.
97. Willems L, Chapuis N, Puissant A, Maciel TT, Green AS, Jacque N, Vignon C, Park S, Guichard S, Herault O, Fricot A, Hermine O, Moura IC, Auberger P, Ifrah N, Dreyfus F, et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia. 2012; 26(6):1195-1202.
98. Bhende PM, Park SI, Lim MS, Dittmer DP and Damania B. The dual PI3K/mTOR inhibitor, NVP-BEZ235, is efficacious against follicular lymphoma. Leukemia. 2010; 24(10):1781-1784.
99. Sunayama J, Matsuda K, Sato A, Tachibana K, Suzuki K, Narita Y, Shibui S, Sakurada K, Kayama T, Tomiyama A and Kitanaka C. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells. 2010; 28(11):1930-1939.
100. Sunayama J, Sato A, Matsuda K, Tachibana K, Suzuki K, Narita Y, Shibui S, Sakurada K, Kayama T, Tomiyama A and Kitanaka C. Dual blocking of mTor and PI3K elicits a prodifferentiation effect on glioblastoma stem-like cells. Neuro-oncology. 2010; 12(12):1205-1219.
101. Buonamici S, Williams J, Morrissey M, Wang A, Guo R, Vattay A, Hsiao K, Yuan J, Green J, Ospina B, Yu Q, Ostrom L, Fordjour P, Anderson DL, Monahan JE, Kelleher JF, et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Science translational medicine. 2010; 2(51):51ra70.
102. Fan QW, Cheng C, Hackett C, Feldman M, Houseman BT, Nicolaides T, Haas-Kogan D, James CD, Oakes SA, Debnath J, Shokat KM and Weiss WA. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Science signaling. 2010; 3(147):ra81.
103. Dubrovska A, Elliott J, Salamone RJ, Kim S, Aimone LJ, Walker JR, Watson J, Sauveur-Michel M, Garcia-Echeverria C, Cho CY, Reddy VA and Schultz PG. Combination therapy targeting both tumor-initiating and differentiated cell populations in prostate carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010; 16(23):5692-5702.
104. Santiskulvong C, Konecny GE, Fekete M, Chen KY, Karam A, Mulholland D, Eng C, Wu H, Song M and Dorigo O. Dual targeting of phosphoinositide 3-kinase and mammalian target of rapamycin using NVP-BEZ235 as a novel therapeutic approach in human ovarian carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011; 17(8):2373-2384.
105. Dormond-Meuwly A, Roulin D, Dufour M, Benoit M, Demartines N and Dormond O. The inhibition of MAPK potentiates the anti-angiogenic efficacy of mTOR inhibitors. Biochemical and biophysical research communications. 2011; 407(4):714-719.
106. Xu CX, Li Y, Yue P, Owonikoko TK, Ramalingam SS, Khuri FR and Sun SY. The combination of RAD001 and NVP-BEZ235 exerts synergistic anticancer activity against non-small cell lung cancer in vitro and in vivo. PloS one. 2011; 6(6):e20899.
107. Fuereder T, Wanek T, Pflegerl P, Jaeger-Lansky A, Hoeflmayer D, Strommer S, Kuntner C, Wrba F, Werzowa J, Hejna M, Muller M, Langer O and Wacheck V. Gastric cancer growth control by BEZ235 in vivo does not correlate with PI3K/mTOR target inhibition but with [18F]FLT uptake. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011; 17(16):5322-5332.
108. Masuda M, Shimomura M, Kobayashi K, Kojima S and Nakatsura T. Growth inhibition by NVP-BEZ235, a dual PI3K/mTOR inhibitor, in hepatocellular carcinoma cell lines. Oncology reports. 2011; 26(5):1273-1279.
109. Xu CX, Zhao L, Yue P, Fang G, Tao H, Owonikoko TK, Ramalingam SS, Khuri FR and Sun SY. Augmentation of NVP-BEZ235’s anticancer activity against human lung cancer cells by blockage of autophagy. Cancer biology & therapy. 2011; 12(6):549-555.
110. Roulin D, Waselle L, Dormond-Meuwly A, Dufour M, Demartines N and Dormond O. Targeting renal cell carcinoma with NVP-BEZ235, a dual PI3K/mTOR inhibitor, in combination with sorafenib. Molecular cancer. 2011; 10:90.
111. Jin N, Jiang T, Rosen DM, Nelkin BD and Ball DW. Synergistic action of a RAF inhibitor and a dual PI3K/mTOR inhibitor in thyroid cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011; 17(20):6482-6489.
112. Roper J, Richardson MP, Wang WV, Richard LG, Chen W, Coffee EM, Sinnamon MJ, Lee L, Chen PC, Bronson RT, Martin ES and Hung KE. The dual PI3K/mTOR inhibitor NVP-BEZ235 induces tumor regression in a genetically engineered mouse model of PIK3CA wild-type colorectal cancer. PloS one. 2011; 6(9):e25132.
113. Awasthi N, Yen PL, Schwarz MA and Schwarz RE. The efficacy of a novel, dual PI3K/mTOR inhibitor NVP-BEZ235 to enhance chemotherapy and antiangiogenic response in pancreatic cancer. Journal of cellular biochemistry. 2012; 113(3):784-791.
114. Gallagher EJ, Fierz Y, Vijayakumar A, Haddad N, Yakar S and LeRoith D. Inhibiting PI3K reduces mammary tumor growth and induces hyperglycemia in a mouse model of insulin resistance and hyperinsulinemia. Oncogene. 2012; 31(27):3213-3222.
115. Acquaviva J, Smith DL, Sang J, Friedland JC, He S, Sequeira M, Zhang C, Wada Y and Proia DA. Targeting KRAS-mutant non-small cell lung cancer with the Hsp90 inhibitor ganetespib. Molecular cancer therapeutics. 2012; 11(12):2633-2643.
116. Issa A, Gill JW, Heideman MR, Sahin O, Wiemann S, Dey JH and Hynes NE. Combinatorial targeting of FGF and ErbB receptors blocks growth and metastatic spread of breast cancer models. Breast cancer research : BCR. 2013; 15(1):R8.
117. Kim A, Lee JE, Lee SS, Kim C, Lee SJ, Jang WS and Park S. Coexistent mutations of KRAS and PIK3CA affect the efficacy of NVP-BEZ235, a dual PI3K/MTOR inhibitor, in regulating the PI3K/MTOR pathway in colorectal cancer. International journal of cancer Journal international du cancer. 2013; 133(4):984-996.
118. Nyfeler B, Chen Y, Li X, Pinzon-Ortiz M, Wang Z, Reddy A, Pradhan E, Das R, Lehar J, Schlegel R, Finan PM, Cao ZA, Murphy LO and Huang A. RAD001 enhances the potency of BEZ235 to inhibit mTOR signaling and tumor growth. PloS one. 2012; 7(11):e48548.
119. Potiron VA, Abderrahmani R, Giang E, Chiavassa S, Di Tomaso E, Maira SM, Paris F and Supiot S. Radiosensitization of prostate cancer cells by the dual PI3K/mTOR inhibitor BEZ235 under normoxic and hypoxic conditions. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 2013; 106(1):138-146.
120. Rahmani M, Aust MM, Attkisson E, Williams DC, Jr., Ferreira-Gonzalez A and Grant S. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. Cancer research. 2013; 73(4):1340-1351.
121. Roberts PJ, Usary JE, Darr DB, Dillon PM, Pfefferle AD, Whittle MC, Duncan JS, Johnson SM, Combest AJ, Jin J, Zamboni WC, Johnson GL, Perou CM and Sharpless NE. Combined PI3K/mTOR and MEK inhibition provides broad antitumor activity in faithful murine cancer models. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012; 18(19):5290-5303.
122. Sano T, Takeuchi S, Nakagawa T, Ishikawa D, Nanjo S, Yamada T, Nakamura T, Matsumoto K and Yano S. The novel phosphoinositide 3-kinase-mammalian target of rapamycin inhibitor, BEZ235, circumvents erlotinib resistance of epidermal growth factor receptor mutant lung cancer cells triggered by hepatocyte growth factor. International journal of cancer Journal international du cancer. 2013; 133(2):505-513.
123. Venkannagari S, Fiskus W, Peth K, Atadja P, Hidalgo M, Maitra A and Bhalla KN. Superior efficacy of co-treatment with dual PI3K/mTOR inhibitor NVP-BEZ235 and pan-histone deacetylase inhibitor against human pancreatic cancer. Oncotarget. 2012; 3(11):1416-1427.
124. Yang F, Qian XJ, Qin W, Deng R, Wu XQ, Qin J, Feng GK and Zhu XF. Dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 has a therapeutic potential and sensitizes cisplatin in nasopharyngeal carcinoma. PloS one. 2013; 8(3):e59879.
125. Yasumizu Y, Miyajima A, Kosaka T, Miyazaki Y, Kikuchi E and Oya M. Dual Phosphatidylinositol-3-Kinase/Mammalian Target of Rapamycin Inhibitor NVP-BEZ235 Sensitizes Docetaxel in Castration Resistant Prostate Cancer. The Journal of urology. 2013.
126. Cassell A, Freilino ML, Lee J, Barr S, Wang L, Panahandeh MC, Thomas SM and Grandis JR. Targeting TORC1/2 enhances sensitivity to EGFR inhibitors in head and neck cancer preclinical models. Neoplasia. 2012; 14(11):1005-1014.
127. Dai C, Zhang B, Liu X, Ma S, Yang Y, Yao Y, Feng M, Bao X, Li G, Wang J, Guo K, Ma W, Xing B, Lian W, Xiao J, Cai F, et al. Inhibition of PI3K/AKT/mTOR pathway enhances temozolomide-induced cytotoxicity in pituitary adenoma cell lines in vitro and xenografted pituitary adenoma in female nude mice. Endocrinology. 2013; 154(3):1247-1259.
128. Falcon BL, Barr S, Gokhale PC, Chou J, Fogarty J, Depeille P, Miglarese M, Epstein DM and McDonald DM. Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer research. 2011; 71(5):1573-1583.
129. Wang Q, Wei F, Li C, Lv G, Wang G, Liu T, Bellail AC and Hao C. Combination of mTOR and EGFR Kinase Inhibitors Blocks mTORC1 and mTORC2 Kinase Activity and Suppresses the Progression of Colorectal Carcinoma. PloS one. 2013; 8(8):e73175.
130. Zhang H, Berel D, Wang Y, Li P, Bhowmick NA, Figlin RA and Kim HL. A comparison of Ku0063794, a dual mTORC1 and mTORC2 inhibitor, and temsirolimus in preclinical renal cell carcinoma models. PloS one. 2013; 8(1):e54918.
131. Hayman TJ, Kramp T, Kahn J, Jamal M, Camphausen K and Tofilon PJ. Competitive but Not Allosteric mTOR Kinase Inhibition Enhances Tumor Cell Radiosensitivity. Translational oncology. 2013; 6(3):355-362.
132. Ducker GS, Atreya CE, Simko JP, Hom YK, Matli MR, Benes CH, Hann B, Nakakura EK, Bergsland EK, Donner DB, Settleman J, Shokat KM and Warren RS. Incomplete inhibition of phosphorylation of 4E-BP1 as a mechanism of primary resistance to ATP-competitive mTOR inhibitors. Oncogene. 2013.
133. Frost P, Berlanger E, Mysore V, Hoang B, Shi Y, Gera J and Lichtenstein A. Mammalian target of rapamycin inhibitors induce tumor cell apoptosis in vivo primarily by inhibiting VEGF expression and angiogenesis. Journal of oncology. 2013; 2013:897025.
134. Hoang B, Frost P, Shi Y, Belanger E, Benavides A, Pezeshkpour G, Cappia S, Guglielmelli T, Gera J and Lichtenstein A. Targeting TORC2 in multiple myeloma with a new mTOR kinase inhibitor. Blood. 2010; 116(22):4560-4568.
135. Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, Vu C, Lilly MB, Mallya S, Ong ST, Konopleva M, Martin MB, Ren P, Liu Y, Rommel C and Fruman DA. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nature medicine. 2010; 16(2):205-213.
136. Bagci-Onder T, Wakimoto H, Anderegg M, Cameron C and Shah K. A dual PI3K/mTOR inhibitor, PI-103, cooperates with stem cell-delivered TRAIL in experimental glioma models. Cancer research. 2011; 71(1):154-163.
137. Bender A, Opel D, Naumann I, Kappler R, Friedman L, von Schweinitz D, Debatin KM and Fulda S. PI3K inhibitors prime neuroblastoma cells for chemotherapy by shifting the balance towards pro-apoptotic Bcl-2 proteins and enhanced mitochondrial apoptosis. Oncogene. 2011; 30(4):494-503.
138. Chaisuparat R, Hu J, Jham BC, Knight ZA, Shokat KM and Montaner S. Dual inhibition of PI3Kalpha and mTOR as an alternative treatment for Kaposi’s sarcoma. Cancer research. 2008; 68(20):8361-8368.
139. Chen JS, Zhou LJ, Entin-Meer M, Yang X, Donker M, Knight ZA, Weiss W, Shokat KM, Haas-Kogan D and Stokoe D. Characterization of structurally distinct, isoform-selective phosphoinositide 3’-kinase inhibitors in combination with radiation in the treatment of glioblastoma. Molecular cancer therapeutics. 2008; 7(4):841-850.
140. Donev IS, Wang W, Yamada T, Li Q, Takeuchi S, Matsumoto K, Yamori T, Nishioka Y, Sone S and Yano S. Transient PI3K inhibition induces apoptosis and overcomes HGF-mediated resistance to EGFR-TKIs in EGFR mutant lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011; 17(8):2260-2269.
141. Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D, Shokat KM and Weiss WA. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer cell. 2006; 9(5):341-349.
142. Guillard S, Clarke PA, Te Poele R, Mohri Z, Bjerke L, Valenti M, Raynaud F, Eccles SA and Workman P. Molecular pharmacology of phosphatidylinositol 3-kinase inhibition in human glioma. Cell Cycle. 2009; 8(3):443-453.
143. Kharas MG, Janes MR, Scarfone VM, Lilly MB, Knight ZA, Shokat KM and Fruman DA. Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor prevents expansion of human BCR-ABL+ leukemia cells. The Journal of clinical investigation. 2008; 118(9):3038-3050.
144. Werzowa J, Koehrer S, Strommer S, Cejka D, Fuereder T, Zebedin E and Wacheck V. Vertical inhibition of the mTORC1/mTORC2/PI3K pathway shows synergistic effects against melanoma in vitro and in vivo. The Journal of investigative dermatology. 2011; 131(2):495-503.
145. Opel D, Naumann I, Schneider M, Bertele D, Debatin KM and Fulda S. Targeting aberrant PI3K/Akt activation by PI103 restores sensitivity to TRAIL-induced apoptosis in neuroblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011; 17(10):3233-3247.
146. Gupta M, Hendrickson AE, Yun SS, Han JJ, Schneider PA, Koh BD, Stenson MJ, Wellik LE, Shing JC, Peterson KL, Flatten KS, Hess AD, Smith BD, Karp JE, Barr S, Witzig TE, et al. Dual mTORC1/mTORC2 inhibition diminishes Akt activation and induces Puma-dependent apoptosis in lymphoid malignancies. Blood. 2012; 119(2):476-487.
147. Marone R, Erhart D, Mertz AC, Bohnacker T, Schnell C, Cmiljanovic V, Stauffer F, Garcia-Echeverria C, Giese B, Maira SM and Wymann MP. Targeting melanoma with dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitors. Molecular cancer research : MCR. 2009; 7(4):601-613.
148. Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, Poulikakos PI, Scaltriti M, Moskatel E, Baselga J, Guichard S and Rosen N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer discovery. 2011; 1(3):248-259.
149. Zhuang G, Yu K, Jiang Z, Chung A, Yao J, Ha C, Toy K, Soriano R, Haley B, Blackwood E, Sampath D, Bais C, Lill JR and Ferrara N. Phosphoproteomic analysis implicates the mTORC2-FoxO1 axis in VEGF signaling and feedback activation of receptor tyrosine kinases. Science signaling. 2013; 6(271):ra25.
150. Xue Q, Hopkins B, Perruzzi C, Udayakumar D, Sherris D and Benjamin LE. Palomid 529, a novel small-molecule drug, is a TORC1/TORC2 inhibitor that reduces tumor growth, tumor angiogenesis, and vascular permeability. Cancer research. 2008; 68(22):9551-9557.
151. Diaz R, Nguewa PA, Diaz-Gonzalez JA, Hamel E, Gonzalez-Moreno O, Catena R, Serrano D, Redrado M, Sherris D and Calvo A. The novel Akt inhibitor Palomid 529 (P529) enhances the effect of radiotherapy in prostate cancer. British journal of cancer. 2009; 100(6):932-940.
152. Gravina GL, Marampon F, Petini F, Biordi L, Sherris D, Jannini EA, Tombolini V and Festuccia C. The TORC1/TORC2 inhibitor, Palomid 529, reduces tumor growth and sensitizes to docetaxel and cisplatin in aggressive and hormone-refractory prostate cancer cells. Endocrine-related cancer. 2011; 18(4):385-400.
153. Syed F, Sherris D, Paus R, Varmeh S, Pandolfi PP and Bayat A. Keloid disease can be inhibited by antagonizing excessive mTOR signaling with a novel dual TORC1/2 inhibitor. The American journal of pathology. 2012; 181(5):1642-1658.
154. Yilmaz OH, Katajisto P, Lamming DW, Gultekin Y, Bauer-Rowe KE, Sengupta S, Birsoy K, Dursun A, Yilmaz VO, Selig M, Nielsen GP, Mino-Kenudson M, Zukerberg LR, Bhan AK, Deshpande V and Sabatini DM. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature. 2012; 486(7404):490-495.
155. Aoki K, Tamai Y, Horiike S, Oshima M and Taketo MM. Colonic polyposis caused by mTOR-mediated chromosomal instability in Apc+/Delta716 Cdx2+/- compound mutant mice. Nature genetics. 2003; 35(4):323-330.
156. Zhang YJ, Dai Q, Sun DF, Xiong H, Tian XQ, Gao FH, Xu MH, Chen GQ, Han ZG and Fang JY. mTOR signaling pathway is a target for the treatment of colorectal cancer. Annals of surgical oncology. 2009; 16(9):2617-2628.
157. Fujishita T, Aoki K, Lane HA, Aoki M and Taketo MM. Inhibition of the mTORC1 pathway suppresses intestinal polyp formation and reduces mortality in ApcDelta716 mice. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105(36):13544-13549.
158. Shao J, Evers BM and Sheng H. Roles of phosphatidylinositol 3’-kinase and mammalian target of rapamycin/p70 ribosomal protein S6 kinase in K-Ras-mediated transformation of intestinal epithelial cells. Cancer research. 2004; 64(1):229-235.
159. Roulin D, Cerantola Y, Dormond-Meuwly A, Demartines N and Dormond O. Targeting mTORC2 inhibits colon cancer cell proliferation in vitro and tumor formation in vivo. Molecular cancer. 2010; 9:57.
160. Gulhati P, Cai Q, Li J, Liu J, Rychahou PG, Qiu S, Lee EY, Silva SR, Bowen KA, Gao T and Evers BM. Targeted inhibition of mammalian target of rapamycin signaling inhibits tumorigenesis of colorectal cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009; 15(23):7207-7216.
161. Gulhati P, Bowen KA, Liu J, Stevens PD, Rychahou PG, Chen M, Lee EY, Weiss HL, O’Connor KL, Gao T and Evers BM. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer research. 2011; 71(9):3246-3256.
162. Zhang Y and Zheng XF. mTOR-independent 4E-BP1 phosphorylation is associated with cancer resistance to mTOR kinase inhibitors. Cell Cycle. 2012; 11(3):594-603.
163. Alain T, Morita M, Fonseca BD, Yanagiya A, Siddiqui N, Bhat M, Zammit D, Marcus V, Metrakos P, Voyer LA, Gandin V, Liu Y, Topisirovic I and Sonenberg N. eIF4E/4E-BP ratio predicts the efficacy of mTOR targeted therapies. Cancer research. 2012; 72(24):6468-6476.
164. Tan J, Lee PL, Li Z, Jiang X, Lim YC, Hooi SC and Yu Q. B55beta-associated PP2A complex controls PDK1-directed myc signaling and modulates rapamycin sensitivity in colorectal cancer. Cancer cell. 2010; 18(5):459-471.
165. Tan J, Li Z, Lee PL, Guan P, Aau M, Lee ST, Feng M, Lim CZ, Lee EY, Wee ZN, Lim YC, Karuturi RK and Yu Q. PDK1 Signaling Towards PLK1-Myc Activation Confers Oncogenic Transformation and Tumor Initiating Cell Activation and Resistance to mTOR-targeted Therapy. Cancer discovery. 2013.
166. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976; 194(4260):23-28.
167. Wicha MS, Liu S and Dontu G. Cancer stem cells: an old idea--a paradigm shift. Cancer research. 2006; 66(4):1883-1890; discussion 1895-1886.
168. Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ and Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009; 457(7229):608-611.
169. Odoux C, Fohrer H, Hoppo T, Guzik L, Stolz DB, Lewis DW, Gollin SM, Gamblin TC, Geller DA and Lagasse E. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer research. 2008; 68(17):6932-6941.
170. Cai Z, Ke J, He X, Yuan R, Chen Y, Wu X, Wang L, Wang J, Lan P and Wu X. Significance of mTOR Signaling and Its Inhibitor Against Cancer Stem-Like Cells in Colorectal Cancer. Annals of surgical oncology. 2013.
171. Francipane MG and Lagasse E. Selective targeting of human colon cancer stem-like cells by the mTOR inhibitor Torin-1. Oncotarget. 2013; 4(11):1948-1962.
172. Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, Hennessy BT, Tseng H, Pochanard P, Kim SY, Dunn IF, Schinzel AC, Sandy P, Hoersch S, Sheng Q, Gupta PB, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer cell. 2009; 16(1):21-32.
173. Day FL, Jorissen RN, Lipton L, Mouradov D, Sakthianandeswaren A, Christie M, Li S, Tsui C, Tie J, Desai J, Xu ZZ, Molloy P, Whitehall V, Leggett BA, Jones IT, McLaughlin S, et al. PIK3CA and PTEN gene and exon mutation-specific clinicopathologic and molecular associations in colorectal cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013; 19(12):3285-3296.
174. Francipane MG and Lagasse E. Genetic Instability and Epigenetic Diversity of Cancer: A Focus On Colorectal Cancer. In: “Cancer Targeted Drug Delivery: Elusive Dream” (YH Bae et al, eds) Springer 2013.
175. Francipane MG, Chandler J and Lagasse E. Cancer Stem Cells: A Moving Target. Current pathobiology reports. 2013; 1(2):111-118.
176. Komori J, Boone L, DeWard A, Hoppo T and Lagasse E. The mouse lymph node as an ectopic transplantation site for multiple tissues. Nature biotechnology. 2012; 30(10):976-983.
177. Ma Q and Lu AY. Pharmacogenetics, pharmacogenomics, and individualized medicine. Pharmacological reviews. 2011; 63(2):437-459.
178. Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN and Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001; 293(5531):876-880.
179. Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, Leversha MA, Jeffrey PD, Desantis D, Singer S, Brennan MF, Maki RG and DeMatteo RP. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005; 11(11):4182-4190.
180. Chen J, Zheng XF, Brown EJ and Schreiber SL. Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proceedings of the National Academy of Sciences of the United States of America. 1995; 92(11):4947-4951.