
INTRODUCTION
Peripheral T-cell lymphomas (PTCLs) are aggressive non-Hodgkin lymphomas of mature T-cell origin that demonstrate marked clinical, pathological, and molecular heterogeneity, with over 20 subtypes currently recognized by the World Health Organization [1]. Outcomes generally are poor following standard combination chemotherapy regimens, most commonly cyclophosphamide, doxorubicin (hydroxydaunorubicin), Oncovin (vincristine), and prednisone (CHOP) [2]. Although these data indicate a pressing need for new therapeutic approaches in PTCL, attempts to improve outcomes using alternative chemotherapy regimens have been disappointing. Targeted therapies offer promise, but data on drug and patient selection are limited [3]. Therefore, the identification and validation of candidate therapeutic targets in PTCL is critical to improving outcomes in this disease.
Retinoic acid receptor alpha (RARA) is a transcription factor that forms heterodimers with retinoid X receptor (RXR) [4]. These heterodimers bind to DNA motifs known as retinoic acid response elements (RAREs) and regulate gene transcription upon interaction with the natural ligand, retinoic acid, resulting in the regulation of genes involved in cellular growth and differentiation. Although it was originally thought that ligand-binding to RARA resulted in transcriptional activation, chromatin immunoprecipitation-sequencing and transcriptome profiling have revealed roles for RARA as both a repressor and activator of transcription [5, 6]. Retinoic acid has demonstrated anti-proliferative effects in many tumor models, and as such, retinoic acid receptors (RARs) have been targeted therapeutically through the use of natural and synthetic retinoids.
Retinoid therapy has been used most notably for the treatment of acute promyelocytic leukemia, a myeloid neoplasm expressing RARA fusion proteins [4, 7]. In addition, retinoids have been used effectively in some cutaneous T-cell lymphomas (CTCLs), a group of mature T-cell lymphomas originating in the skin, for which the synthetic RXR retinoid, bexarotene, is approved by the United States Food and Drug Administration (FDA) as a second-line therapy [8]. Response rates to RAR and RXR retinoids as monotherapy in CTCL are around 50% [9, 10]. Though experience remains limited, occasional partial or complete responses to retinoids also have been observed in relapsed/refractory systemic PTCLs [11, 12]. However, the mechanism(s) of action of retinoids in PTCL, the specific role of RARA, and a means to identify patients most likely to respond to retinoids are unknown.
Individualized medicine approaches have been employed to use the results of high-throughput sequencing to identify drug-target combinations specific for each patient. Recently, we evaluated a patient with PTCL in the Mayo Clinic Center for Individualized Medicine, whose tumor bore a non-synonymous somatic mutation, RARAR394Q, in the ligand-binding region of the RARA gene (non-synonymous mutations summarized in Supplementary Table 1). Since this mutation had not been previously reported and the role of RARA in PTCL had not been characterized, we investigated the role of RARA in the growth and chemosensitivity to retinoids in T-cell lymphoma cells.
RESULTS
Wild-type and mutant RARA proteins drive T-cell lymphoma cell growth
To investigate the role of wild-type RARA (RARAwt) and RARAR394Q, we utilized three mature T-cell lymphoma cell lines (see Materials and Methods) with varied native RARA expression: one RARAhigh cell line (Mac-1) and two RARAlow cell lines (Karpas 299 and HuT78; Figure 1A). We used the two RARAlow cell lines to examine the effects of overexpressing RARAwt or RARAR394Q on cell growth, compared to an empty-vector control (pCI). RARAwt increased growth of Karpas 299 by 22% (P < 0.001) and of HuT78 by 36% (P < 0.001), while RARAR394Q increased growth of Karpas 299 by 36% (P < 0.001) and of HuT78 by 42% (P < 0.001; Figure 1B). The difference in the increase in growth between RARAR394Q and RARAwt was statistically significant in Karpas 299 (P = 0.04) but not in HuT78 (P = 0.17). Because both RARAR394Q and RARAwt increased cell growth but the R394Q mutation conferred only a mild growth advantage over wild-type, we focused our efforts preferentially on understanding the growth-promoting role of RARA in general, rather than characterizing the specific effects of the R394Q mutation on RARA function. In keeping with the growth-promoting role of RARA, siRNA knockdown of RARA in RARAhigh Mac-1 cells resulted in a 22% inhibition of cell growth (P = 0.0002; Figure 1C).
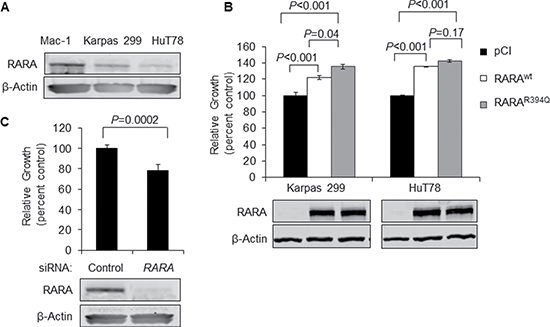
Figure 1: Overexpression of RARAwt or RARAR394Q drives T-cell lymphoma cell growth. (A) Native RARA is expressed strongly in Mac-1 and to a lesser degree in Karpas 299 and HuT78 cell lines. (B) Cell growth is increased upon overexpression of RARAwt or RARAR394Q in Karpas 299 and HuT78 cell lines with low native RARA expression. (C) Knockdown of RARA inhibits cell growth in Mac-1 cells with high native RARA expression. RARA, retinoic acid receptor alpha; wt, wild-type; siRNA, small interfering RNA.
RARA drives cyclin-dependent kinase expression and G1-S transition in T-cell lymphoma cells
Having identified a role for RARA in driving T-cell lymphoma cell growth, we next examined the effect of RARA on the cell cycle. siRNA knockdown of RARA in RARAhigh Mac-1 cells resulted accumulation of cells in G1 (120% of control, P = 0.004), with corresponding decreases in the fractions of cells in S-phase and G2/M (P = 0.02; Figure 2A). To explore this finding further, we evaluated the expression of the cyclin-dependent kinases (CDKs), CDK6, CDK4, and CDK2, which are involved in the regulation of the G1-S transition [13]. Indeed, RARA knockdown in Mac-1 cells inhibited CDK6, CDK4, and CDK2 protein expression by 65%, 32%, and 14%, respectively (Figure 2B). Correspondingly, overexpression of RARAwt increased CDK6, CDK4, and CDK2 protein expression by 52%, 39%, and 39% respectively; overexpression of RARAR394Q caused similar increases in CDK expression (60%, 30%, and 42% respectively; Figure 2C).
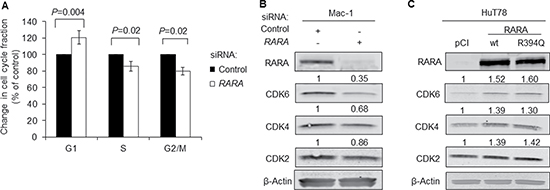
Figure 2: RARA drives expression of cyclin-dependent kinases. (A) Knockdown of RARA causes G1 cell cycle arrest (P = 0.004) in Mac-1 cells. (B) Expression of the regulators of cell cycle progression, CDK6, CDK4, and to a lesser extent, CDK2, is inhibited by RARA knockdown in Mac-1 cells. (C) Expression of CDK6, CDK4, and CDK2 is increased following overexpression of RARAwt and RARAR394Q in HuT78 cells. RARA, retinoic acid receptor alpha; wt, wild-type; siRNA, small interfering RNA; CDK, cyclin-dependent kinase.
Retinoids cause RARA degradation and cell-cycle arrest in T-cell lymphoma cells
Because we showed that RARA drove T-cell lymphoma cell growth and cell-cycle progression, we next examined the ability of retinoids to reverse these effects. We evaluated the activity of two retinoids that act as ligands for RARA. All-trans retinoic acid (ATRA) is a ligand for all RARs [14], while the synthetic retinoid, AM80 (4-[(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]benzoic acid or tamibarotene), preferentially targets RARA and retinoic acid receptor beta (RARB). However, RARB is not expressed in the cell lines used in this study (Supplementary Figure 1). Therefore, we used AM80 as a relatively specific RARA ligand and ATRA as a less specific ligand also predicted to target retinoic acid receptor gamma (RARG), which is expressed in our cell lines, albeit at somewhat lower levels than RARA. In addition, we evaluated the ability of the RXR ligand, bexarotene, to target RARA because of its clinical application in cutaneous T-cell lymphoma.
Treatment of T-cell lymphoma cell lines with AM80, ATRA, or bexarotene resulted in dose-dependent inhibition of cell growth (Figures 3A, 3B and Supplementary Figure 3A). Importantly, the RARAhigh cell line, Mac-1, was most chemosensitive to retinoids; the RARAlow cell lines, Karpas 299 and HuT78, were less chemosensitive, with the cell line expressing the lowest level of RARA protein, HuT78, demonstrating the least chemosensitivity. Cell-cycle analysis revealed dose-dependent G1 arrest following treatment with AM80 or ATRA (Figure 3C–3E). Again, these effects were most pronounced in the RARAhigh cell line, Mac-1. RARA ligands previously have been shown to lead to RARA degradation through the ubiquitin-proteasome pathway [15]. Consistent with this known process, RARA protein was markedly reduced after treatment with AM80, ATRA, or bexarotene (Figure 3F, 3G and Supplementary Figure 3B). Furthermore, treatment with either AM80 or ATRA resulted in dose-dependent decreases in the expression of CDK4 and CDK6 proteins.
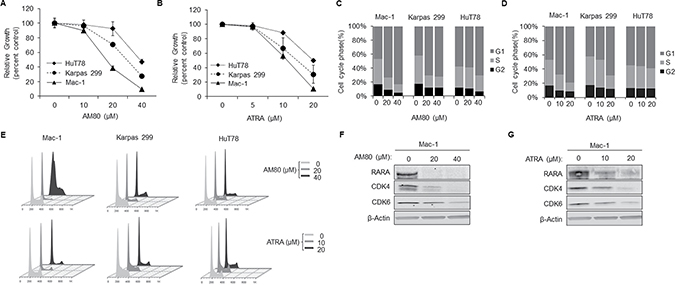
Figure 3: Retinoids cause G1-arrest and concurrent inhibition of RARA and CDK4/6 expression. (A, B) AM80 and ATRA cause dose-dependent growth inhibition in T-cell lymphoma cells that is proportional to native RARA expression (as shown in Figure 1A). (C, D) AM80 and ATRA cause G1 arrest in T-cell lymphoma cells that is proportional to native RARA expression. (E) Representative cell cycle plots of T-cell lymphoma cells after treatment with AM80 or ATRA. (F ,G) AM80 and ATRA cause RARA degradation and down-regulation of CDK4 and CDK6 in RARAhigh Mac-1 cells. RARA, retinoic acid receptor alpha; CDK, cyclin-dependent kinase; ATRA, all-trans retinoic acid.
Retinoids suppress a cell cycle gene expression program in T-cell lymphoma cells
To characterize the effects of retinoids on T-cell lymphoma cells further, we performed RNA sequencing and analyzed the transcriptomes of Mac-1, Karpas 299, and HuT78 cell lines treated with AM80, ATRA, or bexarotene at varying doses. We then identified genes consistently up- or down-regulated in a dose-dependent manner compared to vehicle-treated control cells. Of 235 genes differentially expressed after AM80 treatment, 156 (66%) were down-regulated and 79 (34%) were up-regulated (Figure 4A). Down-regulated genes included several key cell cycle regulators including CDK1, CDK2, TYMS (thymidylate synthetase), and CCNE2 (cyclin E2). Gene set enrichment analysis (GSEA) confirmed that genes down-regulated by AM80 were highly enriched for regulators of the cell cycle (P < 0.001) and, more specifically, the G1-S transition (P = 0.004; Figure 4B). In addition, comprehensive network analysis identified that the most prominent network (score = 56) in the list of genes differentially expressed by AM80 was composed of genes associated with cell cycle, cellular assembly and organization, and DNA replication (Figure 4C). Finally, functional annotation of differentially expressed genes identified cellular processes involving cell replication and cell cycle progression (Figure 4D). Taken together with the relative specificity of AM80 for RARA, these findings strongly support and extend our in vitro functional observations that RARA drives cell cycle progression and, more specifically, G1-S transition, in T-cell lymphoma cells.
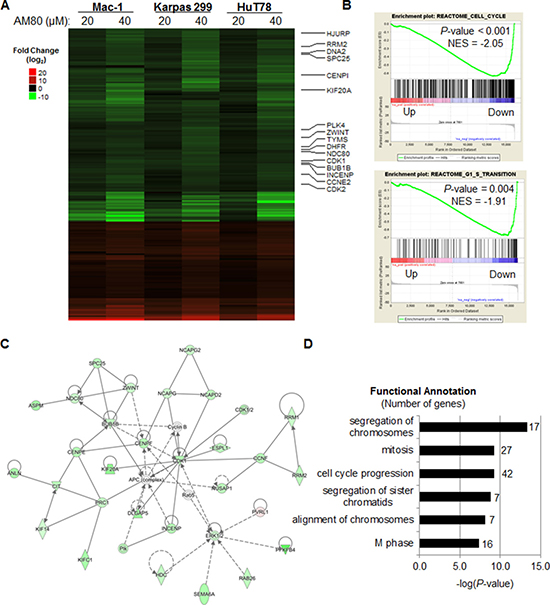
Figure 4: The RARA-specific retinoid, AM80, down-regulates expression of cell cycle gene in T-cell lymphoma cells. (A) AM80 treatment results in a consistent gene expression signature across T-cell lymphoma cell lines that includes down-regulation of multiple cell cycle genes. The heat map displays log2 fold changes from control (0 μM dose) at 24 hours. (B) Gene set enrichment analysis (GSEA) plots show enrichment for cell cycle and G1-S transition gene sets in genes down-regulated by AM80. (C) Pathway analysis of genes differentially expressed upon AM80 treatment identifies a top regulatory network (score = 56) comprising cell cycle, cellular assembly and organization, and DNA replication-associated genes. (D) Functional annotation of genes differentially expressed by AM80 identifies cell cycle-related functions as top hits. NES, Normalized Enrichment Score.
The pan-RAR agonist, ATRA, as well as the RXR agonist, bexarotene, also down-regulated expression of genes involved in cell cycle regulation, including genes identical to those down-regulated by AM80 (Supplementary Figures 2A and 3C). However, consistent with the broader array of RAR and RXR targets for ATRA and bexarotene, respectively, the functions of the differentially expressed genes were more diverse. Network analysis for both ATRA and bexarotene identified the top networks to involve lipid metabolism and small molecule biochemistry rather than cell cycle (Supplementary Figure 2B and 2D).
RARA overexpression increases chemosensitivity to the synthetic retinoid, AM80
Because of our observation that the cell line with the highest native RARA expression, Mac-1, exhibited the most chemosensitivity to retinoids, we explored the role of RARA overexpression on retinoid chemosensitivity in cell lines with lower RARA expression. Using doses of AM80 to which the RARAlow cell lines, HuT78 and Karpas 299, were relatively resistant, we found that overexpression of RARAwt significantly increased chemosensitivity compared to empty vector control (HuT78: 5 μM, 18% growth inhibition, P = 0.01; 10 μM, 19% growth inhibition, P = 0.04; and Karpas 299: 5 μM, 16% growth inhibition, P = 0.04; 10 μM, 23% growth inhibition, P = 0.02; Figure 5A, 5B). Overexpressing RARAR394Q conferred slightly less chemosensitivity than RARAwt in both cell lines, but these differences were not statistically significant.
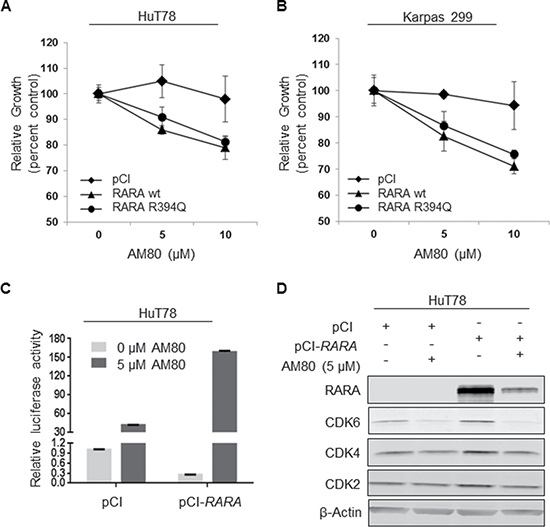
Figure 5: RARA overexpression increases chemosensitivity to AM80. (A, B) Overexpression of either RARAwt or RARAR394Q increases chemosensitivity of (A) HuT78 and (B) Karpas 299 cells to AM80. The presence of the R394Q mutation does not reduce chemosensitivity significantly. (C) RARAwt overexpression increases retinoic acid response element (RARE) activity (indicated by luciferase) in HuT78 cells treated with AM80. (D) RARAwt overexpression increases the degree of CDK2, CDK4 and CDK6 inhibition caused by AM80. wt, wild-type; CDK, cyclin-dependent kinase.
Because RARs regulate transcription by binding to RARE motifs in target gene promoters, we examined the effect of RARAwt overexpression with and without AM80 treatment on RARE activity using a luciferase assay. As expected, in the absence of AM80, RARE activity was lower in HuT78 cells overexpressing RARA than in cells with low basal RARA expression (Figure 5C). However, AM80 increased RARE activity in RARA-overexpressing cells to a much greater degree (382% of control) than in RARAlow cells. Finally, RARAwt overexpression augmented the degree of CDK2, CDK4, and CDK6 inhibition caused by AM80 treatment (Figure 5D). Taken together, these findings suggest that T-cell lymphoma cells expressing high levels of RARA are particularly chemosensitive to RARA-directed retinoids, and that measuring RARA protein expression—but not necessarily identifying RARA mutations—might be used predict this degree of chemosensitivity.
DISCUSSION AND CONCLUSIONS
In this study, we used a combined approach of transcriptome sequencing, functional studies, and inhibitor assays to characterize the role of RARA in mature T-cell lymphoma cells. Following identification of a RARAR394Q mutation in a PTCL patient being evaluated in an individualized medicine clinic, we demonstrated that both RARAR394Q and RARAwt accelerated growth of T-cell lymphoma cells, with modestly enhanced function of the mutant compared to the wild-type form. We then demonstrated that the growth induction caused by RARA was due to promotion of the cell cycle, specifically G1-S transition. Finally, we demonstrated that these effects could be reversed by retinoids that target RARA, and that indeed high RARA expression increased chemosensitivity to these agents. These findings suggest the possibility of therapeutic approaches including retinoids for some patients with PTCL, and the potential use of determining RARA expression to identify those patients most likely to derive benefit from retinoid therapy.
Clinical genomic sequencing approaches to individualize treatment have become an important new strategy for the management of patients with cancer and other diseases [16]. While sequence data from some patients point to genetic abnormalities that encode protein products known to be targetable with existing drugs, the significance of many findings remains unclear, prompting functional investigations in disease-specific experimental models [3]. Here, we examined the potential role of RARAR394Q in T-cell lymphoma. This mutation was chosen as a candidate for further study because amino acid R394 of RARA has been demonstrated to be important in retinoid binding [17]. However, we observed only modest functional differences between RARAwt or RARAR394Q overexpression in T-cell lymphoma cells either in the presence or absence of retinoids. Interestingly, another substitution affecting this same amino acid, RARAR394W, was reported in two patients with relapsed acute promyelocytic leukemia with PML-RARA fusions, and was shown to be associated with only modest effects on ATRA binding and RARE reporter transactivation; however, a 12-nucleotide deletion in this same region, ΔR394-L398/394M, had pronounced effects in these assays [18]. Therefore, amino acid substitutions alone at RARA R394 do not appear to have immediate clinical relevance with regard to retinoid chemosensitivity.
Although the role of RARAR394Q remains unclear, overexpression of either RARAR394Q or RARAwt increased T-cell lymphoma cell growth significantly, and enhanced chemosensitivity was observed in RARAhigh cells as well as RARAlow cells experimentally manipulated to overexpress RARA. Importantly, clinical responses to retinoids have been reported in various T-cell malignancies, including PTCL, CTCL, and adult T-cell leukemia (ATL) [9–12, 19–22]. For example, Cheng et al reported 5 complete responses and one partial response among 12 PTCL patients treated with 13-cis retinoic acids [12]. We found RARA to be the predominantly expressed RAR in PTCL cell lines, but also noted that its expression varied among the lines tested. As predicted, the relationship between RARA expression and chemosensitivity was more marked for the relatively RARA-specific retinoid, AM80, than for the pan-RAR ligand ATRA or the RXR-ligand bexarotene. Of note, a relationship between RARA expression and AM80 sensitivity also has been reported in acute myeloid leukemia cells [23]. Taken together, these data suggest the possibility that, while clinical response rates to retinoids in patients with systemic PTCLs have been modest, these response rates might be improved by using RARA expression as a biomarker to select patients more likely to be chemosensitive.
Our data demonstrate a critical role for RARA in cell cycle regulation of T-cell lymphoma cells, and particularly in G1-S transition. In myeloid cells, retinoic acid has been shown to induce G1 arrest with corresponding downregulation of cyclins D and E and decreased CDK activity [24–26]. Similarly, AM80 has been shown to cause G1 arrest in ATL cells [27]. Although chromatin immunoprecipitation studies have demonstrated binding of RARA to CDK2 and CDK6, a direct effect on transcriptional regulation has not been demonstrated; thus, inhibition of CDKs may occur as an indirect, downstream effect of retinoid activity [28, 29]. Of note, CDKs including CDK6 play a critical role in physiological development and tumorigenesis of thymocytes [30]. In addition, recurrent amplifications of CDK6 as well as deletions of CNDK2A encoding the CDK4/6 inhibitor, p16INK4A, have been reported in PTCLs [31–33]. Clinical CDK4/6 inhibitors have shown activity in B-cell lymphomas and other cancers [13], and might be an alternative means to block CDK up-regulation in RARAhigh T-cell lymphomas.
MATERIALS AND METHODS
Retinoids
ATRA (Sigma-Aldrich, Milwaukee, WI, USA), AM80 (Tocris, Ellisville, MS, USA), and bexarotene (Selleck Chemicals, Houston, TX, USA) were dissolved in dimethyl sulfoxide and stock solutions and stored at –80°C. Retinoids were protected from light during handling.
Cell lines, culture conditions, and transfection
Karpas 299 (ALK-positive anaplastic large cell lymphoma, a PTCL subtype) and HuT78 (CTCL) cell lines were obtained from ATCC, Gaithersburg, MD, USA. Mac-1 (developed by M.E.K.) was derived from circulating CTCL cells from a patient with multiple T-cell neoplasms [34]. Karpas 299 and Mac-1 were maintained in RPMI 1640 (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS; Clontech Laboratories, Inc, Mountain View, CA, USA) and 1% penicillin/streptomycin (Gibco, Grand Island, NY, USA). HuT78 was maintained in Iscove’s Modified Dulbecco’s Medium (Gibco, Grand Island, NY, USA) containing 15% FBS and 1% penicillin/streptomycin. All cell lines were electroporated with 300V for 10 msec in antibiotic-free medium using the ECM 830 electroporation system (BTX Harvard Apparatus).
siRNA gene knockdown
Control (AllStars Negative Control; Qiagen, Valencia, CA, USA) and RARA (ON-TARGETplus SMARTpool; GE Dharmacon, Waltham, MA, USA) siRNAs were transfected by electroporation and protein expression was assessed at 72 hours. Pooled RARA siRNA sequences were: GCAAAUACACUACGAACAA; CCA AGGAGUCUGUGAGAAA; GAGCAGCAGUUCUGA AGAG; GAACAACGUGUCUCUCUGG.
Expression plasmids
Human RARA was subcloned from pcDNA6-RARA (Plasmid# 35555 [35], Addgene, Cambridge, MA, USA; NheI and XbaI restriction sites) into pCI (Promega, Madison, WI, USA). The R394Q mutation was introduced into pCI-RARA using site-directed mutagenesis (QuickChange II, Agilent Technologies, Santa Clara, CA, USA). Expression vectors were transiently transfected into Karpas 299 and HuT78 cells by electroporation and protein expression was assessed at 48hours. For retinoid treatment, cells were cultured in media containing 2% charcoal-filtered FBS immediately following transfection, and cell treatments were administered after a 4-hour post-electroporation recovery period.
Cell growth
The CellTiter Aqueous Non-Radioactive Cell Proliferation Assay Kit (MTS; Promega) was used to assess cell growth following the manufacturer’s instructions. Briefly, 10,000 cells per well were seeded in a 96-well plate and cultured in RPMI containing 2% charcoal-stripped FBS and indicated retinoid concentrations (Gibco, Grand Island, NY, USA) for 72 hours. At the end of the treatment period, the MTS reagent was added, cells were incubated an additional 2–4 hours, and absorbance was measured at 490 nanometers.
Cell cycle analysis
Cell cycle analysis was performed 72 h post- transfection or drug treatment. Approximately 1 × 106 cells were pelleted, washed in phosphate-buffered saline, and fixed in 75% ethanol overnight at 4°C. Cells then were washed in phosphate-buffered saline, re-suspended in a 1× saponin-based permeabilization buffer and wash reagent (Thermo Fisher Scientific, Waltham, MA, USA), and stained with FxCycle™ Far Red stain (Thermo Fisher) for 30 minutes at room temperature. Samples were evaluated on a FACSCalibur cytometer (Becton Dickinson, Mountain View, CA, USA). Data were analyzed using FlowJo 7.6.5 software (Tree Star Inc., Ashland, OR, USA).
Immunoblotting
Immunoblotting was performed as previously described [36] using antibodies to RARA (sc-551, Santa Cruz Biotechnology, Inc, Santa Cruz, CA, USA), CDK2 (78B2/ #2546, Cell Signaling Technology, Danvers, MA, USA), CDK4 (D9G3E/#12790, Cell Signaling), CDK6 (D4S8S/#13331, Cell Signaling) or β-actin (NB600-501, Novus Biologicals, LLC, Littleton, CO, USA). Proteins were detected using goat anti-mouse (sc-2005, Santa Cruz) or donkey anti-rabbit (NA934, Amersham, Little Chalfont, United Kingdom) antibodies and West Pico Super Signal chemiluminescence (Thermo Fisher), or using IRDye 800CW and 680RD antibodies (LI-COR Biosciences, Lincoln, NE, USA) on the LI-COR Odyssey CLx Imaging System.
Luciferase reporter assays
RARE reporter (Cignal RARE reporter; SABiosciences, Frederick, MD, USA) and pCI empty vector or pCI-RARA (wild-type) were co-transfected into HuT78 cells by electroporation. After recovery for 24 hours, the transfected cells were treated with AM80 or vehicle (dimethyl sulfoxide) in RPMI containing 2% charcoal-stripped FBS for 24 hours. Luciferase expression was measured using the Dual-Luciferase Reporter Assay System (Promega) following manufacturer’s protocol using the Centro XS3 LB 960 microplate luminometer (Berthold Technologies, Zug, Switzerland) as previously described [36]. Total protein concentrations of each lysate were used for normalization.
Transcriptome analysis
Mac-1, Karpas 299 and HuT78 cells were treated with AM80 (0 μM, 20 μM, or 40 μM), ATRA (0 μM, 10 μM, or 20 μM), or bexarotene (0 μM, 10 μM, or 20 μM) in medium containing 2% charcoal-stripped FBS. Cell pellets were collected at 24 hours and immediately stored at −80°C pending RNA extraction. RNA sequencing was performed as previously described [3]. Briefly, following extraction of total RNA, mRNA libraries were constructed (Illumina, TruSeq Technology, San Diego, CA, USA) and 100 base-pair paired-end sequencing was performed on an Illumina HiSeq instrument. The Sequencing data were mapped to hg19 and genes with a minimum of 100 reads in at least one sample were retained for analysis. Dose-dependent effects of each drug were determined using paired t-tests (EdgeR) for medium-versus-low and high-versus-medium comparisons. Genes were considered differentially expressed if they met fold-change and P-value criteria (log2 fold change ≥ 0.5 or ≤ −0.5 and P-value ≤ 0.05) for both comparisons. GSEA was performed on a ranked dataset generated by the product of the fold-change sign and –log(P-value) for the 0 μM versus 40 μM AM80 comparison [37]. Pathway analysis was performed using Ingenuity Pathway Analysis software (Qiagen, Redwood City, CA, USA) on differentially expressed genes.
Statistical analysis
Statistical analysis was performed using Student’s t-test. Statistical significance was defined as P < 0.05.
Authors’ contributions
X.W. and R.L.B. performed experiments; S.D., A.L.F., and R.L.B. analyzed data; G.S.N., K.N.L., and E.D.W. provided patient samples and interpreted clinical and genomic data; M.E.K. provided critical reagents; and A.L.F. and R.L.B. designed the study and wrote the manuscript.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
GRANT SUPPORT
This work was supported by the Center for Individualized Medicine, Mayo Clinic; by award numbers R01 CA177734 (ALF), P30 CA15083 (Mayo Clinic Cancer Center), and P50 CA97274 (University of Iowa/Mayo Clinic Lymphoma SPORE) from the National Cancer Institute; by Research Scholar Grant Number RSG-12-193-01-TBE from the American Cancer Society (ALF); by the Fraternal Order of Eagles Cancer Research Fund, Mayo Clinic Cancer Center (RLB); by CTSA Grant Number UL1 TR000135 from the National Center for Advancing Translational Science (NCATS); by award number CI-48-09 from the Damon Runyon Cancer Research Foundation (ALF); by a scholarship award from the China Scholarship Council (X.W.); by the Department of Laboratory Medicine and Pathology, Mayo Clinic; by the Predolin Foundation; and by the Martin and Dorothy Spatz Charitable Foundation (MEK).
REFERENCES
1. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, Jaffe ES. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016; 127:2375–90. doi: 10.1182/blood-2016-01-643569.
2. Savage KJ. Prognosis and primary therapy in peripheral T-cell lymphomas. Hematology Am Soc Hematol Educ Program. 2008:280–8. doi: 10.1182/asheducation-2008.1.280.
3. Boddicker RL, Razidlo GL, Dasari S, Zeng Y, Hu G, Knudson RA, Greipp PT, Davila JI, Johnson SH, Porcher JC, Smadbeck JB, Eckloff BW, Billadeau DD, et al. Integrated mate-pair and RNA sequencing identifies novel, targetable gene fusions in peripheral T-cell lymphoma. Blood. 2016; 128:1234–45. doi: 10.1182/blood-2016-03-707141.
4. Ablain J, de The H. Retinoic acid signaling in cancer: The parable of acute promyelocytic leukemia. Int J Cancer. 2014; 135:2262–72. doi: 10.1002/ijc.29081.
5. Hua S, Kittler R, White KP. Genomic antagonism between retinoic acid and estrogen signaling in breast cancer. Cell. 2009; 137:1259–71. doi: 10.1016/j.cell.2009.04.043.
6. Tang Q, Chen Y, Meyer C, Geistlinger T, Lupien M, Wang Q, Liu T, Zhang Y, Brown M, Liu XS. A comprehensive view of nuclear receptor cancer cistromes. Cancer Res. 2011; 71:6940–7. doi: 10.1158/0008-5472.CAN-11-2091.
7. Melnick A, Licht JD. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood. 1999; 93:3167–215.
8. Panchal MR, Scarisbrick JJ. The utility of bexarotene in mycosis fungoides and Sezary syndrome. Onco Targets Ther. 2015; 8:367–73. doi: 10.2147/OTT.S61308.
9. Zhang C, Duvic M. Retinoids: therapeutic applications and mechanisms of action in cutaneous T-cell lymphoma. Dermatol Ther. 2003; 16:322–30.
10. Kempf W, Kettelhack N, Duvic M, Burg G. Topical and systemic retinoid therapy for cutaneous T-cell lymphoma. Hematol Oncol Clin North Am. 2003; 17:1405–19.
11. Huang CL, Lin ZZ, Su IJ, Chao TY, Tien HF, Chang MC, Huang MC, Kao WY, Tang JL, Yeh KH, Wang CH, Hsu CH, Liu MY, et al. Combination of 13-cis retinoic acid and interferon-alpha in the treatment of recurrent or refractory peripheral T-cell lymphoma. Leuk Lymphoma. 2002; 43:1415–20. doi: 10.1080/1042819022386806.
12. Cheng AL, Su IJ, Chen CC, Tien HF, Lay JD, Chen BR, Pu YS, Hong RL, Shen MC, Wang CH. Use of retinoic acids in the treatment of peripheral T-cell lymphoma: a pilot study. J Clin Oncol. 1994; 12:1185–92.
13. O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016; 13:417–30. doi: 10.1038/nrclinonc.2016.26.
14. Idres N, Marill J, Flexor MA, Chabot GG. Activation of retinoic acid receptor-dependent transcription by all-trans-retinoic acid metabolites and isomers. J Biol Chem. 2002; 277:31491–8. doi: 10.1074/jbc.M205016200.
15. Kopf E, Plassat JL, Vivat V, de The H, Chambon P, Rochette-Egly C. Dimerization with retinoid X receptors and phosphorylation modulate the retinoic acid-induced degradation of retinoic acid receptors alpha and gamma through the ubiquitin-proteasome pathway. J Biol Chem. 2000; 275:33280–8. doi: 10.1074/jbc.M002840200.
16. Lazaridis KN, McAllister TM, Babovic-Vuksanovic D, Beck SA, Borad MJ, Bryce AH, Chanan-Khan AA, Ferber MJ, Fonseca R, Johnson KJ, Klee EW, Lindor NM, McCormick JB, et al. Implementing individualized medicine into the medical practice. Am J Med Genet C Semin Med Genet. 2014; 166C:15–23. doi: 10.1002/ajmg.c.31387.
17. Lamour FP, Lardelli P, Apfel CM. Analysis of the ligand-binding domain of human retinoic acid receptor alpha by site-directed mutagenesis. Mol Cell Biol. 1996; 16:5386–92.
18. Gallagher RE, Schachter-Tokarz EL, Zhou DC, Ding W, Kim SH, Sankoorikal BJ, Bi W, Livak KJ, Slack JL, Willman CL. Relapse of acute promyelocytic leukemia with PML-RARα mutant subclones independent of proximate all-trans retinoic acid selection pressure. Leukemia. 2006; 20:556–62. doi: 10.1038/sj.leu.2404118.
19. Chou WC, Su IJ, Tien HF, Liang DC, Wang CH, Chang YC, Cheng AL. Clinicopathologic, cytogenetic, and molecular studies of 13 Chinese Patients with Ki-1 anaplastic large cell lymphoma. Special emphasis on the tumor response to 13-cis retinoic acid. Cancer. 1996; 78:1805–12.
20. Chow JM, Cheng AL, Su IJ, Wang CH. 13-cis-retinoic acid induces cellular differentiation and durable remission in refractory cutaneous Ki-1 lymphoma. Cancer. 1991; 67:2490–4.
21. Maeda Y, Kawauchi M, Miyatake J, Hirase C, Yamaguchi T, Matsumura I. Effects of tamibarotene for the treatment of adult T cell leukemia. Ann Hematol. 2012; 91:629–31. doi: 10.1007/s00277-011-1290-4.
22. Maeda Y, Yamaguchi T, Hijikata Y, Tanaka M, Hirase C, Takai S, Morita Y, Sano T, Miyatake J, Tatsumi Y, Kanamaru A. Clinical efficacy of all-trans retinoic acid for treating adult T cell leukemia. J Cancer Res Clin Oncol. 2008; 134:673–7. doi: 10.1007/s00432-007-0334-6.
23. Jimi S, Mashima K, Matsumoto T, Hara S, Suzumiya J, Tamura K. RARalpha is a regulatory factor for Am-80-induced cell growth inhibition of hematologic malignant cells. Int J Oncol. 2007; 31:397–404.
24. Dimberg A, Bahram F, Karlberg I, Larsson LG, Nilsson K, Oberg F. Retinoic acid-induced cell cycle arrest of human myeloid cell lines is associated with sequential down-regulation of c-Myc and cyclin E and posttranscriptional up-regulation of p27(Kip1). Blood. 2002; 99:2199–206.
25. DiPietrantonio AM, Hsieh TC, Olson SC, Wu JM. Regulation of G1/S transition and induction of apoptosis in HL-60 leukemia cells by fenretinide (4HPR). Int J Cancer. 1998; 78:53–61.
26. Shimizu T, Awai N, Takeda K. Complex regulation of CDKs and G1 arrest during the granulocytic differentiation of human myeloblastic leukemia ML-1 cells. Oncogene. 2000; 19:4640–6. doi: 10.1038/sj.onc.1203821.
27. Nakazato T, Okudaira T, Ishikawa C, Nakama S, Sawada S, Tomita M, Uchihara JN, Taira N, Masuda M, Tanaka Y, Ohshiro K, Takasu N, Mori N. Anti-adult T-cell leukemia effects of a novel synthetic retinoid, Am80 (Tamibarotene). Cancer Sci. 2008; 99:2286–94. doi: 10.1111/j.1349-7006.2008.00917.x.
28. Delacroix L, Moutier E, Altobelli G, Legras S, Poch O, Choukrallah MA, Bertin I, Jost B, Davidson I. Cell-specific interaction of retinoic acid receptors with target genes in mouse embryonic fibroblasts and embryonic stem cells. Mol Cell Biol. 2010; 30:231–44. doi: 10.1128/MCB.00756-09 MCB.00756-09.
29. Hu XT, Zuckerman KS. Role of cell cycle regulatory molecules in retinoic acid- and vitamin D3-induced differentiation of acute myeloid leukaemia cells. Cell Prolif. 2014; 47:200–10. doi: 10.1111/cpr.12100.
30. Hu MG, Deshpande A, Enos M, Mao D, Hinds EA, Hu GF, Chang R, Guo Z, Dose M, Mao C, Tsichlis PN, Gounari F, Hinds PW. A requirement for cyclin-dependent kinase 6 in thymocyte development and tumorigenesis. Cancer Res. 2009; 69:810–8. doi: 10.1158/0008-5472.CAN-08-2473.
31. Nagel S, Leich E, Quentmeier H, Meyer C, Kaufmann M, Drexler HG, Zettl A, Rosenwald A, MacLeod RA. Amplification at 7q22 targets cyclin-dependent kinase 6 in T-cell lymphoma. Leukemia. 2008; 22:387–92.
32. Vasmatzis G, Johnson SH, Knudson RA, Ketterling RP, Braggio E, Fonseca R, Viswanatha DS, Law ME, Kip NS, Ozsan N, Grebe SK, Frederick LA, Eckloff BW, et al. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas. Blood. 2012; 120:2280–9. doi: 10.1182/blood-2012-03-419937.
33. Laharanne E, Chevret E, Idrissi Y, Gentil C, Longy M, Ferrer J, Dubus P, Jouary T, Vergier B, Beylot-Barry M, Merlio JP. CDKN2A-CDKN2B deletion defines an aggressive subset of cutaneous T-cell lymphoma. Mod Pathol. 2010; 23:547–58. doi: 10.1038/modpathol.2009.196.
34. Davis TH, Morton CC, Miller-Cassman R, Balk SP, Kadin ME. Hodgkin’s disease, lymphomatoid papulosis, and cutaneous T-cell lymphoma derived from a common T-cell clone. N Engl J Med. 1992; 326:1115–22. doi: 10.1056/NEJM199204233261704.
35. Srinivas H, Juroske DM, Kalyankrishna S, Cody DD, Price RE, Xu XC, Narayanan R, Weigel NL, Kurie JM. c-Jun N-terminal kinase contributes to aberrant retinoid signaling in lung cancer cells by phosphorylating and inducing proteasomal degradation of retinoic acid receptor alpha. Mol Cell Biol. 2005; 25:1054–69.
36. Boddicker RL, Kip NS, Xing X, Zeng Y, Yang ZZ, Lee JH, Almada LL, Elsawa SF, Knudson RA, Law ME, Ketterling RP, Cunningham JM, Wu Y, et al. The oncogenic transcription factor IRF4 is regulated by a novel CD30/NF-kappaB positive feedback loop in peripheral T-cell lymphoma. Blood. 2015; 125:3118–27. doi: 10.1182/blood-2014-05-578575.
37. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005; 102:15545–50. doi: 10.1073/pnas.0506580102.