
INTRODUCTION
Glioblastoma (GBM), one of the most common primary brain tumor in adults, is highly malignant with exhibiting aggressive clinical manifestation and a median life expectancy of less than 2 years [1]. Biologically, GBM is characterized by its high proliferative and invasive activity [2]. Only a few drugs were prescribed for GBM: temozolomide (TMZ) [3], biodegradable carmustine (BCNU) wafer [4, 5], and bevacizumab [6, 7]. Till date, no effective therapeutic for GBM has emerged despite the development of a variety of molecular-targeted agents such as tyrosine kinase inhibitors [8].
Glycogen synthase kinase (GSK)-3β is a serine/threonine protein kinase that regulates various cellular pathways depending on its substrates by phosphorylation [9]. Emerging evidence suggests that GSK3β promotes GBM progression by interacting with the pivotal molecules that participate in tumor cell survival, proliferation, invasion, and resistance to chemoradiation therapy [10, 11]. We and others have reported that GSK3β inhibition attenuates the proliferation, migration and invasion of GBM cells by modulating distinct molecular pathways, and sensitizes them to chemotherapeutic agents and radiation [12–19]. These studies have substantiated GSK3β as therapeutic target of GBM.
Despite experimental evidence supporting the therapeutic effect of GSK3β inhibition, none of GSK3β inhibitors was approved for treatment of diseases such as diabetes mellitus, Alzheimer’s disease, and cancer. The development and approval of new drug is a time-consuming and expensive process with high rate of failure. Recently, drug repositioning to find new applications for existing drugs has emerged to overcome this challenge [20]. Drug repositioning attempts to discover new therapeutic options for already approved drugs, as their pharmacokinetics, pharmacodynamics and post-marketing surveillance safety data are readily available [21]. Drugs such as cimetidine, lithium carbonate, olanzapine, and valproate, prescribed for other diseases inhibited GSK3β activity [22–24]. In the present study, these GSK3β-inhibiting drugs were examined for therapeutic effect against GBM in experimental settings. We also explored whether GSK3β activity in clinical tumor tissues influences the survival of GBM patients to rationalize the targeting of GSK3β. Based on these experimental and clinical findings, we conducted a clinical study to investigate the safety and therapeutic effect of repositioning GSK3β-inhibiting medicines in combination with TMZ, in patients with recurrent GBM.
RESULTS
Activated GSK3β is an unfavorable prognostic marker in GBM
First, we investigated the expression of GSK3β and pGSK3βY216 (active form) in clinical samples. Interestingly, survival analysis of 57 patients with GBM showed a significant correlation between pGSK3βY216 expression and both progression-free survival (PFS) and overall survival (OS) (Figure 1). Subgroup analysis of age, Karnofsky Performance Scale (KPS), and O6-methylguanine-DNA-methyltransferase (MGMT) promoter status, and extent of resection (EOR) showed no statistical difference between GSK high group and GSK low group (Supplementary Table 1). Univariate analysis by Kaplan-Meier method showed methylated MGMT promoter, less age, and high EOR as predictive factors in our cohort (Supplementary Figure 1). Multivariate analysis revealed that activation of GSK3β was an independent unfavorable predictive factor (Table 1).
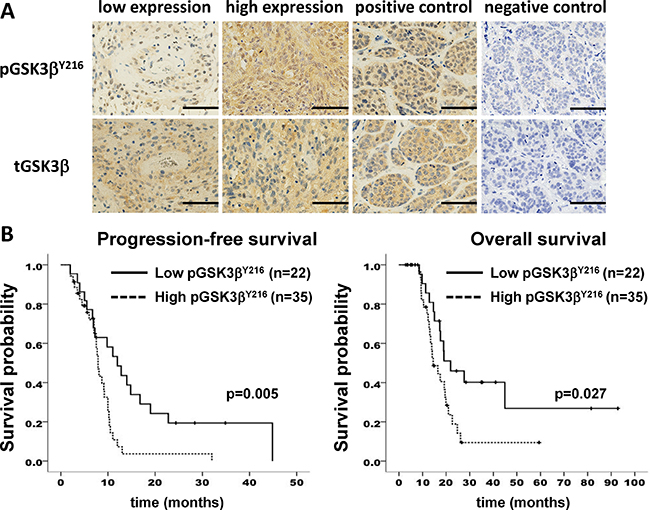
Figure 1: GSK3β activation correlates with poor survival in patients with glioblastoma. (A) immunohistochemical analysis of human glioblastoma samples for pGSK3βY216 and total (t) GSK3β. Positive and negative control was a case of metastatic breast carcinoma. Scale bar, 50 μm. (B) Kaplan-Meier curves showing survival of glioblastoma patients based on expression level of pGSK3βY216. n, number of patients.
Table 1: Multivariate predictors of survival in our cohort
Variable |
Progression-free survival |
Overall survival |
||||
|---|---|---|---|---|---|---|
RR |
95% CI |
p* |
RR |
95% CI |
p* |
|
Low pGSK3βY216 |
2.15 |
1.08–4.31 |
0.030 |
2.41 |
1.12–5.25 |
0.026 |
Age < 65 |
1.66 |
0.81–3.41 |
0.166 |
1.90 |
0.78–4.60 |
0.155 |
KPS ≥ 80% |
1.49 |
0.76–2.92 |
0.247 |
1.06 |
0.47 –2.41 |
0.890 |
Methylated MGMT promoter |
2.30 |
1.17–4.52 |
0.015 |
0.99 |
0.49–2.01 |
0.977 |
EOR ≥ 80% |
1.63 |
0.76–3.51 |
0.212 |
1.94 |
0.79–4.78 |
0.151 |
Abbreviations: CI, confidence interval; EOR, extent of resection; GSK3β, glycogen synthase kinase 3β; KPS, Karnofsky Performance Scale; MGMT, O6-methylguranine-DNA-methyltransferase; pGSK3βY216, GSK3β phosphorylated at tyrosine (Y) 216 residue; RR, relative risk.
*Cox regression model.
Inhibition of GSK3β activity by existing drugs
We examined whether the existent drugs (cimetidine, lithium, olanzapine and valproate) inhibit GSK3β activity in cells by monitoring the level of pGSS641, the primary substrate of GSK3β (reviewed in ref. [9]). The levels of pGSS641 in all GBM cells decreased following treatment with each drug (Supplementary Figure 2). Hereafter the combination of these drugs is referred as “CLOVA cocktail” by the initial letters of each drug. While there were some differences in the GSK3β-inhibitory effect among the drugs and cell lines, the CLOVA cocktail decreased the level of pGSS641 in all GBM cells more efficiently than each of the cocktail constituents (Figure 2A). The results indicate that doses of these four medicines mixed in the CLOVA cocktail are within the ranges of clinical use nevertheless this cocktail inhibits GSK3β activity in GBM cells.
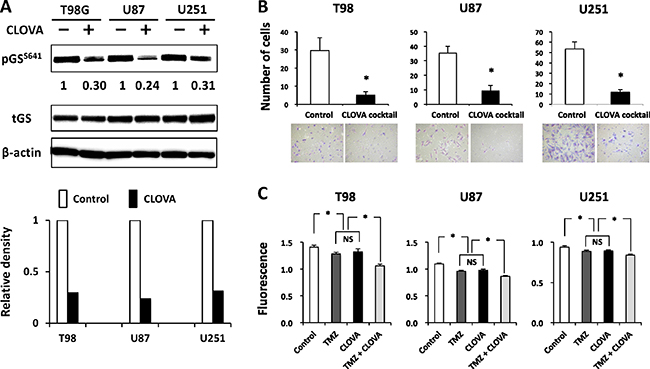
Figure 2: Effect of treatment with the CLOVA cocktail on the level of serine 641 phosphorylation of glycogen synthase (pGSS641). Expression level of pGSS641 (A), invasion (B) and proliferation (C) in GBM cells. Cells were treated with the CLOVA cocktail (the mixture of cimetidine, 0.1 mM; lithium, 1 mM; olanzapine, 0.1 μM; and valproate, 1 mM). A, Western blotting analysis for changes in the levels of pGSS641 in GBM cells following treatment with the CLOVA cocktail for 24 hours. The value below the blots shows relative level of pGSS641 quantified by densitometry and normalized to that of total (t) GS. All blots were cropped for clarity and conciseness of the images. B, Invading cells through a Matrigel-coated Transwell chamber were scored for cells treated with or without CLOVA cocktail for 8 hours. The mean number of cells in eight high-power microscopic fields was calculated with standard deviations (SDs). C, Growth analysis of GBM cell lines treated with the indicated reagents or without treatment (Control) for 96 h. The plate was read on fluorescence plate reader. B and C: NS, not significant; *p < 0.05.
Effects of the GSK3β-inhibiting drugs on invasion and proliferation of GBM cells
The Transwell assay showed that all 4 drugs inhibited the invasion of GBM cells to extracellular matrix (Supplementary Figure 3A). Lithium showed proliferation-inhibition at 5 mM and 10 mM in T98 and U87, while only at 10 mM in U251 cells. Valproate at 5 mM and 10 mM inhibited proliferation of all the 3 cell lines. However, neither cimetidine nor olanzapine inhibited cell proliferation at the indicated concentrations (Supplementary Figure 3B), and this issue was discussed later.
Next, we investigated the effect of CLOVA cocktail that consists of the 4 drugs as a mixture at the lowest concentrations (cimetidine, 0.1 mM; lithium, 1 mM; olanzapine, 0.1 μM; and valproate, 1 mM) concomitant with TMZ on GBM invasion and proliferation. The concentrations of cimetidine, lithium, and valproate used in culture medium were determined according to their blood concentration at the administration of maximum daily dose, because the blood-brain barrier (BBB) is damaged in GBM tissue. The dose of olanzapine was calculated based on its intracerebral concentration at the administration of maximum daily dose and its high BBB permeability and accumulation in brain tissue [25]. As expected, CLOVA cocktail itself showed additive inhibitory effect on cell invasion (Figure 2B) while TMZ showed no effect at the same time point (data not shown). CLOVA cocktail and TMZ inhibited cell proliferation equally, and moreover the combination of them showed remarkable inhibitory effect on cell proliferation (Figure 2C). The results shown in Figure 2, Supplementary Figures 2 and 3 collectively suggest a causal association between the effects of CLOVA cocktail against the GSK3β activity of GBM cells and their proliferative and invasive capacity.
Effect of CLOVA cocktail on GBM cell invasion and proliferation in a brain tumor model
The tumor histology of our animal model demonstrated several features, characteristic of human GBM, including high proliferative and invasive nature. Inhibition of GSK3β activity by treatment with each drug and the CLOVA cocktail was confirmed by the decreased level of pGSS641 in tumor cells, especially in satellite lesions, and the effect of CLOVA cocktail was strongest (Figure 3A, Supplementary Figure 4). Similarly, the number of diffusely infiltrating tumor cells that stained positive for nestin significantly decreased in CLOVA cocktail-treated mice (Figure 3A and 3B). Consequently, well-demarcated border between the tumor and adjacent normal brain tissues was observed both in mice treated with each drug alone and the combination (Supplementary Figure 4, Figure 3). Previous studies showed that focal adhesion kinase (FAK) and Rac1 interact [26] and facilitate GBM invasion [27, 28]. The attenuated invasive capacity of GBM tumors following treatment with CLOVA cocktail was associated with decrease in activating phosphorylation of (pFAKY397 and pFAKY861; Supplementary Figure 5) and with alteration in subcellular localization of active Rac1 in tumor cells (Figure 3C). These findings are consistent with our earlier study showing that GSK3β inhibition decreased active Rac1 fraction and FAK phosphorylation in human GBM cell lines [29].
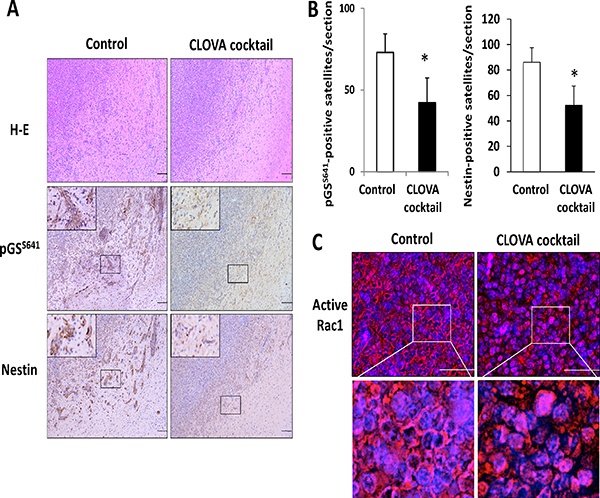
Figure 3: Effect of CLOVA cocktail on the glioblastoma animal model. (A) Representative histological and immunohistochemical sections of brain tumors treated with or without CLOVA cocktail. Both activity of GSK3β (estimated by the level of pGSS641) and expression of nestin decreased in the satellite lesions. Mice treated with CLOVA cocktail showed a well-demarcated border between the tumor and adjacent normal brain tissue. The magnified image of the area in the square is shown in the left upper corner in each panel of pGSS641 and nestin. Scale bars, 100 μm. (B) Effects of CLOVA cocktail on the number of pGSS641- and nestin-positive cell clusters. *p < 0.05. (C) Immunofluorescence microscopic findings of tumor tissues for active Rac1 (red). Cell nuclei were counterstained with Hoechst 33342 (blue). Subcellular localization of active Rac1 changed from cellular rim to whole cytoplasm following treatment with CLOVA cocktail. Magnified images of the area in the square are shown in the lower panels. pGSS641, glycogen synthase (GS) phosphorylated at serine 641 residue.
To compare the tumor proliferative potential, MIB-1 immunostaining was performed. Interestingly, MIB-1 staining index remarkably decreased in CLOVA cocktail treated-mice (Figure 4A), while less marked reduction was observed in mice receiving individual drug treatment (Supplementary Figure 4). TMZ alone and in combination with the CLOVA cocktail reduced the tumor size while the effects were not statistically significant (Supplementary Figure 6). Of note, CLOVA cocktail significantly prolonged survival of the mouse model (Figure 4B).
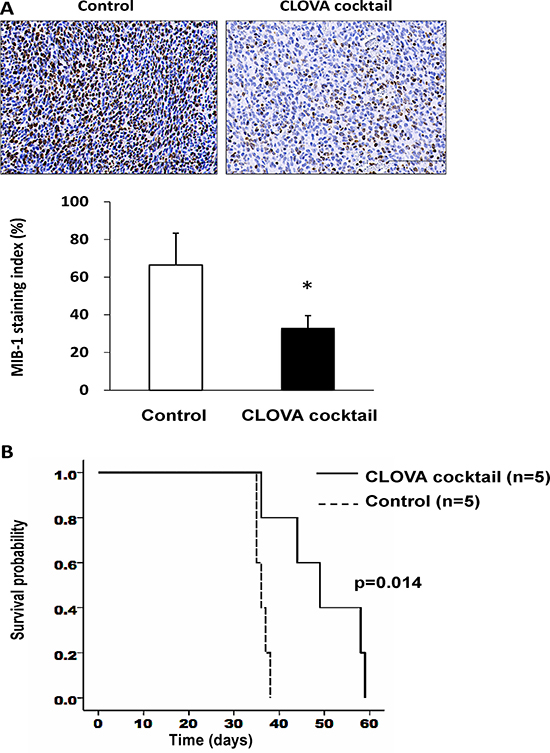
Figure 4: Effect of CLOVA cocktail on the glioblastoma animal model. (A) Immunohistochemistry of MIB-1 and the staining index (%) indicating activity of cell proliferation. Scale bars, 100 μm. *p < 0.05. (B) Survival of mice treated with CLOVA cocktail and those treated with DMSO/H2O (Control). Log-rank test, p = 0.014. n, number of mice.
Phase I/II clinical study
Seven patients were enrolled in this study (Figure 5A). All patients were eligible following radiological disease progression (Macdonald criteria [30]) and classified as Recursive Partitioning Analysis (RPA) class 7 [31]. Median age was 66 years and median KPS was 50. MGMT promoter methylation was determined in 4 patients (57%). Three patients took CLOVA cocktail on the day before death and 4 patients discontinued the oral medication because of aspiration pneumonitis, 2 weeks before death. Median OS after first recurrence was 11.2 (95% CI, 3.8–18.6) months in patients treated with concomitant CLOVA cocktail with maintenance TMZ although they were classified into RPA class 7 wherein the median survival duration was estimated as 4.9 months [31], compared to median OS of 4.3 (95% CI, 2.5–6.1) months in historical control group (log-rank test, p = 0.004) (Figure 5B). The concomitant CLOVA cocktail with TMZ was safe and well tolerated. Most common treatment-related AE was grade 1/2 somnolence caused by antipsychotic drug constituents of CLOVA cocktail (Table 2). A representative case showed marked reduction of the recurrent tumor assessed as partial response (PR) 9 months after beginning of CLOVA cocktail (Figure 5C). Immunohistochemistry using the respective antibodies (Supplementary Table 2) of pretreatment and autopsy tissue showed decrease in pGSS641, nestin, MIB-1 index (pretreatment 32.9% to autopsy 8.0%) and MGMT levels (Figure 5D), suggesting that the CLOVA cocktail treatment inhibited GSK3β activity, attenuated tumor invasion and enhanced TMZ effect, similar to in vivo experiment (Figures 3 and 4).
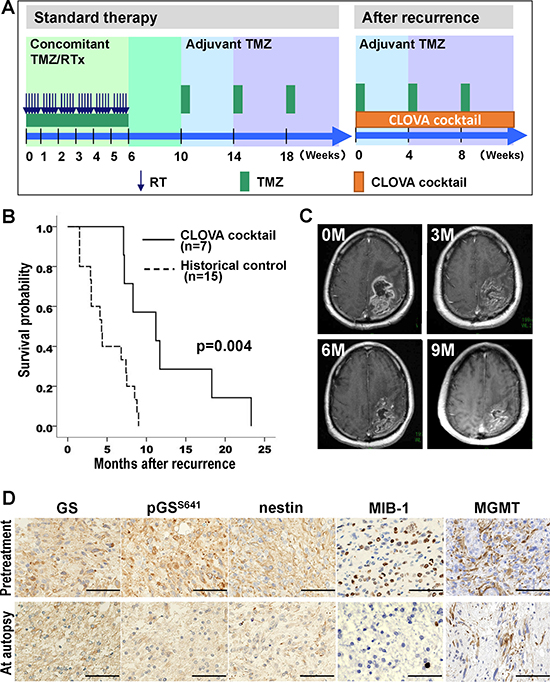
Figure 5: Phase I/II clinical study. (A) Treatment protocol in the clinical study. (B) Overall survival of the patients treated with or without CLOVA cocktail in combination with temozolomide. (C) MRI images of a representative case (Case No. 6 in Table 2) of 77 y.o. male with recurrent GBM of the left parietal lobe. Three (3M), six (6M), and nine months (9M) after beginning of CLOVA cocktail (0M), the recurrent tumor adjacent to the resection cavity shrank continuously. All MRI images were post-contrast axial T1-weighted. (D) Immunohistochemical findings of the recurrent tumor before the treatment and the tumor obtained by autopsy in the case shown in C. Scale bar 50 μm; GS, glycogen synthase; pGSS641, GS phosphorylated at serine 641 residue; MGMT, O6-methylguanine-DNA methyltransferase; RTx, radiotherapy; TMZ, temozolomide.
Table 2: Patient characteristic enrolled and the results of the clinical study
Case |
Age |
Sex |
KPS |
MGMT* |
Response† |
RPA class |
OS after first recurrence (week) |
AEs¶ |
|---|---|---|---|---|---|---|---|---|
1 |
70 |
Male |
20 |
M |
PR |
7 |
73.1 |
2 |
2 |
66 |
Male |
50 |
U |
SD |
7 |
28.6 |
1 |
3 |
64 |
Female |
30 |
M |
SD |
7 |
28.4 |
1 |
4 |
75 |
Male |
40 |
U |
SD |
7 |
93.1 |
1 |
5 |
55 |
Male |
50 |
U |
SD |
7 |
33.0 |
1 |
6 |
77 |
Male |
50 |
M |
SD |
7 |
46.7 |
2 |
7 |
60 |
Female |
50 |
M |
SD |
7 |
44.9 |
1 |
Median |
66 |
Male 5 Female 2 |
50 |
M 4 U 3 |
PR 1 SD 6 |
7 |
44.9 |
|
Historical control |
72 (median) |
Male 8 Female 7 |
40 |
M 8 U 6 ND 1 |
SD 3 PD 12 |
7 (median) |
17.3 (median) |
Abbreviations: AE, adverse event; KPS, Karnofsky Performance Status; MGMT, O6-methylguanine-DNA-methyltransferase; PD, progressive disease; PFS, progression-free survival; PR, partial response; OS, overall survival; RPA, recursive partitioning analysis; SD, stable disease.
*MGMT promoter status; ND, not detectable; M, methylated; U, unmethylated.
†Response was assessed according to Macdonald criteria.
¶AE score was based on Common Terminology Criteria for Adverse Events (CTCAE) version 4.0.
DISCUSSION
In this study, we first demonstrated the independent prognostic relevance of tumor-GSK3β activity as detected by the increased level of pGSK3βY216 that inversely associated with survival of GBM patients, while no correlation was found between the expression level of mRNA and total protein and the prognosis (data not shown). This suggests aberrant GSK3β activity in the tumor cells as a new prognostic factor in GBM and clinically rationalizes the strategy of targeting of this kinase for treatment of GBM. We also found therapeutic effects of the existent GSK3β-inhibiting medicines against GBM in the experimental and preclinical settings as well as a clinical study for the chemotherapy-resistant refractory patients. Previously, we and others have reported that inhibition of GSK3β activity attenuated proliferation and invasion of GBM cells and sensitized them to chemotherapeutic agents and radiation by modulating distinctive molecular pathways [19, 29]. Consistent with our earlier study [29], we confirmed that inactivation of GSK3β suppressed the molecular axis involving FAK and Rac1 in the present preclinical study. It is notable that the cocktail of GSK3β-inhibiting medicines tested in this study attenuated survival, migration and invasion of GBM cells and animal-model tumors. This cocktail also revitalized TMZ against recurrent GBMs and this effect was associated with decreased MGMT expression in the patient, thereby resulting in improvement of prognosis in patients of GSK high group in spite of low EOR. Our present study reinforces the cancer therapeutic effect of GSK3β inhibition by repurposing these medicines for GBM. It also suggests the necessity for the development and approval of specific GSK3β inhibitors to achieve efficient therapeutic effect for diseases including cancer.
Combination of multiple drugs with different mechanisms of action and/or different binding sites of a same target is often required for cancer chemotherapy to optimize the therapeutic effects, minimize the adverse effects, and prevent the development of treatment resistance [32, 33]. CLOVA cocktail showed additive effect on GBM proliferation and invasion (Figures 2 and 3; Supplementary Figures 3 and 4) but no adverse effect in the treated mice. Notably, cell viability showed no difference between control group and the group treated with CLOVA cocktail at the point of 8 hr when invasion assay was evaluated (data not shown). Nowicki et al also demonstrated that the effect of GSK3β inhibition on migration occurred earlier time points than effects on cell viability [34]. There are several ways of inhibiting GSK3β activity. They include up-regulation of pGSK3βS9 (inactive form), down-regulation of pGSK3βY216 (active form) and targeting the catalytic domain and/or the adenosine triphosphate (ATP)-binding pocket structure in this enzyme (Supplementary Figure 7) [9]. Cimetidine potently blocks the catalytic domain of GSK3β. Lithium has dual inhibitory effects on GSK3β to induce phosphorylation of serine 9 (S9) residue and to compete with Mg2+ that is required for GSK3β-dependent substrate phosphorylation. Olanzapine also increases the level of S9 phosphorylation and docks with the ATP-binding pocket of GSK3β. Valproate directly inhibits the activated GSK3β wherein Y216 residue is phosphorylated. Aberrant regulation of pGSK3βY216 could promote the malignant features of GBM [18, 19, 35] since it is an independent prognostic factor for GBM patient in the present study. Elevated pGSK3βY216 fraction contributed the malignant feature of pancreatic cancer [36] and inhibition of pGSK3βY216 enhanced the effect of imatinib inducing apoptosis in chronic myeloid leukemia [35]. These knowledge and observations support the present study showing that lithium and valproate inhibiting pGSK3βY216 exhibited stronger antitumoral effect than cimetidine and olanzapine that induce pGSK3βS9.
We define or regard the CLOVA cocktail as a single drug (the mixture of four constituents) basically in this study, and showed its effects against GBM cells proliferation and invasion. This effect is consistent with a substantial number of previous studies including ours showing the tumor-promoting roles of GSK3β, therapeutic effect of its inhibition and the underlying biological mechanisms in various cancer types including GBM [9–16]. The less effect of cimetidine and olanzapine (both repurposed, but not specific GSK3β inhibitors) on GBM cell proliferation at their limited dose ranges does not always make a denial of the solid evidence for the critical role of GSK3β in promoting proliferation of tumor cells including GBM.
Each of the GSK3β-inhibiting medicines has unique cancer therapeutic mechanisms other than its GSK3β-inhibitory effect. Cimetidine is a histamine H2 receptor antagonist prescribed for gastroduodenal ulcers and its anti-tumor activity was reported in many cancer types including glioma. It was shown to attenuate cell proliferation and migration by blocking histamine that promotes tumor angiogenesis [37, 38]. Lithium is a classical, non-competent GSK3β inhibitor [23] and prescribed for the treatment and prophylaxis of bipolar mood disorders and depression [39]. A previous study showed a reversible dose-dependent effect of lithium on attenuation of glioma cell invasion via inhibition of GSK3β activity [34]. Olanzapine is one of the most frequently prescribed anti-psychotic agents for schizophrenia and bipolar disorder [40]. It has attracted the attention of neuro-oncologists because of its ability to enhance the cytotoxic effects of TMZ on GBM [40]. GSK3β inhibition by olanzapine stimulated adenosine monophosphate (AMP)-activated protein kinase catabolic action followed by induction of p53-dependent autophagy, which enhances the pro-apoptotic effect of TMZ [41]. Valproate is approved for treatment of epileptic seizure, bipolar disorder, and migraine. It acts as a histone deacetylase inhibitor [42] that alters the chromatin structure, consequently increasing DNA accessibility to anticancer drugs and enhancing the effect of radiation [43, 44], in addition to other anticancer mechanisms [45]. In addition to their effects on GSK3β, lithium and valproate induced autophagy via inhibition of inositol monophosphatase [46] and modulation of oxidative stress [47], respectively, resulting in cancer therapeutic effect. Anti-tumor effects of valproate were tested by a number of clinical trials [48] and some studies showed significant survival in GBM patients treated with valproate [43, 45, 47]. Recently, a meta-analysis invalidated the use of VPA for reasons other than seizure control in patients with newly diagnosed GBM outside clinical trials [50], suggesting the limitations of single medication with GSK3β-inhibitory drug.
Based on the results of our experimental and preclinical studies shown here, we conducted a clinical study for the TMZ-resistant recurrent GBM patients by repositioning of the GSKβ-inhibiting medicines in combination with TMZ (UMIN:00005111). Patients treated with this regimen showed significant longer survival (median survival 11.2 months after recurrence) than historical control (median survival 4.4 months) treated with TMZ alone even though all the former patients were classified as RPA 7 in which the bad prognosis is predicted [31]. Immunohistochemical analysis of the patients’ tumors (Figure 4) suggested that the repurposed drugs inhibited GSK3β activity in the tumor cells, decreased expression of MIB-1 cell proliferation marker, and reduced invasion by the residual tumor cells. Although this clinical study has limitations such as single institution and small number of patients enrolled, it was first to provide the evidence of GSK3β-targeted cancer therapy based on drug repositioning. Not all, but combination of some GSK3β-inhibiting medicines might be enough for inhibition of aberrant GSK3β in the tumors. The next step toward clinical translation of this therapeutic strategy is to plan a large-scale, prospective multicenter trial to establish the optimum combination of the GSK3β-inhibiting medicines. A subsequent trial is also important to compare the effects of them in combination with TMZ and with bevacizumab. It will clarify whether the GSK3β-targeted therapy can prevent the GBM invasion enhanced in the bevacizumab-resistant tumors [51, 52].
Accumulating evidence has shown that inhibition of GSK3β provides dual benefits for the treatment of GBM patients by attenuation of tumor progression [17, 29] and protection from neurodegenerative effects of irradiation [53, 54]. This study also supports and promotes future clinical translation of the GSK3β-targeted therapy by drug repositioning. To ascertain the effect of GSK3β-targeted therapy with repurposing drugs, large multicenter clinical trial is currently in progress to validate the efficacy and safety of CLOVA cocktail in combination with TMZ, and best combination of the drugs composed of the cocktail in patients with recurrent GBM.
MATERIALS AND METHODS
Established predictive factors and GSK3β activity in the tumor of GBM patients
We examined 57 patients with GBM (Supplementary Table 1) for influence of tumor-GSK3β activity on their survival. This study was approved by Kanazawa University Medical Ethics Committee. Fresh tumor tissues were obtained by biopsy and surgery and the remaining tissue samples were fixed in 4% paraformaldehyde and embedded in paraffin for routine histopathologic examination and immunohistochemical analysis. The histological diagnosis was determined according to the revised World Health Organization criteria [55].
Established predictive factors including age [56], Karnofsky Performance Status (KPS) [56], EOR [57], and methylation in O6-methylguanine-DNA methyltransferase (MGMT) gene promoter [58] were analyzed in these patients. Progression-free survival (PFS) and overall survival (OS) were calculated from the initial operation to last magnetic resonance imaging (MRI) and to death, respectively. Log-rank test for univariate analysis was performed to determine statistical significance of Kaplan-Meier survival curve. Cox proportional hazard regression model was used to identify the multivariate predictors of survival.
Methylation of MGMT promoter was examined by methylation-specific PCR. Genomic DNA was extracted from paraffin sections of tumor by using QIAamp DNA FFPE Tissue kit™ (QIAGEN, Hilden, Germany). Sodium bisulfite conversion of 1 μg DNA was performed using an EpiTect Bisulfite Kit (QIAGEN) according to the manufacture’s protocol. Methylation-specific PCR of bisulfite-converted DNA was carried out by a nested, two-stage PCR approach as described previously [59] using GeneAmp PCR System 2700 (Applied Biosystems, Foster city, CA, USA). U87 and U138 cell lines were used as methylated and unmethylated controls, respectively. Amplified PCR products were separated by 3% agarose gel electrophoresis and visualized with ethidium bromide.
Tumor GSK3β activity was examined immunohistochemically for the level of phosphorylation at tyrosine (Y) 216 residue (pGSK3βY216). Representative paraffin sections of the tumors were immunostained with GSK3β and pGSK3βY216 antibodies (BD Biosciences, San Jose, CA, USA) (Supplementary Table 2), using Envision+ Kit (DAKO Japan, Kyoto, Japan) as described previously [60]. Images were acquired with a BZ-X700 microscope (Keyence, Osaka, Japan) and digitally processed with the Keyence Analysis Software. The patients were classified into 2 groups depending on the level of pGSK3βY216 as “GSK high” (≥ 50% of the tumor cells were labeled) and “GSK low” (< 50% of the tumor cells were labeled). Immunohistochemical evaluation was carried out by neuropathologist (H.S.) and oncologist (T.M.) in independent readings.
Cell culture
Human glioma cell lines T98, U87, U251, and U138 were obtained from American Type Culture Collection (ATCC) in 2009. These cell lines were characterized in the resource institute by short tandem repeat profile analysis. Authentication of the cell lines was unnecessary because cells were expanded by culturing them for less than two passages and stored at −80°C. Low-passage cells were used for experiments within the period of 6 months after resuscitation. They were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37°C with 5% CO2. All cells were Mycoplasma free.
Existent GSK3β-inhibiting medicines and temozolomide
Six GSK3β-inhibitory drugs were reported; lithium [61, 62], cimetidine [63], gemifloxacin [63], hydroxychloroquine [63], olanzapine [64], and valproate [24, 65]. Of these, 4 drugs were selected because of their general usage for the symptoms of the patients suffering from brain tumor; cimetidine for gastroduodenal ulcer, lithium and olanzapine for mood disorder, and valproate for seizure.
imetidine, lithium, olanzapine and valproate were purchased from WAKO Pure Chemicals (Osaka, Japan). For stock solutions, cimetidine and olanzapine were dissolved in dimethyl sulfoxide (DMSO), and lithium and valproate were dissolved in distilled water. As described in the Results, the concentrations of these drugs used in culture medium were as follows: cimetidine: 0.1, 0.5, and 1 mM; lithium: 1, 5, and 10 mM; olanzapine: 0.1, 0.5, and 1.0 μM; and valproate: 1, 5, and 10 mM. The combination of these drugs at their lowest concentration was referred as “CLOVA cocktail” by taking the initial letters of each drug.
Temozolomide (TMZ) was purchased from Sigma-Aldrich (St Louis, MO, USA). The concentration of this agent was 200 μM for cell culture and 100 mg/kg for animal model experiment.
Western blot analysis
Cellular protein was extracted from cultured cells following the treatment using lysis buffer (Sigma-Aldrich) containing a mixture of protease and phosphatase inhibitors (Sigma-Aldrich). A 15-μg aliquot of whole protein extract was analyzed by Western immunoblot for the protein of interest (Supplementary Table 2), as described previously [66]. β-actin was used as loading control. Immunoblot signals were measured using the CS analyzer (version 2.0; ATTO, Tokyo, Japan).
Cell invasion assay
Cell invasion assays were performed using modified Boyden chambers consisting of Transwell with pre-coated Matrigel membrane filter inserts in 24-well tissue culture plates (BD Biosciences) as described previously [66]. Serum-deprived cells suspended in DMEM containing 0.1% fetal bovine serum (FBS) were added to each Transwell. After incubation at 37°C for 8 h, non-invading cells were removed by wiping the upper side of the membrane, and the invading cells were fixed with methanol and stained with Diff-Quick Kit (Sysmex, Kobe, Japan). The invading cells on the filter were counted from 8 randomly selected high-power microscopic fields. The mean number of cells and standard deviations were calculated.
Cell proliferation assay
Alamar Blue assay (Biosource, Camarillo, CA, USA) was performed according to manufacturer’s manual. Practically, 1,000 cells of each population were seeded in wells of 96-well plastic plates in 200 μl of culture medium supplemented with 0.1% FBS. The plates were incubated for 4 h at 37°C, and 20 μl Alamar Blue (10% of total volume) was added to the cells and incubated. The plate was read on a fluorescence plate reader (excitation, 30 nm; emission, 590 nm) at 0, 24, 48, 72, and 96 h. Averages of the fluorescence values were calculated and plotted. To investigate the influence of drugs on cell proliferation, cells were serum starved for 24 h and seeded in the proliferation assay format. The cells were then treated with various concentrations of each drug, CLOVA cocktail, TMZ, or with the latter two in combination.
Mouse model of GBM and treatment
Following an institutional review board-approved protocol, we generated a mouse brain tumor model of human GBM by retrovirus-mediated introduction of the mutant K-ras gene (K-rasG12V) in neurospheres derived from the brain of p16Ink4A−/−/p19Arf−/− mouse and transplantation of them into the brain of wild-type mice according to our previous study [67]. The activation of GSK3β was validated both in the cultured sphere and the in vivo developed tumor by Western blot (data not shown). These mice were treated with the CLOVA cocktail or TMZ either alone or in combination. Practically, the 20 mice were randomly assigned to four groups for treatment with H2O/DMSO as control (n = 5), CLOVA cocktail alone (n = 5), TMZ alone (n = 5), and CLOVA cocktail and TMZ in combination (n = 5). Each drug of CLOVA cocktail at dose equivalent to the human dose was normalized to mice weights as follows; cimetidine 13.3 mg/kg; lithium 6.7 mg/kg; olanzapine 167 μg/kg; and valproate 13.3 mg/kg. CLOVA cocktail was orally administered every day and TMZ was administered intraperitoneally at 100 mg/kg for first 5 days. Following 2 weeks of treatment, all 20 mice were euthanized. For survival study, the 10 mice were treated with H2O/DMSO (n = 5) or CLOVA cocktail (n = 5). All animal experiments followed the Guidelines for the Care and Use of Laboratory Animals at Kanazawa University that covers the national guideline.
Representative paraffin sections of the tumors were immunostained for glycogen synthase phosphorylated at S641 residue (pGSS641), MIB-1, nestin, and focal adhesion kinase phosphorylated at Y397 and Y861 residue (pFAKY397 and pFAKY861) using the respective antibodies (Supplementary Table 2) as described above. Nestin-positive tumor cell clusters were scored to evaluate the degree of invasion as described previously [68]. Representative paraffin sections of the tumors were immunostained with a 1:50 dilution of anti-active Rac1 monoclonal antibody (NewEast Biosciences, King of Prussia, PA, USA) and then incubated in biotinylated horse anti-mouse IgG antibody (1:50; Vector Laboratories, Burlingame, CA, USA), followed by incubation with Alexa Fluor 594-labelled streptavidin (1:500; Vector Laboratories). Non-immune mouse serum at 1:50 dilution was used as negative control in immunostaining. The stained sections were mounted with the mounting medium for fluorescence with 4′,6′-diamidino-2-phenylindole (DAPI; Santa Cruz Biotechnology, Heidelberg, Germany). Images were captured with a BZ-X700 microscope (Keyence).
Design of clinical study
The single center, single-arm, clinical study investigated the efficacy and safety of CLOVA cocktail in combination with TMZ in patients with recurrent GBM in Kanazawa University Hospital from January 2009 to October 2010 under approval by Medical Ethics Committee of Kanazawa University (UMIN Clinical Trial Registry: UMIN000005111). Written informed consent was obtained from all patients. Primary endpoint was OS after first recurrence. Secondary endpoint was safety compared with matched historical control population treated with TMZ alone.
Patients
Adult patients (age ≥ 18 years) with histologically proven recurrent GBM after standard chemoradiation therapy with TMZ were eligible for the study (Table 2).
Treatment
CLOVA cocktail comprising 800 mg cimetidine, 400 mg lithium, 10 mg olanzapine, and 800 mg valproate was orally administered daily. Dose of each drug was determined based on medical package insert, and therapeutic drug monitoring of lithium and valproate was performed every month. Maintenance TMZ at 200 mg/m2/day was administered for 5 consecutive days every 4 weeks (Figure 5A).
Efficacy and safety assessments
A baseline gadolinium-enhanced MRI scan was performed within 1 week before beginning the treatment. MRI was repeated every 1 month. Progression was based on investigators’ clinical and radiological assessment according to Macdonald criteria [30]. Safety was evaluated by descriptively summarizing adverse events (AEs), laboratory assessments, and physical examinations. All AEs were recorded according to Common Terminology Criteria for Adverse Events (CTCAE) version 4.0.
Matched historical controls
Matched historical controls were identified from records of patients with recurrent GBM who had received treatment with maintenance TMZ at 200 mg/m2/day in Kanazawa University Hospital (Supplementary Table 3) same as the patients enrolled the study.
Statistical analyses
Statistical significance was determined using Student’s t-test, Mann-Whitney U test and Fisher’s exact test for comparison of two groups as appropriate. Log-rank analysis was used to determine statistical significance of Kaplan-Meier survival curve. All analyses were performed using SPSS statistical package version 19 for Macintosh (IBM Japan Ltd., Tokyo, Japan). The significance level was set at p = 0.05.
ACKNOWLEDGMENTS
The authors are grateful for Erika Komura for help with immunohistochemistry.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
GRANT SUPPORT
Grant-in-Aids for Scientific Research (M. Nakada; T. Minamoto) from the Japanese Ministry of Education, Science, Sports, Technology and Culture; from the Ministry of Health, Labour and Welfare; from the Japan Society for the Promotion of Science; the grants from Foundation for Promotion of Cancer Research (M. Nakada); Mochida Memorial Foundation for Medical and Pharmaceutical Research (M. Nakada); Princess Takamatsu Cancer Research Fund (M. Nakada); and the Extramural Collaborative Research Grant of Cancer Research Institute, Kanazawa University (T. Furuta; T. Minamoto).
REFERENCES
1. Slotty PJ, Siantidis B, Beez T, Steiger HJ, Sabel M. The impact of improved treatment strategies on overall survival in glioblastoma patients. Acta Neurochir (Wien). 2013; 155:959–963.
2. Xie Q, Mittal S, Berens ME. Targeting adaptive glioblastoma: an overview of proliferation and invasion. Neuro Oncol. 2014; 16:1575–1584.
3. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352:987–996.
4. Westphal M, Hilt DC, Bortey E, Delavault P, Olivares R, Warnke PC, Whittle IR, Jaaskelainen J, Ram Z. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003; 5:79–88.
5. Brem H, Piantadosi S, Burger PC, Walker M, Selker R, Vick NA, Black K, Sisti M, Brem S, Mohr G, Muller P, Morawetz R, Schold SC, et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. The Polymer-brain Tumor Treatment Group. Lancet. 1995; 345:1008–1012.
6. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, Jeraj R, Brown PD, Jaeckle KA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014; 370:699–708.
7. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, Brandes AA, Hilton M, Abrey L, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014; 370:709–722.
8. Nakada M, Kita D, Watanabe T, Hayashi Y, Hamada J. The mechanism of chemoresistance against tyrosine kinase inhibitors in malignant glioma. Brain Tumor Pathol. 2014; 31:198–207.
9. Nakada M, Minamoto T, Pyko IV, Hayashi Y, Hamada JI. The pivotal role of GSK3β in glioma biology. Molecular Targets of CNS Tumors, ed. Miklos Garami, InTech, 2011; 567–590.
10. Miyashita K, Nakada M, Shakoori A, Ishigaki Y, Shimasaki T, Motoo Y, Kawakami K, Minamoto T. An emerging strategy for cancer treatment targeting aberrant glycogen synthase kinase 3β. Anticancer Agents Med Chem. 2009; 9:1114–1122.
11. Atkins RJ, Stylli SS, Luwor RB, Kaye AH, Hovens CM. Glycogen synthase kinase-3β (GSK-3β) and its dysregulation in glioblastoma multiforme. J Clin Neurosci. 2013; 20:1185–1192.
12. Domoto T, Pyko IV, Furuta T, Miyashita K, Uehara M, Shimasaki T, Nakada M, Minamoto T. Glycogen synthase kinase 3β is a pivotal mediator in cancer invasion and resistance to therapy. Cancer Sci. 2016; 107:1363–1372.
13. McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL, Montalto G, D'Assoro AB, Libra M, Nicoletti F, Maestro R, Basecke J, Rakus D, et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget. 2014; 5:2881–911. doi: 10.18632/oncotarget.2037.
14. Osolodkin DI, Palyulin VA, Ze rov NS. Glycogen synthase kinase 3 as an anticancer drug target: novel experimental findings and trends in the design of inhibitors. Curr Pharm Des. 2013; 19:665–79.
15. Minamoto T, Kotake M, Nakada M, Shimasaki T, Motoo Y, Kawakami K. Distinct pathologic role for glycogen synthase kinase 3β in colorectal cancer progression. Colorectal Cancer Biology-From Genes to Tumor, ed. Rajunor Ettarh, In Tech. 2012; 107–134.
16. Ougolkov AV, Billadeau DD. Targeting GSK-3: a promising approach for cancer therapy? Future Oncol. 2006; 2:91–100.
17. Pyko IV, Nakada M, Sabit H, Teng L, Furuyama N, Hayashi Y, Kawakami K, Minamoto T, Fedulau AS, Hamada J. Glycogen synthase kinase 3β inhibition sensitizes human glioblastoma cells to temozolomide by affecting O6-methylguanine DNA methyltransferase promoter methylation via c-Myc signaling. Carcinogenesis. 2013; 34:2206–2217.
18. Miyashita K, Kawakami K, Nakada M, Mai W, Shakoori A, Fujisawa H, Hayashi Y, Hamada J, Minamoto T. Potential therapeutic effect of glycogen synthase kinase 3β inhibition against human glioblastoma. Clin Cancer Res. 2009; 15:887–897.
19. Kotliarova S, Pastorino S, Kovell LC, Kotliarov Y, Song H, Zhang W, Bailey R, Maric D, Zenklusen JC, Lee J, Fine HA. Glycogen synthase kinase-3 inhibition induces glioma cell death through c-MYC, nuclear factor-κB, and glucose regulation. Cancer Res. 2008; 68:6643–6651.
20. Strittmatter SM. Overcoming drug development bottlenecks with repurposing: old drugs learn new tricks. Nat Med. 2014; 20:590–591.
21. Tobinick EL. The value of drug repositioning in the current pharmaceutical market. Drug News Perspect. 2009; 22:119–125.
22. Taha MO, Bustanji Y, Al-Ghussein MA, Mohammad M, Zalloum H, Al-Masri IM, Atallah N. Pharmacophore modeling, quantitative structure-activity relationship analysis, and in silico screening reveal potent glycogen synthase kinase-3β inhibitory activities for cimetidine, hydroxychloroquine, and gemifloxacin. J Med Chem. 2008; 51:2062–2077.
23. Jope RS. Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends in Pharmacological Sciences. 2003; 24:441–443.
24. Chen G, Huang LD, Jiang YM, Manji HK. The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. J Neurochem. 1999; 72:1327–1330.
25. Arakawa R, Okumura M, Ito H, Takano A, Takahashi H, Takano H, Maeda J, Okubo Y, Suhara T. Positron emission tomography measurement of dopamine D receptor occupancy in the pituitary and cerebral cortex: relation to antipsychotic-induced hyperprolactinemia. J Clin Psychiatry. 2010; 71:1131–1137.
26. Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014; 14:598–610.
27. Fortin Ensign SP, Mathews IT, Symons MH, Berens ME, Tran NL. Implications of Rho GTPase signaling in glioma cell invasion and tumor progression. Front Oncol. 2013; 3:241.
28. Golubovskaya VM, Huang G, Ho B, Yemma M, Morrison CD, Lee J, Eliceiri BP, Cance WG. Pharmacologic blockade of FAK autophosphorylation decreases human glioblastoma tumor growth and synergizes with temozolomide. Mol Cancer Ther. 2013; 12:162–172.
29. Chikano Y, Domoto T, Furuta T, Sabit H, Kitano-Tamura A, Pyko IV, Takino T, Sai Y, Hayashi Y, Sato H, Miyamoto K, Nakada M, Minamoto T. Glycogen synthase kinase 3β sustains invasion of glioblastoma via the focal adhesion kinase, Rac1, and c-Jun N-terminal kinase-mediated pathway. Mol Cancer Ther. 2015; 14:564–574.
30. Macdonald DR, Cascino TL, Schold SC, Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990; 8:1277–1280.
31. Carson KA, Grossman SA, Fisher JD, Shaw EG. Prognostic factors for survival in adult patients with recurrent glioma enrolled onto the new approaches to brain tumor therapy CNS consortium phase I and II clinical trials. J Clin Oncol. 2007; 25:2601–2606.
32. Woodcock J, Griffin JP, Behrman RE. Development of novel combination therapies. N Engl J Med. 2011; 364:985–987.
33. Lynce F, Swain SM. Pertuzumab for the treatment of breast cancer. Cancer Invest. 2014; 32:430–438.
34. Nowicki MO, Dmitrieva N, Stein AM, Cutter JL, Godlewski J, Saeki Y, Nita M, Berens ME, Sander LM, Newton HB, Chiocca EA, Lawler S. Lithium inhibits invasion of glioma cells; possible involvement of glycogen synthase kinase-3. Neuro Oncol. 2008; 10:690–699.
35. Reddiconto G, Toto C, Palama I, De Leo S, de Luca E, De Matteis S, Dini L, Passerini CG, Di Renzo N, Maffia M, Coluccia AM. Targeting of GSK3β promotes imatinib-mediated apoptosis in quiescent CD34+ chronic myeloid leukemia progenitors, preserving normal stem cells. Blood. 2012; 119:2335–2345.
36. Shimasaki T, Ishigaki Y, Nakamura Y, Takata T, Nakaya N, Nakajima H, Sato I, Zhao X, Kitano A, Kawakami K, Tanaka T, Takegami T, Tomosugi N, et al. Glycogen synthase kinase 3β inhibition sensitizes pancreatic cancer cells to gemcitabine. J Gastroenterol. 2012; 47:321–333.
37. Lefranc F, Yeaton P, Brotchi J, Kiss R. Cimetidine, an unexpected anti-tumor agent, and its potential for the treatment of glioblastoma (review). Int J Oncol. 2006; 28:1021–1030.
38. Van der Ven LT, Prinsen IM, Jansen GH, Roholl PJ, Defferrari R, Slater R, Den Otter W. Growth of cultured human glioma tumour cells can be regulated with histamine and histamine antagonists. Br J Cancer. 1993; 68:475–483.
39. Curran G, Ravindran A. Lithium for bipolar disorder: a review of the recent literature. Expert Rev Neurother. 2014; 14:1079–1098.
40. Karpel-Massler G, Kast RE, Westhoff M, Dwucet A, Welscher N, Nonnenmacher L, Hlavac M, Siegelin MD, Wirtz CR, Debatin K, Halatsch M. Olanzapine inhibits proliferation, migration and anchorage-independent growth in human glioblastoma cell lines and enhances temozolomide's antiproliferative effect. J Neurooncol. 2014; 122:21–33.
41. Kast RE, Karpel-Massler G, Halatsch ME. Can the therapeutic effects of temozolomide be potentiated by stimulating AMP-activated protein kinase with olanzepine and metformin? Br J Pharmacol. 2011; 164:1393–1396.
42. Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. Embo J. 2001; 20:6969–6978.
43. Guthrie GD, Eljamel S. Impact of particular antiepileptic drugs on the survival of patients with glioblastoma multiforme. J Neurosurg. 2013; 118:859–865.
44. Van Nifterik KA, Van den Berg J, Slotman BJ, Lafleur MV, Sminia P, Stalpers LJ. Valproic acid sensitizes human glioma cells for temozolomide and gamma-radiation. J Neurooncol. 2012; 107:61–67.
45. Kerkhof M, Dielemans JC, van Breemen MS, Zwinkels H, Walchenbach R, Taphoorn MJ, Vecht CJ. Effect of valproic acid on seizure control and on survival in patients with glioblastoma multiforme. Neuro Oncol. 2013; 15:961–967.
46. Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005; 170:1101–1111.
47. Fu J, Shao CJ, Chen FR, Ng HK, Chen ZP. Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro Oncol. 2010; 12:328–340.
48. Krauze AV, Myrehaug SD, Chang MG, Holdford DJ, Smith S, Shih J, Tofilon PJ, Fine HA, Camphausen K. A phase 2 study of concurrent radiation therapy, temozolomide, and the histone deacetylase inhibitor valproic acid for patients with glioblastoma. Int J Radiat Oncol Biol Phys. 2015; 92:986–92.
49. Felix FH, Trompieri NM, de Araujo OL, da Trindade KM, Fontenele JB. Potential role for valproate in the treatment of high risk brain tumors of childhood results from a retrospective observational cohort study. Pediatr Hematol Oncol. 2011; 28:556–570.
50. Happold C, Gorlia T, Chinot O, Gilbert MR, Nabors LB, Wick W, Pugh SL, Hegi M, Cloughesy T, Roth P, Reardon DA, Perry JR, Mehta MP, et al. Does valproic acid or Levetiracetam improve survival in glioblastoma? A pooled analysis of prospective clinical trials in newly diagnosed glioblastoma. J Clin Oncol. 2016; 34:731–739.
51. Fack F, Espedal H, Keunen O, Golebiewska A, Obad N, Harter PN, Mittelbronn M, Bahr O, Weyerbrock A, Stuhr L, Miletic H, Sakariassen PO, Stieber D, et al. Bevacizumab treatment induces metabolic adaptation toward anaerobic metabolism in glioblastomas. Acta Neuropathol. 2015; 129:115–131.
52. Piao Y, Liang J, Holmes L, Henry V, Sulman E and de Groot JF. Acquired resistance to anti-VEGF therapy in glioblastoma is associated with a mesenchymal transition. Clin Cancer Res. 2013; 19:4392–4403.
53. Thotala DK, Hallahan DE, Yazlovitskaya EM. Inhibition of glycogen synthase kinase 3 beta attenuates neurocognitive dysfunction resulting from cranial irradiation. Cancer Res. 2008; 68:5859–5868.
54. Yang ES, Nowsheen S, Wang T, Thotala DK, Xia F. Glycogen synthase kinase 3β inhibition enhances repair of DNA double-strand breaks in irradiated hippocampal neurons. Neuro Oncol. 2011; 13:459–470.
55. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007; 114:97–109.
56. Lacroix M, Abi-Said D, Fourney DR, Gokaslan ZL, Shi W, DeMonte F, Lang FF, McCutcheon IE, Hassenbusch SJ, Holland E, Hess K, Michael C, Miller D, et al. A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. J Neurosurg. 2001; 95:190–198.
57. Sanai N, Polley MY, McDermott MW, Parsa AT, Berger MS. An extent of resection threshold for newly diagnosed glioblastomas. J Neurosurg. 2011; 115:3–8.
58. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005; 352:997–1003.
59. Suzuki T, Nakada M, Yoshida Y, Nambu E, Furuyama N, Kita D, Hayashi Y, Hamada J. The correlation between promoter methylation status and the expression level of O6-methylguanine-DNA methyltransferase in recurrent glioma. Jpn J Clin Oncol. 2011; 41:190–196.
60. Furuta T, Nakada M, Misaki K, Sato Y, Hayashi Y, Nakanuma Y, Hamada J. Molecular analysis of a recurrent glioblastoma treated with bevacizumab. Brain Tumor Pathol. 2014; 31:32–39.
61. Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996; 93:8455–8459.
62. Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996; 6:1664–1668.
63. Taha MO, Bustanji Y, Al-Ghussein MA, Mohammad M, Zalloum H, Al-Masri IM, Atallah N. Pharmacophore modeling, quantitative structure-activity relationship analysis, and in silico screening reveal potent glycogen synthase kinase-3β inhibitory activities for cimetidine, hydroxychloroquine, and gemifloxacin. J Med Chem. 2008; 51:2062–2077.
64. Mohammad MK, Al-Masri IM, Taha MO, Al-Ghussein MA, Alkhatib HS, Najjar S, Bustanji Y. Olanzapine inhibits glycogen synthase kinase-3β: an investigation by docking simulation and experimental validation. Eur J Pharmacol. 2008; 584:185–191.
65. Kim AJ, Shi Y, Austin RC, Werstuck GH. Valproate protects cells from ER stress-induced lipid accumulation and apoptosis by inhibiting glycogen synthase kinase-3. J Cell Sci. 2005; 118:89–99.
66. Nakada M, Niska JA, Miyamori H, McDonough WS, Wu J, Sato H, Berens ME. The phosphorylation of EphB2 receptor regulates migration and invasion of human glioma cells. Cancer Res. 2004; 64:3179–3185.
67. Tamase A, Muraguchi T, Naka K, Tanaka S, Kinoshita M, Hoshii T, Ohmura M, Shugo H, Ooshio T, Nakada M, Sawamoto K, Onodera M, Matsumoto K, et al. Identification of tumor-initiating cells in a highly aggressive brain tumor using promoter activity of nucleostemin. Proc Natl Acad Sci USA. 2009; 106:17163–17168.
68. Wild-Bode C, Weller M, Rimner A, Dichgans J, Wick W. Sublethal irradiation promotes migration and invasiveness of glioma cells: implications for radiotherapy of human glioblastoma. Cancer Res. 2001; 61:2744–2750.