
INTRODUCTION
Pulmonary artery hypertension (PAH) is a complicated and fatal disease. Although progress achieved during last two decades with the introduction of oral medical therapies, the prognosis of PAH remains poor, especially in patients with advanced right heart failure [1]. It is imperative to seek potentially new therapies that prevent the development and progression of PAH. In the past few years, many experiments have indicated an important role of the renin-angiotensin-aldosterone system (RAAS) and sympathetic nervous system (SNS), in particular aldosterone [2–7]. Recently, de Man F.S. et al. found that the progression of PAH was accompanied by systemic RAAS overactivity [2].
We [8] and other investigators [9, 10] have reported that renal denervation (RDN) has the ability to reduce SNS activity and rebalance RAAS. It is reasonable to investigate the impact of RDN on PAH. And limited experiments have discussed the optimal time for RDN to treat PAH. Using a rat model of monocrotaline (MCT) induced pulmonary remodeling, we sought to study these isssues.
RESULTS
Right ventricular function
At day 35, echocardiography revealed significant increases in right ventricular anterior wall thickness (RVAW, 1.56 ± 0.20 mm vs. 1.00 ± 0.25 mm, p < 0.05), Non-filling time of right ventricle (NFT; r, 133.33 ± 30.97 ms vs. 106.44 ± 18.23 ms, p < 0.05), and pulmonary ejection time (PET, 88.89 ± 4.63 vs. 73.78 ± 5.59, p < 0.05) in MCT-induced PAH group compared with the RDN24h group. Compared with MCT group, RVAW in RDN2w group (1.03 ± 0.15 mm vs. 1.56 ± 0.20 mm, p < 0.05) was significantly reduced (Table 1).
Table 1: Echocardiographic parameters at day 35
Group |
TV E/A |
RVAW(mm) |
NFT; r (ms) |
PA VTI (mm) |
PV peak Vel (mm/s) |
PET (ms) |
|---|---|---|---|---|---|---|
Control |
0.61 ± 0.08 |
0.51 ± 0.05 |
97.22 ± 5.67 |
41.96 ± 11.01 |
–960.4 ± 98.0 |
73.78 ± 7.88 |
RDN24h |
0.82 ± 0.27 |
1.00 ± 0.25*# |
106.44 ± 18.23* |
26.66 ± 2.97# |
–816.6 ± 102.6# |
73.78 ± 5.59* |
RDN2w |
0.89 ± 0.33 |
1.03 ± 0.15*# |
124.58 ± 7.91# |
26.10 ± 4.91# |
–694.69 ± 94.5# |
79.63 ± 8.98 |
MCT |
1.17 ± 0.36# |
1.56 ± 0.20# |
133.33 ± 30.97# |
23.75 ± 5.03# |
–699.9 ± 108.1# |
88.89 ± 4.63# |
TV E: Tricuspid valve E wave velocity;
TV A: Tricuspid valve A wave velocity;
TV E/A: Ratio of tricuspid valve E' to A';
RVAW: Right ventricular anterior wall;
NFT; r: Non-filling time (right ventricle);
PA VTI: Pulmonary Artery velocity time integral;
PV peak vel: Pulmonary Artery peak velocity;
PET:Pulmonary Ejection Time.
*P < 0.05, vs. MCT; #P < 0.05 vs. Control (n = 5 per group).
Body weight (BW), heart weight (HW) and the ratio of HW/BW
Compared with Control, MCT significantly decreased the body weight (BW) (287.33 ± 57.08 g vs. 348.36 ± 46.25 g, p < 0.05) of the rats, while there was no significant differences between RDN (RDN24h and RDN2w) groups and Control group. But RDN therapy had no influence on the ratio of HW/BW (Table 2).
Table 2: BW, HW, HW/BW
Group |
Day |
N |
BW (g) |
HW (mg) |
HW/BW (mg/g) |
|---|---|---|---|---|---|
Control |
35 |
8 |
348.36 ± 46.25 |
1084.0 ± 159.55 |
3.12 ± 0.30 |
RDN24h |
35 |
6 |
309.33 ± 22.29 |
1460.2 ± 133.42# |
4.73 ± 0.38# |
RDN2w |
35 |
6 |
298.02 ± 49.97 |
1486.9 ± 91.70# |
5.13 ± 1.04# |
MCT |
35 |
7 |
287.33 ± 57.08# |
1405.2 ± 174.15# |
5.06 ± 1.21# |
N: the number of rats; BW: body weight; HW: heart weight; Num: number; HW/BW: the ratio of heart weight to body weight; MCT: monocrotaline.
*P < 0.05, vs. MCT; #P < 0.05 vs. Control.
Fibrosis of lung tissue and pulmonary vessel
With regard to pulmonary fibrosis, collagen deposition was observed to be significantly higher in MCT challenged animals as measured by Masson’s trichrome staining. These changes were completely manifested 35 days after MCT injection and without intervention with RDN procedure. Remarkably, RDN treatment after monocrotaline injection 24 hours reversed lung tissue fibrosis (5.64% ± 1.57% RDN24h vs. 10.18% ± 3.90% MCT, p < 0.05) and pulmonary vascular fibrosis (8.87% ± 5.22% RDN24h vs. 14.92% ± 5.52% MCT, p < 0.05). But, when we performed RDN procedure after monocrotaline injection for 2 weeks, the improvements in lung tissue fibrosis (9.19% ± 2.27% RDN2w vs. 10.18% ± 3.90% MCT, p = 0.375) and pulmonary vascular fibrosis (12.05% ± 5.40% RDN2w vs. 14.92% ± 5.52% MCT, p = 0.303) effects of RDN were blunted (Figures 1 and 2).
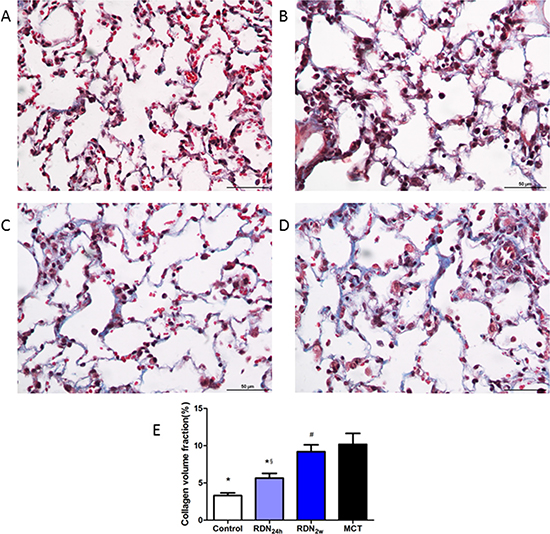
Figure 1: Effects of RDN on MCT-induced lung fibrosis histological section of lung. Masson’s Trichrome Staining (magnification× 400), the blue stands for fibrosis. (A) Control group (n = 8), (B) RDN24h group (n = 6), (C) RDN2w group (n = 6), (D) MCT group (n = 7). Comparison of collagen volume fraction (CVF,%) between four groups (E) *P < 0.05, vs. MCT; #P < 0.05, vs. Control; §P < 0.05, RDN24h vs. RDN2w.
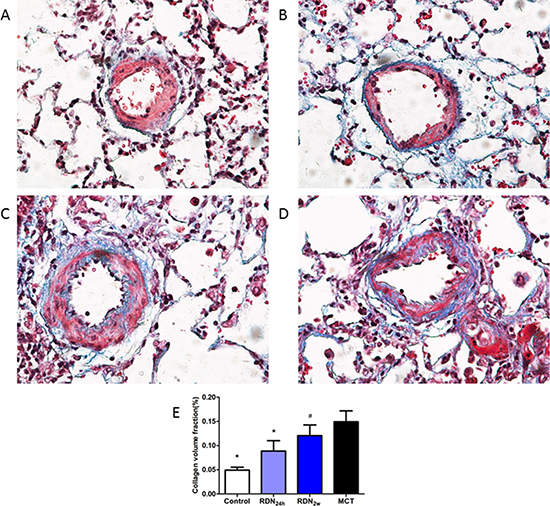
Figure 2: Effects of RDN on MCT-induced pulmonary vascular fibrosis histological section of pulmonary vascular. Masson’s Trichrome Staining (magnification× 400), the blue stands for fibrosis. (A) Control group (n = 8), (B) RDN24h group (n = 6), (C) RDN2w group (n = 6), (D) MCT group (n = 7). Comparison of collagen volume fraction (CVF,%) between four groups (E). *P < 0.05, vs. MCT; #P < 0.05, vs. Control.
Fibrosis of right ventricle and cross-sectional area of heart
RDN surgery performed in the earlier stage of PAH resulted in significantly reduced RVAW thickness (1.00 ± 0.25 mm vs. 1.56 ± 0.20 mm, p < 0.05) compared with MCT group (Figure 3). Monocrotaline injection results in pulmonary arterial hypertension and right heart failure, which were accompanied by myocardial and pulmonary fibrosis. The cross-sectional area of myocardial interstitial fibrosis was inhibited by earlier RDN therapy (11.39% ± 5.00% MCT vs. 6.96% ± 4.09% RDN24h, p < 0.05) which was increased by monocrotaline injection. However, there are no significant differences between RDN2w group and MCT group (Figure 4).
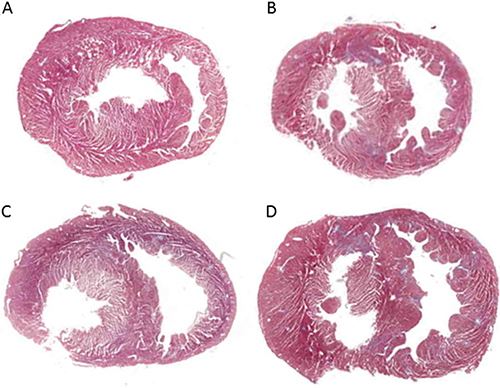
Figure 3: The Heart cross section. (A) Control group (n = 8), (B) RDN24h group (n = 6), (C) RDN2w group (n = 6), (D) MCT group (n = 7). Chamber on the left side is left ventricular (LV), chamber on the right side is right ventricular (RV), tissue between two chambers is interventricular septal (IVS).
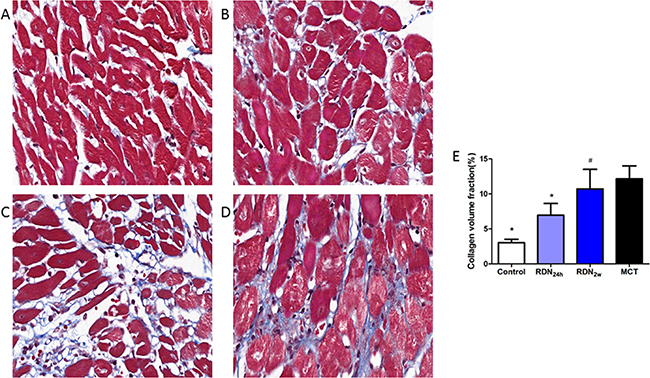
Figure 4: Right ventricular fibrosis histological section of right ventricular. Masson’s Trichrome Staining (magnification× 400), the blue stands for fibrosis. (A) Control group (n = 8), (B) RDN24h group (n = 6), (C) RDN2w group (n = 6), (D) MCT group (n = 7). Comparison of collagen volume fraction (CVF,%) between four groups (E). *P < 0.05, vs. MCT; #P < 0.05, vs. Control.
RDN reduces RAAS and SNS activity
Ang II, which regulates fibroblast proliferation, differentiation and extracellular matrix deposition, is an important pro-fibrosis factor. Compared with MCT group, RDN24h group significantly decreased plasma Ang II concentration (277.94 ± 110.00 pg/ml vs. 154.96 ± 55.98 pg/ml, p < 0.05), while only decreasing tendency of plasma Ang II concentration was observed in RDN2w group. ALD, a member of RAAS, was also significantly reduced in RDN24h (26.97 ± 6.53 pg/ml vs. 14.64 ± 3.70 pg/ml, p < 0.05) and RDN2w (26.97 ± 6.53 pg/ml vs. 16.87 ± 8.44 pg/ml, p < 0.05) group. To response to overactive SNS, postganglionic neurons release high concentration NE, RDN blunted this increase. Compared with MCT, NE concentration significantly decreased in RDN24h (84.98 ± 15.06 pg/ml vs. 56.90 ± 25.39 pg/ml, p < 0.05) and RDN2w (84.98 ± 15.06 pg/ml vs. 57.16 ± 30.00 pg/ml, p < 0.05) group (Figure 5).
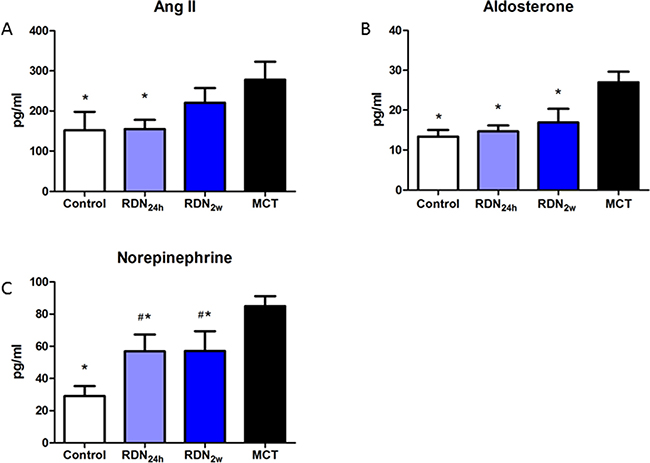
Figure 5: RDN Reduces RAAS and SNS activity effects of RDN on the changes of plasma Ang II. (A) aldosterone (B) noradrenaline (C) levels in the four groups. *P < 0.05, vs. MCT group; #P < 0.05, vs. Control group (n = 6 per group). Ang II, angiotensin II; RAAS, Renin–angiotensin–aldosterone system.
Kaplan-Meier survival analysis
During the follow-up in the current experiment, 4 of 10 rats died both in the RDN24h and RDN2w group, and 13 of 20 animals died in the MCT group. None rats in the Control group died. On Kaplan–Meier survival analysis, after 35 days of follow-up, the rats treated with RDN (RDN24h and RDN2w) had higher survival rate (60% vs. 35%) than MCT group (Figure 6).
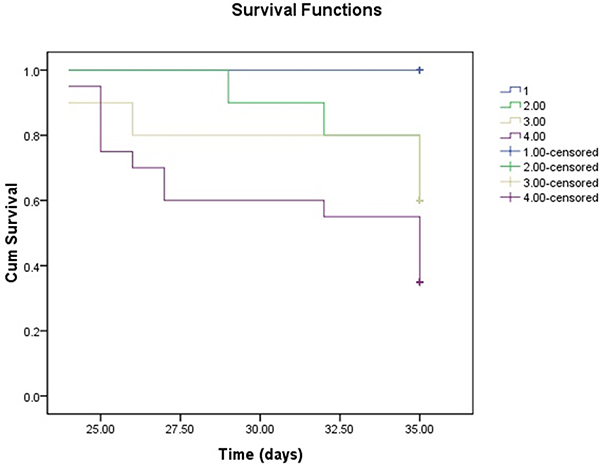
Figure 6: Kaplan-Meier survival analysis survival over the 35-day experiment was analyzed according to the daily recording of deaths by the standard Kaplan-Meier analysis with the log rank test.
DISCUSSION
The present study mainly demonstrated that: 1) MCT induces remarkable fibrosis in lung tissue, pulmonary vascular and right ventricular; 2) RDN decreases the activity of RAAS and SNS; 3) RDN prevents lung tissue, pulmonary vascular and right ventricular fibrosis and results in a time-dependent improvement in cardio-pulmonary remodeling.
MCT-induced pulmonary remodeling is a process from lung tissue and pulmonary vascular fibrosis to pulmonary hypertension, and then causes right ventricular remodeling and failure [11, 12]. In this process, neurohormonal systems such as the RAAS and SNS are up-regulated [2]. At 35 days, echocardiography revealed significant increases in right ventricular anterior wall thickness (RVAW) in MCT-induced pulmonary remodeling group compared with control. Compared with Control, MCT significantly increased the the Ratio of HW/BW of the rats. We observed high concentration plasma NE, out-of-balanced RAAS axis, and excessive collagen deposition of cardio-pulmonary. While RDN surgery performed in the earlier stage resulted in significant attenuation of MCT-induced disease progression.
Pulmonary remodeling is a transitional phase to pulmonary hypertension and right heart failure. Thus, it is of great importance to find a way to slow or even reverse the progression of pulmonary remodeling. Velez-Roa S et al. have reported that the activity of SNS was increased in patients with pulmonary hypertension, and sympathetic activation participated in pulmonary arteriolar remodeling [7]. Previous data revealed that β-blockers could delay the progression toward right heart failure, and partially preserves right ventricular function in pulmonary hypertension [13, 14]. Linz D et al. had reported that RDN reduced tyrosine hydroxylase-positive sympathetic nerve staining and resulted in lower norepinephrine levels [15]. In recent studies, they had reported that pulmonary artery denervation induces permanent sympathetic nerve injury and subsequent improvements in hemodynamics and pulmonary remodeling in humans and animals with pulmonary hypertension [11, 16, 17]. In our prior study, we also found that RDN could regulate the expression of β1,2-receptors, which contribute to inhibit cardiac remodeling [8]. In accordance with these studies, we concluded that RDN significantly reduced NE level. The involvement of the RAAS in myocardial fibrosis is evident in several pathological conditions such as hypertensive heart disease [18], congestive heart failure [19] and myocardial infarction [20]. There is experimental and clinical evidence indicating that activated RAAS is involved in the pathophysiology of pulmonary hypertension [2, 21–23]. Ang II, the major effector peptide of the RAAS, promotes vasoconstriction, inflammation, proliferation and fibrosis within the lung [24, 25]. And plasma aldosterone levels were elevated in experimental models of pulmonary hypertension and humans with the disease [26, 27]. The persistently elevated level of aldosterone initiates signaling pathways that promote vascular remodeling, impair vascular reactivity, and contributes to right ventricular dysfunction [28]. We [8] and others [9, 10] had reported that RDN could reduce SNS activity and rebalance RAAS. In this regard, RDN has been of extreme significance when evaluating the effects of direct reducing SNS and RAAS activity. In our current study, we found a prominently beneficial effect of RDN in the earlier phase of disease process. What of particular interest was that RDN inhibited cardiopulmonary fibrosis and slowed down the progression of right heart failure. But treatment with RDN in the later period of MCT-induced pulmonary hypertension had no significant influence on pulmonary hemodynamics and remodeling of cardiopulmonary.
The prominent reversal of cardiopulmonary fibrosis is a remarkable and noteworthy effect of RDN treatment. Fibrosis is a common pathway to organ injury and failure [29]. We observed a 44.6% reduction of lung fibrosis, 40.5% reduction in pulmonary vascular fibrosis and 38.9% reduction in right ventricular fibrosis following RDN at the 35-day time point. As deposition of fibrous tissue in heart or lung is associated with the increasing risk of adverse cardiovascular events [30], the ability of RDN preventing fibrosis might contribute to a better prognosis of PAH.
Pulmonary hypertension is commonly associated with increased afterload, which causes maladaptive remodeling of the right ventricle, characterized by hypertrophy and fibrosis [31]. In addition, persistent pressure overload induces cardiomyocyte death and contractile dysfunction, which eventually leads to end-organ failure. The beneficial outcomes of RDN treatment were associated with decreased right ventricular remodeling and improved right heart function. But these beneficial effects were blunted when RDN performed in the later period.
Earlier RDN treatment in the process of PAH disease could improve right ventricular function and reduce cardio-pulmonary fibrosis, and it also improved survival rate in MCT-induced PAH. These experimental findings suggest that RDN should be further investigated as a potential approach for treating earlier stage of PAH.
Study limitation
We should acknowledge the limitations in our study. Firstly, we failed to measure mean pulmonary arterial pressure at the end of this study. At day 35, we tried to test RV pressure and pulmonary artery pressure using right heart catheterization, but many rats in MCT group died when we opened their chests, and the collected data were too few to be analyzed. However, MCT-induced PAH is a well-established model, and both the mortality and echocardiographic data indicated that MCT had worked. Meanwhile, pulmonary remodeling was substantiated by histological analysis. Secondly, whether RDN is superior to oral drugs needs future investigation.
MATERIALS AND METHODS
Animals and experimental design
All procedures in this study were performed in accordance with the Guide for The Care and Use of Laboratory Animals (National Institutes of Health publication 8th edition, 2011) and were approved by the Nanjing Medical University Experimental Animal Care and Use Committee. The experiment was performed in male Sprague-Dawley rats weighing 200 ± 20 g (Nanjing Medical University Laboratory Animal Center), caged individually at controlled temperature and humidity with a 12-hour light/dark cycle. Echocardiography was performed at baseline and week 5. MCT injection was performed after echocardiography at baseline. Forty-eight male Sprague Dawley rats were randomized into 4 groups. Group 1 consisted of 8 rats that received normal saline and sham RDN procedure (Control). Group 2 comprised 10 rats that received MCT and RDN after 24 hour of intraperitoneally injected with 60 mg/kg MCT (MCT+RDN24h). Group 3 comprised 10 rats that received MCT and renal denervation after 2 weeks of intraperitoneally injected with 60 mg/kg MCT (MCT+RDN2w). Group 4 comprised 20 rats that received MCT and sham renal denervation procedure (MCT). At the end of the study, after the second echocardiography work-up and blood collection, all animals were euthanized with an overdose of pentobarbital sodium (200 mg/kg) by intraperitoneal injection.
MCT-Induced PAH
Forty-eight male Sprague Dawley rats were randomly assigned to four groups: Control group (n = 8), MCT+RDN24h group (n = 10), MCT+RDN2w group (n = 10) and MCT group (n = 20). MCT-induced PAH rats were intraperitoneally injected with 60 mg/kg monocrotaline [32–34] (Sigma, Switzerland), which was dissolved in 1 mol/L HCL, the PH was adjusted to 7.4 and diluted with PBS.
Renal denervation
With pentobarbital sodium (60 mg/kg intraperitoneal injection) anesthesia, bilateral renal denervation was performed in RDN24h and RDN2w group, whereas sham RDN procedure was performed in MCT and Control group. RDN was implemented as described previously [8]. Visible nerves along the renal arteries and veins were stripped and picked with 10× magnification. Chemical denervation was conducted by daubing the bilateral renal artery with 20% phenol solution in absolute alcohol. Then the arteries and veins were washed with isotonic saline. For sham RDN procedure, the operation was the same, but the renal arteries and veins were not isolated and the nerves were left intact.
Echocardiography
Cardiac structure and function were evaluated by echocardiography with Vevo2100-a high resolution imaging system (VisualSonics, Canada) with a MS-250, 16.0–21.0 MHZ imaging transducer at baseline and week 5. All of the rats were anesthetized by aether during the process of echocardiography work-up.
Histopathology
After perfusion with ice-cold PBS, the heart and lung were cut and fixed in 4% phosphate buffered formalin for 48–72 h at 4°C, then tissues were dehydrated and embedded in paraffin. The heart and lung tissues were fast frozen by liquid nitrogen, then moved to –80°C. Masson’s trichrome staining was performed to detect right ventricular and lung fibrosis. Five fields of each sample were randomly selected and collagen volume fraction (CVF) was assessed by Image-Pro Plus 6.0.
ELISA
Blood samples were obtained by EDTA-tubes, and then centrifuged at 3000 rpm at 4°C for 10 minutes to separate the plasma which stored at –80°C for later use. Plasma norepinephrine (NE), aldosterone (ALD) and angiotensin II (Ang II) levels at day 35 were measured using enzyme linked immunosorbent assay (ELISA) kits, according to the manufacturer’s instructions (Uscn Life Science Inc, Wuhan, China).
Kaplan-Meier survival analysis
Survival over the 35-day experiment was analyzed according to the daily recording of deaths by the standard Kaplan-Meier analysis with the log rank test.
Statistics
Data are expressed as mean ± SEM and analyzed by SPSS 16.0 (SPSS Inc, Chicago, IL, USA). For two-group comparison, data were analyzed with two-tailed unpaired t tests. For multiple-groups comparisons, data were performed using one-way ANOVA with LSD test. P < 0.05 was considered statistically significant.
CONCLUSIONS
In conclusions, RDN significantly prevents the overactivity of RAAS and SNS, and attenuates lung tissue, pulmonary vascular and right ventricular fibrosis in the earlier stage of pulmonary hypertension. Simultaneously, RDN improves the survival rate of PAH.
ACKNOWLEDGMENTS AND FUNDING
This study was supported by the Natural Science Fund of Health Administration of Jiangsu Province (No.H201302).
CONFLICTS OF INTEREST
All the authors declared no competing interests.
REFERENCES
1. Liu QQ, Jing ZC. The limits of oral therapy in pulmonary arterial hypertension management. Ther Clin Risk Manag. 2015; 11:1731–41. doi: 10.2147/TCRM.S49026.
2. de Man FS, Tu L, Handoko ML, Rain S, Ruiter G, Francois C, Schalij I, Dorfmuller P, Simonneau G, Fadel E, Perros F, Boonstra A, Postmus PE, et al. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012; 186:780–9. doi: 10.1164/rccm.201203-0411OC.
3. Shenoy V, Ferreira AJ, Qi Y, Fraga-Silva RA, Diez-Freire C, Dooies A, Jun JY, Sriramula S, Mariappan N, Pourang D, Venugopal CS, Francis J, Reudelhuber T, et al. The angiotensin-converting enzyme 2/angiogenesis-(1–7)/Mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med. 2010; 182:1065–72. doi: 10.1164/rccm.200912-1840OC.
4. Vasan RS, Evans JC, Larson MG, Wilson PW, Meigs JB, Rifai N, Benjamin EJ, Levy D. Serum aldosterone and the incidence of hypertension in nonhypertensive persons. N Engl J Med. 2004; 351:33–41. doi: 10.1056/NEJMoa033263.
5. Ciarka A, Doan V, Velez-Roa S, Naeije R, van de Borne P. Prognostic significance of sympathetic nervous system activation in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010; 181:1269–75. doi: 10.1164/rccm.200912-1856OC.
6. Nootens M, Kaufmann E, Rector T, Toher C, Judd D, Francis GS, Rich S. Neurohormonal activation in patients with right ventricular failure from pulmonary hypertension: relation to hemodynamic variables and endothelin levels. J Am Coll Cardiol. 1995; 26:1581–5. doi: 10.1016/0735-109700399-1.
7. Velez-Roa S, Ciarka A, Najem B, Vachiery JL, Naeije R, van de Borne P. Increased sympathetic nerve activity in pulmonary artery hypertension. Circulation. 2004; 110:1308–12. doi: 10.1161/01.CIR.0000140724.90898.D3.
8. Li ZZ, Jiang H, Chen D, Liu Q, Geng J, Guo JQ, Sun RH, Zhu GQ, Shan QJ. Renal sympathetic denervation improves cardiac dysfunction in rats with chronic pressure overload. Physiol Res. 2015; 64:653–62.
9. Liang Z, Shi XM, Liu LF, Chen XP, Shan ZL, Lin K, Li J, Chen FK, Li YG, Guo HY, Wang YT. Renal denervation suppresses atrial fibrillation in a model of renal impairment. PLoS One. 2015; 10:e0124123. doi: 10.1371/journal.pone.0124123.
10. Linz D, Hohl M, Schutze J, Mahfoud F, Speer T, Linz B, Hubschle T, Juretschke HP, Dechend R, Geisel J, Rutten H, Bohm M. Progression of kidney injury and cardiac remodeling in obese spontaneously hypertensive rats: the role of renal sympathetic innervation. Am J Hypertens. 2015; 28:256–65. doi: 10.1093/ajh/hpu123.
11. Zhou L, Zhang J, Jiang XM, Xie DJ, Wang JS, Li L, Li B, Wang ZM, Rothman AM, Lawrie A, Chen SL. Pulmonary Artery Denervation Attenuates Pulmonary Arterial Remodeling in Dogs With Pulmonary Arterial Hypertension Induced by Dehydrogenized Monocrotaline. JACC Cardiovasc Interv. 2015; 8:2013–23. doi: 10.1016/j.jcin.2015.09.015.
12. Correia-Pinto J, Henriques-Coelho T, Roncon-Albuquerque R Jr, Lourenco AP, Melo-Rocha G, Vasques-Novoa F, Gillebert TC, Leite-Moreira AF. Time course and mechanisms of left ventricular systolic and diastolic dysfunction in monocrotaline-induced pulmonary hypertension. Basic Res Cardiol. 2009; 104:535–45. doi: 10.1007/s00395-009-0017-3.
13. de Man FS, Handoko ML, van Ballegoij JJ, Schalij I, Bogaards SJ, Postmus PE, van der Velden J, Westerhof N, Paulus WJ, Vonk-Noordegraaf A. Bisoprolol delays progression towards right heart failure in experimental pulmonary hypertension. Circ Heart Fail. 2012; 5:97–105. doi: 10.1161/CIRCHEARTFAILURE.111.964494.
14. Perros F, Ranchoux B, Izikki M, Bentebbal S, Happe C, Antigny F, Jourdon P, Dorfmuller P, Lecerf F, Fadel E, Simonneau G, Humbert M, Bogaard HJ, et al. Nebivolol for improving endothelial dysfunction, pulmonary vascular remodeling, and right heart function in pulmonary hypertension. J Am Coll Cardiol. 2015; 65:668–80. doi: 10.1016/j.jacc.2014.11.050.
15. Linz D, van Hunnik A, Hohl M, Mahfoud F, Wolf M, Neuberger HR, Casadei B, Reilly SN, Verheule S, Bohm M, Schotten U. Catheter-based renal denervation reduces atrial nerve sprouting and complexity of atrial fibrillation in goats. Circ Arrhythm Electrophysiol. 2015; 8:466–74. doi: 10.1161/CIRCEP.114.002453.
16. Chen SL, Zhang FF, Xu J, Xie DJ, Zhou L, Nguyen T, Stone GW. Pulmonary artery denervation to treat pulmonary arterial hypertension: the single-center, prospective, first-in-man PADN-1 study (first-in-man pulmonary artery denervation for treatment of pulmonary artery hypertension). J Am Coll Cardiol. 2013; 62:1092–100. doi: 10.1016/j.jacc.2013.05.075.
17. Rothman AM, Arnold ND, Chang W, Watson O, Swift AJ, Condliffe R, Elliot CA, Kiely DG, Suvarna SK, Gunn J, Lawrie A. Pulmonary artery denervation reduces pulmonary artery pressure and induces histological changes in an acute porcine model of pulmonary hypertension. Circ Cardiovasc Interv. 2015; 8:e002569. doi: 10.1161/CIRCINTERVENTIONS.115.002569.
18. Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res. 1990; 67:1355–64.
19. Weber KT, Brilla CG, Janicki JS. Myocardial fibrosis: functional significance and regulatory factors. Cardiovasc Res. 1993; 27:341–8.
20. Huang BS, Leenen FH. The brain renin-angiotensin-aldosterone system: a major mechanism for sympathetic hyperactivity and left ventricular remodeling and dysfunction after myocardial infarction. Curr Heart Fail Rep. 2009; 6:81–8.
21. Antoniu SA. Targeting the angiotensin pathway in idiopathic pulmonary fibrosis. Expert Opin Ther Targets. 2008; 12:1587–90. doi: 10.1517/14728220802515459.
22. Aras O, Dilsizian V. Targeting tissue angiotensin-converting enzyme for imaging cardiopulmonary fibrosis. Curr Cardiol Rep. 2008; 10:128–34.
23. Yuan YM, Luo L, Guo Z, Yang M, Ye RS, Luo C. Activation of renin-angiotensin-aldosterone system (RAAS) in the lung of smoking-induced pulmonary arterial hypertension (PAH) rats. J Renin Angiotensin Aldosterone Syst. 2015; 16:249–53. doi: 10.1177/1470320315576256.
24. Duprez DA. Role of the renin-angiotensin-aldosterone system in vascular remodeling and inflammation: a clinical review. J Hypertens. 2006; 24:983–91. doi: 10.1097/01.hjh.0000226182.60321.69.
25. Uhal BD, Li X, Piasecki CC, Molina-Molina M. Angiotensin signalling in pulmonary fibrosis. Int J Biochem Cell Biol. 2012; 44:465–8. doi: 10.1016/j.biocel.2011.11.019.
26. Maron BA, Opotowsky AR, Landzberg MJ, Loscalzo J, Waxman AB, Leopold JA. Plasma aldosterone levels are elevated in patients with pulmonary arterial hypertension in the absence of left ventricular heart failure: a pilot study. Eur J Heart Fail. 2013; 15:277–83. doi: 10.1093/eurjhf/hfs173.
27. Maron BA, Zhang YY, White K, Chan SY, Handy DE, Mahoney CE, Loscalzo J, Leopold JA. Aldosterone inactivates the endothelin-B receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide levels and modulate pulmonary arterial hypertension. Circulation. 2012; 126:963–74. doi: 10.1161/CIRCULATIONAHA.112.094722.
28. Maron BA, Leopold JA. The role of the renin-angiotensin-aldosterone system in the pathobiology of pulmonary arterial hypertension (2013 Grover Conference series). Pulm Circ. 2014; 4:200–10. doi: 10.1086/675984.
29. Rockey DC, Bell PD, Hill JA. Fibrosis—A Common Pathway to Organ Injury and Failure. N Engl J Med. 2015; 373:96. doi: 10.1056/NEJMc1504848.
30. Wong TC, Piehler K, Meier CG, Testa SM, Klock AM, Aneizi AA, Shakesprere J, Kellman P, Shroff SG, Schwartzman DS, Mulukutla SR, Simon MA, Schelbert EB. Association between extracellular matrix expansion quantified by cardiovascular magnetic resonance and short-term mortality. Circulation. 2012; 126:1206–16. doi: 10.1161/CIRCULATIONAHA.111.089409.
31. Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest. 2009; 135:794–804. doi: 10.1378/chest.08-0492.
32. Falcao-Pires I, Goncalves N, Henriques-Coelho T, Moreira-Goncalves D, Roncon-Albuquerque R Jr, Leite-Moreira AF. Apelin decreases myocardial injury and improves right ventricular function in monocrotaline-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2009; 296:H2007–14. doi: 10.1152/ajpheart.00089.2009.
33. Bruce E, Shenoy V, Rathinasabapathy A, Espejo A, Horowitz A, Oswalt A, Francis J, Nair A, Unger T, Raizada MK, Steckelings UM, Sumners C, Katovich MJ. Selective activation of angiotensin AT2 receptors attenuates progression of pulmonary hypertension and inhibits cardiopulmonary fibrosis. Br J Pharmacol. 2015; 172:2219–31. doi: 10.1111/bph.13044.
34. Henriques-Coelho T, Correia-Pinto J, Roncon-Albuquerque R Jr, Baptista MJ, Lourenco AP, Oliveira SM, Brandao-Nogueira A, Teles A, Fortunato JM, Leite-Moreira AF. Endogenous production of ghrelin and beneficial effects of its exogenous administration in monocrotaline-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2004; 287:H2885–90. doi: 10.1152/ajpheart.01122.2003.