
INTRODUCTION
Arsenic is an environmental carcinogen associated with human skin, bladder, liver and lung cancers [1, 2]. According to the World Health Organization (WHO), 10 μg/L is the maximum acceptable arsenic concentration in drinking water, however, high levels of arsenic have been found in groundwater in more than 70 countries across 5 continents, including North America, affecting over 200 million people [3-7]. Environmental arsenic in groundwater is predominantly found in the inorganic form (iAs), as pentavalent arsenate (AsV) [8]. The consequences of chronic exposure to low doses lead to deleterious effects in multiple organs and tissues (Figure 1). The oncogenic effect is in part attributed to the production of toxic metabolites in the biotransformation of arsenic (Figure 2).
Among the symptoms of chronic exposure to iAs are changes in skin pigmentation, hyperkeratosis (abnormal thickening of the skin) and other skin lesions [9]. These lesions may be precursors to several types of skin cancer, which is the most prevalent form of arsenic-induced cancer [10, 11]. In addition, iAs exposure also appears to play a role in the development of bladder, liver and lung cancers [12-15] though evidence also points to an increased risk for other tissue types, such as breast, prostate and cervix [14, 16-18]. More recent evidence suggests an increased risk of urinary tract cancer with exposure to arsenic in drinking water at around guideline levels (i.e. 10 μg/L) [19-22]. Furthermore, iAs is reported to be associated with pulmonary disease, cardiovascular diseases, and neurodevelopmental and cognitive impairments, which can even be observed in newborns of mothers previously exposed to arsenic [23-29] (Figure 1).
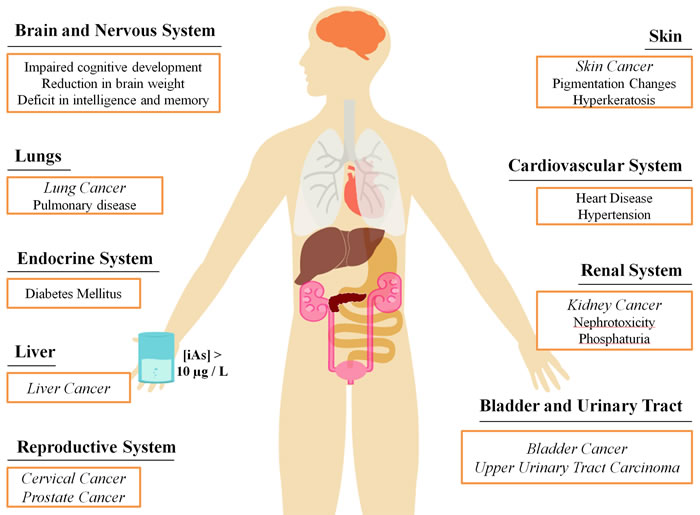
Figure 1: Health effects associated with chronic exposure to inorganic arsenic from contaminated drinking water. Levels of iAs in drinking water near the maximum threshold of 10μg/L can lead to the onset of many diseases in a number of areas in the body. Cancer is a particularly prevalent disease resulting from chronic arsenic exposure, represented in italics.
The occurrence of different organ-specific malignancies associated with arsenic exposure may be a consequence of its transit and storage functions, namely its routes of entry to the human body (e.g. inhalation, adsorption and ingestion), as well as its metabolism and excretion, the latter being correlated with the higher incidence of kidney, urinary tract and bladder cancers [21-22]. Metabolically, cellular intake of AsV occurs through membrane transport proteins including aquaporins and inorganic phosphate (Pi) transporters (Figure 2) [30, 31]. Mitochondrial ATP synthase, which is able to use AsV instead of Pi to produce ATP, conjugates ADP with AsV, which is then reduced to the more cytotoxic AsIII by glutathione (GSH) [32]. The high toxicity of AsIII is partly the result of its strong interaction with protein thiol groups, which can trigger inactivation and proteolysis of key tumour-suppressor proteins [33].
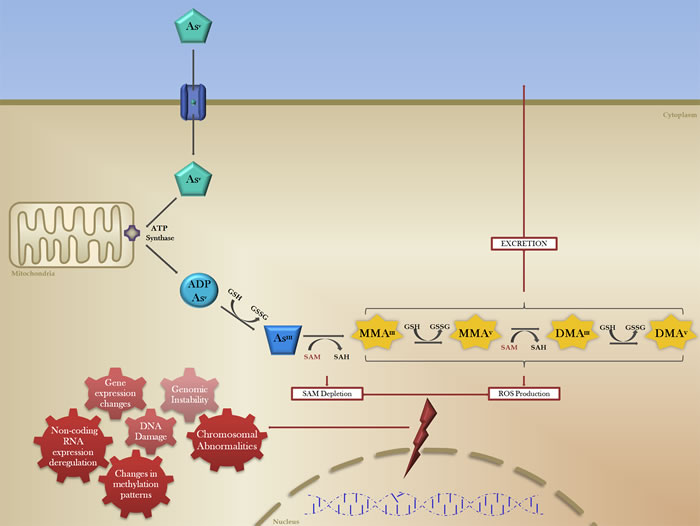
Figure 2: The biotransformation of inorganic arsenic and mechanisms of arsenic-induced carcinogenesis. The reduction, oxidation and methylation of pentavalent arsenic (AsV, green pentagon) occurs after cellular intake via membrane transport proteins (blue cylinder). Mitochondrial ATP synthase (purple) conjugates AsV with ADP, which is then reduced by the electron donor glutathione (GSH) to produce AsIII (blue trapezoid), a more cytotoxic form of arsenic. In order for excretion, AsIII is methylated with methyl groups donated by S-adenosylmethionine methyltransferase (SAM). These methylated arsenic species (MMA, DMA; yellow) all have carcinogenic potential through the induction (red lightning bolt) of a number of genomic and epigenetic effects (red gears), culminating in transcriptomic changes and generalized genomic instability.
Arsenic toxicity is dependent on multiple factors. Molecular alterations at the DNA and RNA level may be at the forefront of this issue, including the disruption of DNA damage-repair mechanisms, coding and non-coding gene expression alterations and changes in mutation patterns [34-36]. This is further complicated by individual factors, such as genetic polymorphisms that may disrupt the intake-excretion balance [37], which may regulate the susceptibility to arsenic-induced damage, as well as lifestyle, which may make individuals with obesity more efficient in the methylation and excretion of arsenic [38]. Interestingly, arsenic trioxide (As2O3) displays anti-tumour activity and as such is currently used as a chemotherapeutic agent in the treatment of acute promyelocytic leukemia (APL), particularly in cases with a translocation between chromosomes 15 and 17 [39]. As2O3 is associated with a number of genetic and epigenetic changes, including alterations in coding and non-coding gene expression levels and abnormal methylation patterns [40, 41]. In light of this, it is important to examine the molecular changes in both the treatment of APL with As2O3 in addition to iAs exposure to fully understand the mechanisms of arsenic-induced carcinogenesis.
As literature relating (epi)genetics to arsenic exposure has been accumulating at an increasing rate (Figure 3), we review the latest advances in the oncogenomic effects of arsenic-induced carcinogenesis.
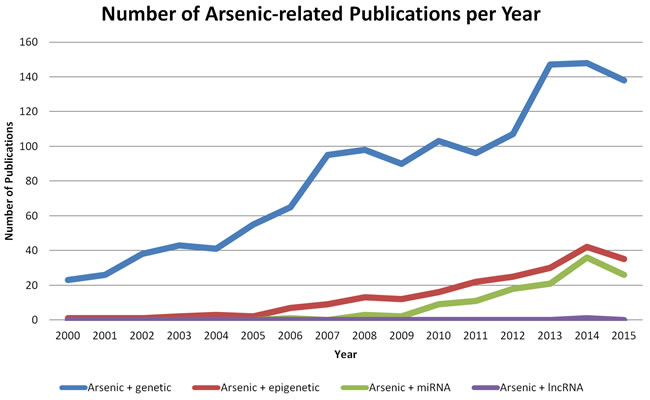
Figure 3: Number of publications relating genetics and epigenetics to arsenic exposure. Search was performed within EndNote (Version 7, Thomson Reuters) and manually filtered. Number of publications are based on a United States National Library of Medicine PubMed search using the terms “arsenic AND genetic” (blue line), “arsenic AND epigenetic” (red line), “arsenic AND miRNA OR microRNA” (green line), or “arsenic AND lncRNA OR lincRNA OR long non-coding RNA” (purple line). 2016 publications were not included in the search, and annual (Jan 1-Dec 31) date limitations were used.
GENOMIC ABERRATIONS ASSOCIATED WITH ARSENIC
Oxidative DNA damage
Carcinogenic aspects of arsenic exposure
Several studies propose that iAs is not able to bind directly to DNA and therefore is not likely to be responsible for mutational damage [42]. However, methylated arsenicals derived from iAs biotransformation have been shown to generate single and double-stranded DNA breaks through the formation of reactive oxygen species (ROS) [43]. Human keratinocytes exposed to arsenic produce two main types of ROS: superoxide anion (O2- •) and hydrogen peroxide (H2O2) [44]. The type of ROS produced in response to arsenic is cell-type specific, highlighting the relevance of this mechanism to arsenic-induced carcinogenesis [45, 46]. In fact, the mechanism of oxidative stress is the most studied features of arsenic toxicity and is recognized as one of the most important [47].
The basis of the carcinogenic aspects of oxidative stress upon exposure to arsenic is that when attacking DNA, ROS produce 8-Hydroxy-29-deoxyguanosine (8-OHdG), which is capable of generating G>T conversions that trigger G>C → T>A transversions [48-50]. 8-OHdG is a biomarker of DNA oxidative damage, shown to be expressed at higher levels in the epidermal nuclei of arsenic-related Bowen’s disease, Bowen’s carcinoma and arsenic keratosis [51-53]. Furthermore, whole-genome sequencing analysis revealed a specific mutational signature that can differentiate arsenic-related lung tumours from tumours unrelated to arsenic, even though they may display the same histological features. Arsenic-related tumours are characterized by low overall number of mutations, high rates of T>G/A>C, and low rates of C>A/G>T transversions [34].
Arsenic-induced mutations can be particularly damaging if they lead to the activation of an oncogene, such as RAS [54]. Mice exposed to arsenic during gestation have higher incidence of liver tumours with a mutation at codon 61 in HRAS compared to liver tumours in mice not exposed to arsenic, and suggests that this mutation might be associated with arsenic-induced oxidative stress [55]. Similarly, it is hypothesized that mutations in the tyrosine kinase domain of the epidermal growth factor receptor (EGFR) are responsible for the activation of the EGFR pathway [56], a common molecular feature of many cancers that is also observed in cell lines exposed to arsenic [57-61].
Oxidative stress can also lead to mutations and instability in mitochondrial DNA (mtDNA), which is associated with the development of skin cancers [62]. Mitochondria are involved in cell proliferation, cell death and abnormal cell differentiation, and therefore alterations in mtDNA structure and function have been correlated with carcinogenesis [63]. Additionally, ROS can also disturb the permeability of the mitochondrial membrane, leading to the aberrant expression of apoptosis related genes [64]. For that reason, As2O3 is used as a therapeutic agent, shown to induce apoptosis in leukemic cells [39].
Chemotherapeutic aspects of arsenic exposure
Interestingly, the carcinogenic and chemotherapeutic effects of arsenic might rely on common mechanisms [65]. In arsenic-induced carcinogenesis, the cells overcome the apoptotic effect that is observed after exposure to As2O3 through the activation of the nuclear factor erythroid-derived factor 2–related factor 2 (NRF2) pathway, responsible for the oxidative stress response, demonstrating that arsenic effects are both dose and time-dependent [66]. Taken together, cellular oxidative stress induced by arsenic exposure contributes to widespread genomic instability, which poses deleterious effects to the cell, and the individual [67, 68].
Chromosomal alterations
Genomic instability resulting from cellular oxidative damage can also lead to further disruptions in chromosome structure and stability, including end-to-end fusion, abnormal sister chromatid separation, and aneuploidy [67]. Doses of arsenic around 10 μg/L have been shown to have an aneuploidogenic effect, illustrating the long-term risk of chronic low-dose exposure to arsenic [69]. Chromosomal aberrations of this sort are implicated in cancer development, possibly through the activation of proto-oncogenes [70]. Arsenic exposure may also disrupt microtubule assembly through interaction with the sulfhydryl groups of tubulin, leading to mitotic spindle complex malfunction [6, 71]. This can result in increased micronuclei formation, which is also associated with the onset of cancer [72, 73]. Another consequence of arsenic-induced genomic instability is the continued progression through the cell cycle despite DNA damage, accomplished through inhibition of the p53 mediated apoptotic response [74].
In addition to chromosomal alterations and genomic instability, arsenic exposure is also related to DNA copy-number alterations (CNAs) (Figure 4), a key feature of tumour progression evidenced by the amplification of oncogenes and the deletion of tumour suppressor genes [75]. It has been demonstrated that lung squamous cell carcinoma exhibits both segmental DNA gains and losses after exposure to arsenic through dietary sources, compared to lung tumour genomes from smokers and non-smokers who have not been exposed to arsenic [76, 77]. Interestingly, this study implicated arsenic-induced DNA losses at the 9q12 locus, which is known to contain a gene from the FOX-gene family [76, 78]. FOX-gene family proteins are DNA-binding proteins that are involved with the regulation of transcription as well as DNA repair, some of which possess tumour suppressive functions while others display oncogenic features, and are frequently deleted or overexpressed through CNAs in many human cancers [79].
Conversely, it has been shown that in CDKN1B and CDKN2A-deleted cells, treatment with As2O3 resulted in increased signal patterns of these genes [80]. As CDKN1B and CDKN2A are members of a cell-cycle-inhibiting gene family, this suggests another possible mechanism of apoptotic induction by As2O3. Furthermore, CNAs may serve as prognostic factors for patients with APL, such as the deletion of the gene encoding CD56 by As2O3, which correlates with higher relapse-free survival [81]. Further characterization of chromosomal abnormalities and CNAs induced by arsenic will help to elucidate its carcinogenic mechanism and potentially implicate novel targets in therapeutic responses.
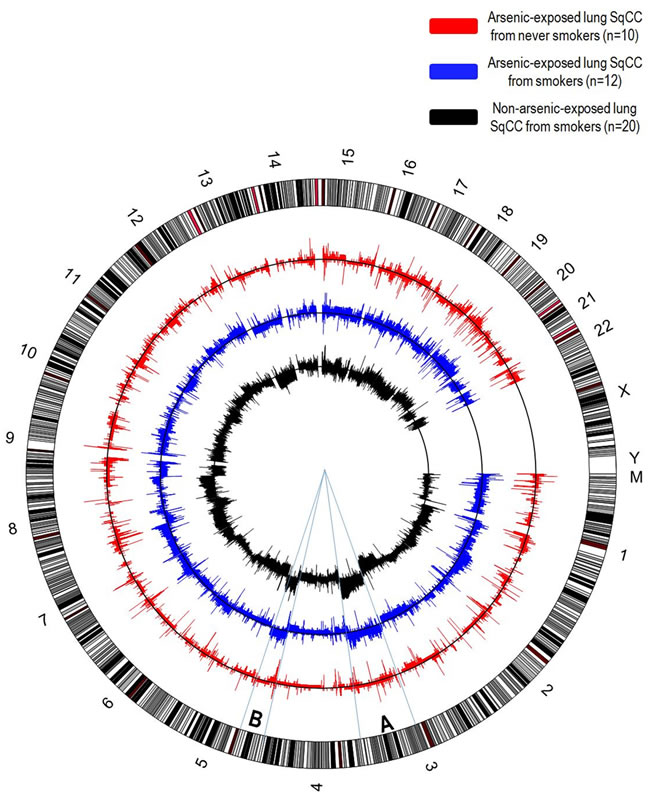
Figure 4: Circular representation of DNA copy-number alterations (CNAs) in lung squamous cell carcinomas. Each chromosome of the human genome (hg19) is represented in the outer circle. Only lung squamous cell carcinomas were considered for this analysis, since this is the histological subtype more strongly associated with arsenic exposure. In arsenic exposed patients, there is an unusually high frequency of lung SqCC among never smokers, while this subtype is almost exclusively associated with smokers in non-arsenic related lung SqCC. CNAs detected in lung SqCCs arsenic-exposed, non-smoker patients (red, n=10), arsenic-exposed, smokers (blue, n=12) and non-arsenic exposed, smokers (dark grey, n=20) are shown. On each chart, the frequency of DNA gains among cases is shown above the black line indicating absence of alterations, while the frequency of DNA losses are shown below. Overall, the number of alterations observed in arsenic-exposed, non-smokers lung SqCCs are significantly lower than smokers. Interestingly, one of the most characteristic alterations described in lung SqCC (DNA gains 3q and 5p) exhibits a remarkable similarity among smokers, regardless of arsenic exposure status, while a low frequency of alterations is observed among non-smokers, arsenic-exposed patients (segments A and B).
EPIGENETIC FEATURES OF ARSENIC-INDUCED CANCER
During arsenic biotransformation, AsIII is known to be methylated by S-adenosylmethionine methyltransferase (SAM) as part of the excretion process (Figure 2), which may lead to the depletion of SAM and consequent epigenetic disruption of the methylome [82-85]. This dependence of cellular detoxification and excretion of iAs on SAM and methyl group availability suggests that there may be epigenetic consequences of arsenic-exposure. Global DNA methylation levels and associated gene methylation changes play a critical role in cancer development, and also provide useful diagnostic and prognostic markers [86-88]. Differential DNA methylation patterns have been observed in individuals with high urinary arsenic concentrations, suggesting that these alterations may be important for non-genotoxic arsenic-induced carcinogenesis [89]. Arsenic exposure has been shown to induce global DNA hypomethylation, as well as specific gene promoter methylation changes through the alteration of CpG methylation status [90].
Global hypomethylation
The methylation of arsenic is necessary for excretion, but this puts a high demand on the activity of several enzymes important in DNA methylation and epigenetic gene regulation, such as SAM and DNA methyltransferases (DNMTs). SAM is a cofactor that acts as a methyl-group donor for many biomolecules [91]. The production of methylated arsenic species leads to the depletion of SAM and a marked decrease in the availability of methyl groups in the cell [92]. Global hypomethylation can lead to chromatin remodeling, allowing for the transcription of previously inaccessible oncogenes and cancer-associated genes. It has been reported that exposure to 5 µM iAs over 29 weeks malignantly transformed cells, and was further correlated with an increased S100P and HYAL1 expression, genes relevant to the malignant process [93]. This was accomplished through hypomethylation near the transcriptional start site of these genes. Evidence of global hypomethylation as a result of iAs exposure has been shown in multiple cancer types, including prostate, breast and liver cancers [36, 94, 95]. Furthermore, widespread DNA hypomethylation in hepatocytes is implicated in the increased expression of pro-growth genes, particularly estrogen receptor-α [95, 96]. Clinically, iAs exposure was observed to be a putative cause of significant DNA hypomethylation in adult peripheral blood mononuclear cells, suggesting possible involvement in lymphatic cancers [97]. Taken together, the current data suggest the significance of global DNA hypomethylation in arsenic-induced carcinogenesis.
Promoter hypermethylation
Global methylation changes may be accentuated by specific promoter methylation alterations in cells exposed to chronic doses of iAs. In a genome-wide study, it was discovered that 2919 genes showed differential DNA methylation profiles when exposed to concentrations of iAs around current WHO guideline levels (at or above 10 µg/L), most of which were identified as CpG islands near the transcription start site [98]. Exposure to higher arsenic concentrations between 250-500 µg/L showed a similar relationship between iAs exposure and promoter hypermethylation [99]. Arsenic levels above 500 µg/L were associated with increased methyl acceptance capacity of promoter DNA, suggesting the onset of widespread hypomethylation at the point where the demand on the global methylation level is no longer sustainable [92, 99]. This displays the existence of a putative threshold at which global hypomethylation may become more prevalent in arsenic-induced carcinogenesis, which may have implications for early diagnosis and treatment of cancers associated with chronic exposure to iAs. These observations suggest that arsenic may be able to induce tumourigenesis and cancer progression through the epigenetic silencing of tumour suppressors as well as the epigenetic activation of oncogenes or associated genes. One of the most notable examples of this is the significant hypermethylation of the TP53 promoter, the level of which was elevated in arsenic-induced skin cancers relative to skin cancers not resulting from arsenic exposure [100]. Evidence of promoter hypermethylation has been shown in a number of cancer types, including prostate, skin, and bladder, although the exact role of this in carcinogenesis has yet to be fully elucidated [101-103].
As2O3 treatment also displays a similar pattern as its therapeutic action may be through inhibition of DNMT expression level, global DNA hypomethylation or alternative epigenetic effects [104]. It can be suggested that this may reflect the carcinogenic mechanism of iAs exposure, but to an extent that leads to targeted cell death in APL cells. This was observed in prostate cancer cell lines, where DNA damage and hypomethylation triggered histone tail modification and chromatin remodeling, leading to the upregulation of pro-apoptotic genes [105]. In liver cells, treatment with As2O3 correlated with hypomethylation in the cis-regulatory sites of the promoter of MYC (a known cancer-associated gene), as well as hypermethylation in the promoter of MAX (a regulator of MYC and cell cycle) [106]. Thus, As2O3-based studies not only further the targeted therapy of cancer, but also help to elucidate the mechanism of arsenic toxicity, and in turn, its role in carcinogenesis.
GENE EXPRESSION CHANGES
The numerous genomic and epigenetic changes resulting from iAs exposure culminate in the deregulation of a variety of genes. In Table 1, we summarize previously described coding-gene expression changes derived from arsenic exposure, demonstrating that this metalloid can alter crucial pathways involved in diverse cellular processes.
Table 1: Coding-gene expression changes linked to carcinogenesis resulting from exposure to arsenic.
mRNA |
Expression Change (non-exposed vs. exposed) |
Arsenic Compound |
Exposure Dose |
Experimental model |
Sample origin |
Reference |
DNA Repair and Stress Response |
||||||
ERCC1 |
Up |
Drinking water |
9.60–46.5µg/L in blood |
Human sample |
Frozen Peripheral Blood Lymphocytes |
[110] |
ERCC1 |
Down |
NaAsO2 |
0.01–10μM |
Cell line |
Jurkat Lymphoblast Cells |
[156] |
POLB |
Up |
NaAsO2 |
2 or 50p.p.m. |
Mice tissue |
Female BALB/c Mice Lung tissue |
[111] |
POLB |
Down |
Drinking water |
9.60–46.5µg/L in blood |
Human sample |
Frozen Peripheral Blood Lymphocytes |
[110] |
POLD2 |
Up |
Drinking water |
9.60–46.5µg/L in blood |
Human sample |
Frozen Peripheral Blood Lymphocytes |
[110] |
PARP1 |
Up |
NaAsO2 |
2 or 50p.p.m. |
Mice tissue |
Female BALB/c Mice Lung tissue |
[111] |
PARP1 |
Down |
MMA(III) or DMA(III) |
0.1μM |
Cell line |
Human HeLa S3 Cells |
[157] |
APEX1 |
Up |
NaAsO2 |
2 or 50p.p.m. |
Mice tissue |
Female BALB/c Mice Lung tissue |
[111] |
APEX1 |
Down |
As2O3 |
0.005 – 5μM |
Cell line |
Normal Human Epidermal Keratinocytes (NHEK) |
[158] |
LIG1 |
Up |
NaAsO2 |
2 or 50p.p.m. |
Mice tissue |
Female BALB/c Mice Lung tissue |
[111] |
OGG1 |
Up |
NaAsO2 |
2 or 50p.p.m. |
Mice tissue |
Female BALB/c Mice Lung tissue |
[111] |
NQO1 |
Up |
NaAsO2 |
2, 5 and 10μM |
Cell line |
Mouse hepa1c1c7 Cells |
[159] |
NQO1 |
Up |
AsIII |
0.005 – 5μM |
Cell line |
Normal Human Epidermal Keratinocytes (NHEK) |
[158] |
XPC |
Down |
AsIII |
0.005 – 5μM |
Cell line |
Normal Human Epidermal Keratinocytes (NHEK) |
[158] |
XBP-1 |
Up |
As2O3 |
5μM |
Cell line |
Murine Neuroblastoma Cells (Neuro-2a) |
[160] |
SESN1 |
Up |
NaAsO2 |
5μM |
Cell line |
Human Breast Cancer Cell MCF-7 (p53+/+) |
[161] |
Cell Proliferation and Growth |
||||||
FOXM1 |
Up |
As2O3 |
1μM |
Cell line |
Human Airway Epithelial Cell (NuLi-1) |
[162] |
GM-CSF |
Up |
NaAsO2 |
0 - 4μM |
Cell line |
Normal Human Epidermal Keratinocytes (NHEK) |
[163] |
PCNA |
Up |
NaAsO2 |
500nM |
Cell line |
Rat Liver Epithelial Cell (TRL1215) |
[164] |
CTBP1 |
Up |
As2O3 |
1µM |
Cell line |
Normal Human Urothelial Cell (HUC1) |
[165] |
FOS |
Up |
AsIII |
50μM |
Cell line |
Human HeLa S3 Cells |
[166] |
TGFB3 |
Up |
NaAsO2 |
500nM |
Cell line |
Rat Liver Epithelial Cell (TRL1215) |
[164] |
Cell Death |
||||||
TNFRSF6 |
Up |
NaAsO2 |
5μM |
Cell line |
Human Newborn Foreskin Cells (HFW) |
[167] |
FADD |
Up |
NaAsO2 |
5μM |
Cell line |
Human Newborn Foreskin Cells (HFW) |
[167] |
MCL1 |
Up |
NaAsO2 |
5μM |
Cell line |
Human Newborn Foreskin Cells (HFW) |
[167] |
BAX |
Up |
As2O3 |
5μM |
Cell line |
Murine Neuroblastoma Cells (Neuro-2a) |
[160] |
BCL2 |
Down |
As2O3 |
5μM |
Cell line |
Murine Neuroblastoma Cells (Neuro-2a) |
[160] |
Cell Cycle |
||||||
ATF3 |
Up |
NaAsO2 |
5μM |
Cell line |
Human Breast Cancer Cell MCF-7 (p53+/+) |
[161] |
CDKN1A |
Down |
NaAsO2 |
0.1μM |
Cell line |
Human Keratinocyte Cell (HaCaT) |
[112] |
TP53 |
Up |
As2O3 |
2μM |
Cell line |
Human Glioma Cells (U87MG and T98G) |
[168] |
MYC |
Up |
NaAsO2 |
500nM |
Cell line |
Rat Liver Epithelial Cell (TRL1215) |
[164] |
MYC |
Up |
NaAsO2 |
0 - 4μM |
Cell line |
Normal Human Epidermal Keratinocytes (NHEK) |
[163] |
RB1 |
Up |
NaAsO2 |
500nM |
Cell line |
Rat Liver Epithelial Cell (TRL1215) |
[164] |
CDC6 |
Up |
As2O3 |
1μM |
Cell line |
Human Airway Epithelial Cell (NuLi-1) |
[162] |
CDK2 |
Up |
As2O3 |
1μM |
Cell line |
Human Airway Epithelial Cell (NuLi-1) |
[162] |
CDK1 |
Up |
As2O3 |
1μM |
Cell line |
Human Airway Epithelial Cell (NuLi-1) |
[162] |
CDC25A |
Up |
As2O3 |
1μM |
Cell line |
Human Airway Epithelial Cell (NuLi-1) |
[162] |
CDC25A |
Up |
NaAsO2 |
5μM |
Cell line |
Human Newborn Foreskin Cells (HFW) |
[167] |
CCND1 |
Up |
As2O3 |
1μM |
Cell line |
Human Airway Epithelial Cell (NuLi-1) |
[162] |
| CCND1 | Up | NaAsO2 |
5μM | Cell line | Human Bronchial Epithelial Cell (Beas-2B) | [56] |
| Cell Signaling | ||||||
| EGFR | Up | As2O3 |
1µM | Cell line | Normal Human Urothelial Cell (HUC1) | [165] |
| TNFα | Up | NaAsO2 |
0 - 4μM | Cell line | Normal Human Epidermal Keratinocytes (NHEK) | [163] |
| TGFα | Up | NaAsO2 |
0 - 4μM | Cell line | Normal Human Epidermal Keratinocytes (NHEK) | [163] |
| H-Ras | Down | NaAsO2 |
50ppb | Mouse tissue | C57BL/6 Mice Offspring Hippocampal Nuclear Fractions | [164] |
| Raf-1 | Down | NaAsO2 |
50ppb | Mouse tissue | C57BL/6 Mice Offspring Hippocampal Nuclear Fractions | [29] |
| VEGF | Up | NaAsO2 |
1 – 10μM | Cell line | Human Uroepithelial Cell (SV-HUC-1) | [169] |
| COX-2 | Up | NaAsO2 |
1 – 10μM | Cell line | Human Uroepithelial Cell (SV-HUC-1) | [169] |
| HIF-1α | Up | NaAsO2 |
1 – 10μM | Cell line | Human Uroepithelial Cell (SV-HUC-1) | [169] |
| ERBB2 | Up | NaAsO2 |
500nM | Cell line | Rat Liver Epithelial Cell (TRL1215) | [164] |
| ERBB2 | Down | As2O3 |
1µM | Cell line | Normal Human Urothelial Cell (HUC1) | [165] |
| MAPK8 | Up | AsIII | 50μM | Cell line | Human HeLa S3 Cells | [115] |
| MAPK8 | Up | NaAsO2 |
500nM | Cell line | Rat Liver Epithelial Cell (TRL1215) | [164] |
| H-RAS | Up | NaAsO2 |
500nM | Cell line | Rat Liver Epithelial Cell (TRL1215) | [164] |
| MET | Up | NaAsO2 |
500nM | Cell line | Rat Liver Epithelial Cell (TRL1215) | [164] |
The disruption of multiple pathways can result in genomic instability and may lead to cancer development. A direct example is the alteration of the expression of genes involved with DNA repair. Arsenic exposure affects the ability of nucleotide excision repair (NER) in cell lines, which can enhance the mutagenicity of other carcinogens such as UV light [107, 108]. Among other factors, NER mechanisms are affected due to reduction of NER-associated genes (Table 1) [6, 109-112]. The poly-(ADP-ribose) polymerase 1 (PARP1) is a protein involved with DNA damage response that controls genomic stability and has been shown to be increased in arsenic exposed cell lines and mice samples [111-113]. Therefore, deregulation of PARP1 may be a possible mechanism of the induction of chromosomal instability and carcinogenesis [113].
Other characteristics of tumour cells that may be increased upon arsenic exposure are growth, proliferation and survival [14, 114]. For example, the PI3K/AKT pathway is affected by arsenic through the phosphorylation of AKT, activation of the JNK-STAT3 pathway and/or suppression of PTEN, an inhibitor of this pathway [6, 115-117]. Therefore, arsenic-induced activation of the PI3K/AKT pathway contributes to cellular transformation due to increased proliferation rates and induction of anchorage-independent growth [118].
The molecular damages caused by arsenic can be so extensive that cells are driven to undergo apoptosis [119]. This effect has been explored by the use of As2O3 as a chemotherapeutic for APL treatment [120]. The production of ROS reduces the mitochondrial membrane potential, leading to an increase in cytochrome-c release and consequent activation of caspases. Consequently, the normal protein ratio between the anti-apoptotic Bcl-2 and the pro-apoptotic Bax is also compromised, triggering apoptosis [64]. Studies show that As2O3 in high doses can induce apoptosis of B-cell leukemic cells, malignant lymphocytes, myeloma cells, and even cell lines derived from esophageal carcinoma and neuroblastoma [121-125]. However, in the case of arsenic-induced cancers, the cells can overcome the apoptotic effect derived from the DNA damage through the activation of factors involved in the antioxidant response, such as NRF2 [66].
NON-CODING RNA EXPRESSION CHANGES
MicroRNAs: Mediators of arsenic–induced carcinogenesis
The discovery that only a small portion of the transcribed human genome is translated into proteins led to a surge of interest in determining the role of non-coding RNAs (ncRNAs) in human diseases, especially regarding small ncRNAs [126-130]. There are three main classes of small ncRNAs: microRNAs (miRNAs), endogenous small interfering RNAs (endo-siRNAs) and PIWI-interacting RNAs (piRNAs) [131]. miRNAs are responsible for the post-transcriptional regulation of mRNAs and mainly repress translation through complementary binding along with RNA-induced silencing complex (RISC) assembly. These molecules have been extensively described and are known to be deregulated in cancer, playing important roles in cancer development and progression [132]. Correspondingly, arsenic studies associated with the deregulation of non-coding RNAs mainly describe alterations in miRNA expression, limiting our understanding of the association between arsenic exposure and the deregulation of long ncRNAs (lncRNAs) [133-135], or other types of small ncRNAs (Figure 3).
There is a strong link between arsenic exposure and the expression of miRNAs (Table 2), which may promote carcinogenesis. Many of these miRNAs are associated with cancer as they are responsible for negatively regulating oncogenes or tumour suppressors that are involved in several important cellular processes (Figure 5) [136]. The genes described in Table 2 and Figure 5 are only a representation of the known miRNAs that have differential expression when exposed to arsenic. In fact, one study showed 36 miRNAs to be consistently deregulated upon exposure to 2 µmol/L of sodium arsenite (NaAsO2) [137]. Of these, many are implicated in cancer. miR-150, for example, has been shown to be a circulating marker of prostate, colorectal, lung and pancreatic cancer [138-141]. In prostate cancer, miR-150 is upregulated, and is additionally correlated with tumour recurrence and metastasis, as well as poor overall survival [142].
Table 2: Selected miRNA expression changes resulting from exposure to iAs linked to important cellular processes.
miRNA |
Expression Change |
Arsenic Exposure |
Putative Target |
Tissue / Cancer Type |
Reference |
miR-143 |
Down |
5µM iAs |
BCL2; BCL-XL |
Prostate cancer |
[17] |
miR-205 |
Down |
1µM As2O3 |
AKT; mTOR |
Urothelial carcinoma |
[165] |
miR-27a |
Down |
Varied As2O3 |
Cell growth; apoptosis; migration |
Breast cancer |
[170] |
miR-200b |
Down |
2.5µM NaAsO2 |
PKCα; |
Human bronchial epithelial cells; lung cancer |
[171] |
miR-21 |
Up |
500µM NaAsO2 |
Cell proliferation promotion; apoptotic inhibition; acts on various tumour suppressors |
Keratinocytes; Skin cancer (Melanoma); glioblastoma; prostate cancer |
|
miR-200a |
Up |
500µM NaAsO2 |
Melanoma development |
Keratinocytes; Skin cancer (Melanoma) |
[172] |
miR-520h |
Down |
Varied As2O3 |
PP2A/C (upregulation of this inhibits NF-κB); metastasis |
Cervical cancer |
[176] |
miR-222 |
Up |
1µM NaAsO2 |
ARID1A, PTEN; cell proliferation, migration |
Lung cancer; Human lung epithelial BEAS-2B cells |
[177] |
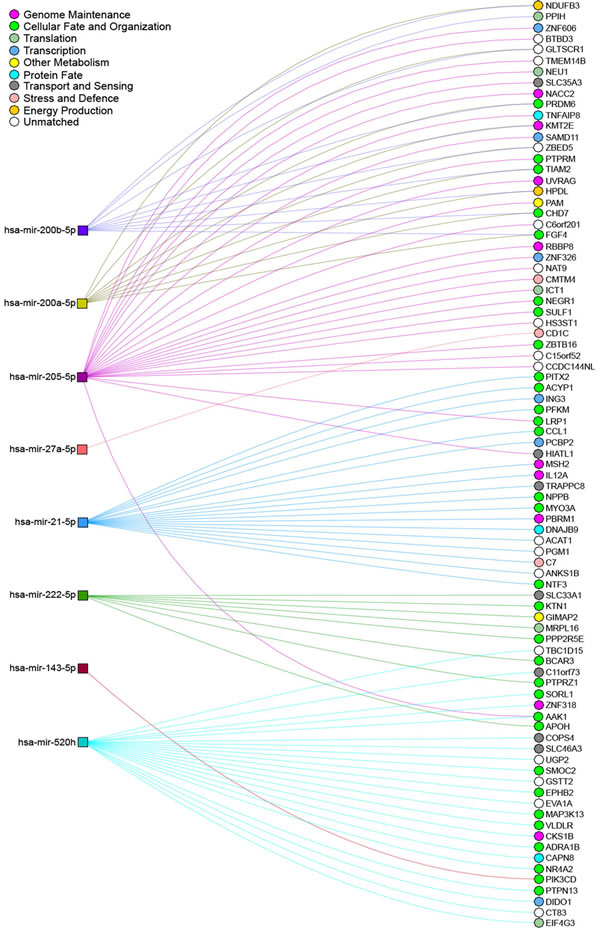
Figure 5: Network interactions between deregulated miRNAs and their predicted targets upon arsenic exposure. miRNAs shown to be deregulated after exposure to arsenic and described in this review were inputted into miRDIP for gene target prediction, using the thresholds of the top 1% of mRNA transcripts predicted by at least 3 different prediction databases. NAViGaTOR [178] was used to visualize the interactions between these miRNAs and their predicted mRNA targets. miRNAs deregulated after exposure to arsenic are depicted by coloured square nodes, while their predicted mRNA targets are represented by circular nodes. Edges indicate predicted miRNA/mRNA interactions and are coloured according to the identity of the selected miRNA. The mRNA-target nodes are coloured as per to their association with Gene Ontology terms. Certain mRNAs appear to be shared by several of the miRNAs identified (i.e. FGF4, AAK1, CHD7, HPDL etc.), representing possible important cellular functions that are affected by arsenic exposure, such as cellular fate and energy production.
Studies looking at the effects of As2O3 on non-coding gene expression show similar results. For example, miR-328 targets hERG, a gene encoding a subunit of a potassium ion channel. In the treatment of breast cancer, As2O3 is an effective therapy partly due to its action where it upregulates miR-328, thereby inducing apoptosis through the inhibition of hERG expression [143]. This highlights the importance of understanding the effects of arsenic exposure on more than the coding portion of the genome. miRNA-based studies have helped to uncover the details of the mechanism of arsenic induced carcinogenesis, which suggests that further characterization of other small non-coding RNAs involved in regulation may be of biological interest.
PIWI-Interacting RNAs: Functions and prospective roles in arsenic-induced carcinogenesis
Although initially thought to be restricted to germ cells, piRNAs have been recently shown to be expressed in somatic tissues, displaying conserved mammalian biological functions [144, 145]. The uniqueness of this class is that they recognize complementary DNA sequences, instead of RNA sequences. Similarly to other classes of small non-coding RNAs, piRNAs are known to regulate gene expression through a small RNA-guided mechanism, in which piRNAs bind to the PIWI proteins of the Argonaute family forming the RISC, which can bind and regulate the expression of transcripts containing complementary sequences [146]. The main described function of piRNAs is the silencing of selfish genetic elements, mainly transposable elements (TEs), in the maintenance of genomic instability [147]. Later studies also demonstrated that piRNAs are able to promote epigenetic activation and even a miRNA-like transcript silencing [148-151]. In Figure 6 we illustrate these known functions, highlighting the importance of piRNAs as regulators of gene expression.
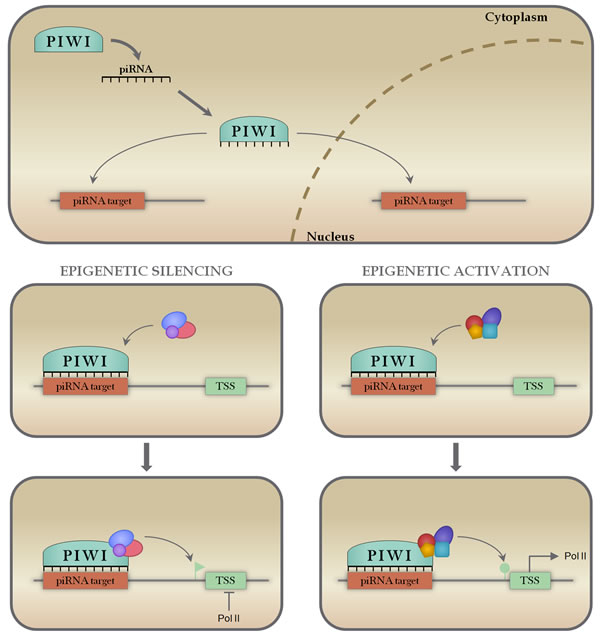
Figure 6: Mechanisms of piRNA action. piRNAs associate with PIWI proteins in the cytoplasm, forming a ribonucleoprotein effector complex that is able to recognize and bind to complementary target sequences on DNA both in the cytoplasm and nucleus (panel A). When bound to the target sequence, piRNA-PIWI complexes can recruit epigenetic remodeling machinery (panels B and D) to either repress transcription through DNA methylation (panel C) or activate transcription through DNA acetylation or methylation removal (Panel E).
Since piRNAs are known to be involved with gene regulation and mainly with the control of genomic stability, it is likely that they are involved in a number of human diseases [126]. In fact, piRNAs display specific expression patterns that are markedly different across tissue types, between non-malignant and tumour tissues and even between different tumour subtypes [144]. As such, piRNAs have emerged as a highly promising area of study that might provide further knowledge on cancer biology and potentially improve tumour diagnosis and therapeutics.
As described here, the most well known and described mechanism of action of arsenic is the induction of oxidative DNA damage and disruption of permeability of the mitochondrial membrane. Numerous piRNAs were found to align with mitochondria specific small RNA sequences in cancer cells and also showed the coexistence of PIWI proteins and piRNAs in mitochondria [152]. Those findings suggest that the piRNA/PIWI complex might be involved in stress response and leads to the assumption that they might be important in arsenic-mediated tumourigenesis. Moreover, the piRNA/PIWI complex is known to be a major epigenetic regulator, being responsible for recruiting epigenetic machinery to binding sites, promoting epigenetic activation or silencing [148, 153]. Since epigenetic changes are another major mechanism for arsenic-induced cancer, this further supports the hypothesis that piRNAs may play important roles in arsenic-induced disease. Interestingly, so far there are no studies that have investigated the relation between piRNAs and arsenic-induced cancers. Therefore, this is an area that with further investigation, could improve our understanding on arsenic toxicology and therapeutics.
CONCLUSION AND EMERGING QUESTIONS
Arsenic contamination of drinking water sources is a major problem worldwide. Clinical implications of the prevalence of arsenic groundwater contamination are evidenced by an impact on the incidence of cancer, even at low exposure levels [154]. This evidence suggests that the current guideline for maximum exposure to arsenic may still present a hazard to exposed populations. Limiting the effects of arsenic exposure on at-risk populations may require the implementation of strategies to manage groundwater concentrations, such as nanofiltration, adsorption and bioremediation [7, 155].
In this review article, we have discussed a spectrum of molecular aberrations induced by arsenic. Arsenic exposure is closely associated with DNA damage through the production of ROS, which may provide a distinct molecular signature. This type of oxidative damage can induce chromosomal instability including copy number alterations that lead to the amplification or deletion of certain loci, which has implications in carcinogenesis when an oncogene or tumour suppressor gene is involved. Arsenic exposure can also induce epigenetic changes, including global hypomethylation by the depletion of the global methyl pool, leading to aberrant gene expression, as well as alterations in promoter CpG island methylation status. Furthermore, arsenic exposure is associated with changes in both coding and non-coding gene expression, which not only affects critical-protein activity in cells, but also the regulation of coding-genes, through disruptions in miRNA and possibly other non-coding gene levels. Interestingly, the regulatory functions of piRNAs overlap with known mechanisms of arsenic toxicity and chemotherapeutic effects, leading to the assumption that piRNAs might play important roles in these mechanisms. However, our current understanding of the precise mechanism of arsenic-induced carcinogenesis is still far from comprehensive, and further work may look to characterize novel biological players involved.
The numerous health effects of arsenic ingestion demonstrate the complexity of the mechanisms linking arsenic exposure to disease. Arsenic has been shown to induce a number of damaging genomic and epigenetic effects, but the scope of these has yet to be determined. The study of these mechanisms will allow for a better understanding of both arsenic-induced cancer and arsenic-based therapies, which may lead to improved approaches for preventing exposure and reducing the onset of cancer, as well as the development of novel cancer therapeutics.
Acknowledgments
Listed authors are the sole contributors in the preparation of this manuscript.
Conflict of Interests
The authors declare that they have no conflicts of interest.
Funding
This work was supported by grants from the Canadian Institutes for Health Research (CIHR FRN-143345).
Authors’ contributions
APS and BCM performed literature search, drafted the manuscript and designed figures and tables. KWN and GLS assisted in writing, contributed in the production of figures and critically revised the manuscript. TJDB provided valuable data and critically revised the manuscript. WLL and VDM participated in the design, writing and analysis of the manuscript.
REFERENCES
1. Sankpal UT, Pius H, Khan M, Shukoor MI, Maliakal P, Lee CM, Abdelrahim M, Connelly SF and Basha R. Environmental factors in causing human cancers: emphasis on tumorigenesis. Tumour biology. 2012; 33:1265-1274.
2. Ng JC, Wang J and Shraim A. A global health problem caused by arsenic from natural sources. Chemosphere. 2003; 52:1353-1359.
3. Huang L, Wu H and van der Kuijp TJ. The health effects of exposure to arsenic-contaminated drinking water: a review by global geographical distribution. International journal of environmental health research. 2015; 25:432-452.
4. Naujokas MF, Anderson B, Ahsan H, Aposhian HV, Graziano JH, Thompson C and Suk WA. The broad scope of health effects from chronic arsenic exposure: update on a worldwide public health problem. Environmental health perspectives. 2013; 121:295-302.
5. Bhattacharjee P, Chatterjee D, Singh KK and Giri AK. Systems biology approaches to evaluate arsenic toxicity and carcinogenicity: an overview. International journal of hygiene and environmental health. 2013; 216:574-586.
6. Hubaux R, Becker-Santos DD, Enfield KS, Rowbotham D, Lam S, Lam WL and Martinez VD. Molecular features in arsenic-induced lung tumors. Molecular cancer. 2013; 12:20.
7. Hubaux R, Becker-Santos DD, Enfield KS, Lam S, Lam WL and Martinez VD. Arsenic, asbestos and radon: emerging players in lung tumorigenesis. Environmental health. 2012; 11:89.
8. Smedley PL and Kinniburgh DG. A review of the source, behaviour and distribution of arsenic in natural waters. Applied Geochemistry. 2002; 17:517-568.
9. Dastgiri S, Mosaferi M, Fizi MA, Olfati N, Zolali S, Pouladi N and Azarfam P. Arsenic exposure, dermatological lesions, hypertension, and chromosomal abnormalities among people in a rural community of northwest Iran. Journal of health, population, and nutrition. 2010; 28:14-22.
10. Banerjee M, Bhattacharjee P and Giri AK. Arsenic-induced Cancers: A Review with Special Reference to Gene, Environment and Their Interaction. Genes and Environment. 2011; 33:128-140.
11. Mayer JE and Goldman RH. Arsenic and skin cancer in the USA: the current evidence regarding arsenic-contaminated drinking water. International journal of dermatology. 2016; 55:e585-e591.
12. Martinez VD, Becker-Santos DD, Vucic EA, Lam S and Lam WL. Induction of human squamous cell-type carcinomas by arsenic. Journal of skin cancer. 2011; 2011:454157.
13. Tsuji JS, Alexander DD, Perez V and Mink PJ. Arsenic exposure and bladder cancer: quantitative assessment of studies in human populations to detect risks at low doses. Toxicology. 2014; 317:17-30.
14. Wang W, Cheng S and Zhang D. Association of inorganic arsenic exposure with liver cancer mortality: A meta-analysis. Environmental research. 2014; 135:120-125.
15. Xie H, Huang S, Martin S and Wise Sr JP. Arsenic is cytotoxic and genotoxic to primary human lung cells. Mutat Res Genet Toxicol Environ Mutagen. 2014; 760:33-41.
16. Romagnolo DF, Daniels KD, Grunwald JT, Ramos SA, Propper CR and Selmin OI. Epigenetics of breast cancer: Modifying role of environmental and bioactive food compounds. Molecular nutrition & food research. 2016; 60:1310-1329.
17. Ngalame NN, Makia NL, Waalkes MP and Tokar EJ. Mitigation of arsenic-induced acquired cancer phenotype in prostate cancer stem cells by miR-143 restoration. Toxicology and applied pharmacology. 2015.
18. Kumar R, Trivedi V, Murti K, Dey A, Singh JK, Nath A and Das P. Arsenic Exposure and Haematological Derangement in Cervical Cancer Cases in India. Asian Pacific journal of cancer prevention. 2015; 16:6397-6400.
19. Saint-Jacques N, Parker L, Brown P and Dummer TJ. Arsenic in drinking water and urinary tract cancers: a systematic review of 30 years of epidemiological evidence. Environmental health. 2014; 13:44.
20. Baris D, Waddell R, Beane Freeman LE, Schwenn M, Colt JS, Ayotte JD, Ward MH, Nuckols J, Schned A, Jackson B, Clerkin C, Rothman N, Moore LE, et al. Elevated Bladder Cancer in Northern New England: The Role of Drinking Water and Arsenic. Journal of the National Cancer Institute. 2016; 108.
21. Colin P, Koenig P, Ouzzane A, Berthon N, Villers A, Biserte J and Roupret M. Environmental factors involved in carcinogenesis of urothelial cell carcinomas of the upper urinary tract. BJU international. 2009; 104:1436-1440.
22. Chiou HY, Chiou ST, Hsu YH, Chou YL, Tseng CH, Wei ML and Chen CJ. Incidence of transitional cell carcinoma and arsenic in drinking water: a follow-up study of 8,102 residents in an arseniasis-endemic area in northeastern Taiwan. American journal of epidemiology. 2001; 153:411-418.
23. Bhattacharyya P, Sen P, Ghosh A, Saha C, Bhattacharya PP, Das A, Majumdar K and Mazumder DG. Chronic lung disease and detection of pulmonary artery dilatation in high resolution computerized tomography of chest in chronic arsenic exposure. J Environ Sci Health A Tox Hazard Subst Environ Eng. 2014; 49:1453-1461.
24. Tsuji JS, Garry MR, Perez V and Chang ET. Low-level arsenic exposure and developmental neurotoxicity in children: A systematic review and risk assessment. Toxicology. 2015; 337:91-107.
25. Tolins M, Ruchirawat M and Landrigan P. The developmental neurotoxicity of arsenic: cognitive and behavioral consequences of early life exposure. Annals of global health. 2014; 80:303-314.
26. Chen Y, Graziano JH, Parvez F, Liu M, Slavkovich V, Kalra T, Argos M, Islam T, Ahmed A, Rakibuz-Zaman M, Hasan R, Sarwar G, Levy D, van Geen A and Ahsan H. Arsenic exposure from drinking water and mortality from cardiovascular disease in Bangladesh: prospective cohort study. Bmj. 2011; 342:d2431.
27. Ameer SS, Engstrom K, Harari F, Concha G, Vahter M and Broberg K. The effects of arsenic exposure on blood pressure and early risk markers of cardiovascular disease: Evidence for population differences. Environmental research. 2015; 140:32-36.
28. Ettinger AS, Arbuckle TE, Fisher M, Liang CL, Davis K, Cirtiu C-M, Bélanger P, LeBlanc A and Fraser WD. Arsenic levels among pregnant women and newborns in Canada: Results from the Maternal-Infant Research on Environmental Chemicals (MIREC) cohort. Environmental research. 2017; 153:8-16.
29. Martinez-Finley EJ, Goggin SL, Labrecque MT and Allan AM. Reduced expression of MAPK/ERK genes in perinatal arsenic-exposed offspring induced by glucocorticoid receptor deficits. Neurotoxicology and teratology. 2011; 33:530-537.
30. Chau D, Ng K, Chan TS, Cheng YY, Fong B, Tam S, Kwong YL and Tse E. Azacytidine sensitizes acute myeloid leukemia cells to arsenic trioxide by up-regulating the arsenic transporter aquaglyceroporin 9. Journal of hematology & oncology. 2015; 8:46.
31. LeBlanc MS, McKinney EC, Meagher RB and Smith AP. Hijacking membrane transporters for arsenic phytoextraction. Journal of biotechnology. 2013; 163:1-9.
32. Nemeti B, Regonesi ME, Tortora P and Gregus Z. Polynucleotide phosphorylase and mitochondrial ATP synthase mediate reduction of arsenate to the more toxic arsenite by forming arsenylated analogues of ADP and ATP. Toxicological sciences. 2010; 117:270-281.
33. Dilda PJ and Hogg PJ. Arsenical-based cancer drugs. Cancer treatment reviews. 2007; 33:542-564.
34. Martinez VD, Thu KL, Vucic EA, Hubaux R, Adonis M, Gil L, MacAulay C, Lam S and Lam WL. Whole-genome sequencing analysis identifies a distinctive mutational spectrum in an arsenic-related lung tumor. Journal of thoracic oncology. 2013; 8:1451-1455.
35. Kligerman AD, Malik SI and Campbell JA. Cytogenetic insights into DNA damage and repair of lesions induced by a monomethylated trivalent arsenical. Mutation research. 2010; 695:2-8.
36. Zhao CQ, Young MR, Diwan BA, Coogan TP and Waalkes MP. Association of arsenic-induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proceedings of the National Academy of Sciences of the United States of America. 1997; 94:10907-10912.
37. Schlawicke Engstrom K, Nermell B, Concha G, Stromberg U, Vahter M and Broberg K. Arsenic metabolism is influenced by polymorphisms in genes involved in one-carbon metabolism and reduction reactions. Mutation research. 2009; 667:4-14.
38. Yu ZM, Fung B, Murimboh JD, Parker L and Dummer TJ. What is the role of obesity in the aetiology of arsenic-related disease? Environment international. 2014; 66:115-123.
39. Alimoghaddam K. A review of arsenic trioxide and acute promyelocytic leukemia. International journal of hematology-oncology and stem cell research. 2014; 8:44-54.
40. Kumagai T, Shih LY, Hughes SV, Desmond JC, O’Kelly J, Hewison M and Koeffler HP. 19-Nor-1,25(OH)2D2 (a novel, noncalcemic vitamin D analogue), combined with arsenic trioxide, has potent antitumor activity against myeloid leukemia. Cancer research. 2005; 65:2488-2497.
41. Wang Y, Zhu XX and Zhu CS. [Abnormal methylation patterns of SFRP1 gene in cells of leukemia and inhibition of arsenic trioxide on the SFRP1 gene]. [Article in Chinese]. Zhonghua xue ye xue za zhi. 2013; 34:157-159.
42. Klein CB, Leszczynska J, Hickey C and Rossman TG. Further evidence against a direct genotoxic mode of action for arsenic-induced cancer. Toxicology and applied pharmacology. 2007; 222:289-297.
43. Kligerman AD and Tennant AH. Insights into the carcinogenic mode of action of arsenic. Toxicology and applied pharmacology. 2007; 222:281-288.
44. Shi H, Hudson LG, Ding W, Wang S, Cooper KL, Liu S, Chen Y, Shi X and Liu KJ. Arsenite causes DNA damage in keratinocytes via generation of hydroxyl radicals. Chemical research in toxicology. 2004; 17:871-878.
45. Liu SX, Athar M, Lippai I, Waldren C and Hei TK. Induction of oxyradicals by arsenic: implication for mechanism of genotoxicity. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98:1643-1648.
46. Kessel M, Liu SX, Xu A, Santella R and Hei TK. Arsenic induces oxidative DNA damage in mammalian cells. Molecular and cellular biochemistry. 2002; 234-235:301-308.
47. Ercal N, Gurer-Orhan H and Aykin-Burns N. Toxic metals and oxidative stress part I: mechanisms involved in metal-induced oxidative damage. Current topics in medicinal chemistry. 2001; 1:529-539.
48. Grollman AP and Moriya M. Mutagenesis by 8-oxoguanine: an enemy within. Trends in genetics. 1993; 9:246-249.
49. Qin XJ, Hudson LG, Liu W, Ding W, Cooper KL and Liu KJ. Dual actions involved in arsenite-induced oxidative DNA damage. Chemical research in toxicology. 2008; 21:1806-1813.
50. Ding W, Hudson LG and Liu KJ. Inorganic arsenic compounds cause oxidative damage to DNA and protein by inducing ROS and RNS generation in human keratinocytes. Molecular and cellular biochemistry. 2005; 279:105-112.
51. Matsui M, Nishigori C, Toyokuni S, Takada J, Akaboshi M, Ishikawa M, Imamura S and Miyachi Y. The role of oxidative DNA damage in human arsenic carcinogenesis: detection of 8-hydroxy-2’-deoxyguanosine in arsenic-related Bowen’s disease. The Journal of investigative dermatology. 1999; 113:26-31.
52. Kasai H. Analysis of a form of oxidative DNA damage, 8-hydroxy-2’-deoxyguanosine, as a marker of cellular oxidative stress during carcinogenesis. Mutation research. 1997; 387:147-163.
53. Kitchin KT. Recent advances in arsenic carcinogenesis: modes of action, animal model systems, and methylated arsenic metabolites. Toxicology and applied pharmacology. 2001; 172:249-261.
54. Bos JL. ras oncogenes in human cancer: a review. Cancer research. 1989; 49:4682-4689.
55. Nohara K, Tateishi Y, Suzuki T, Okamura K, Murai H, Takumi S, Maekawa F, Nishimura N, Kobori M and Ito T. Late-onset increases in oxidative stress and other tumorigenic activities and tumors with a Ha-ras mutation in the liver of adult male C3H mice gestationally exposed to arsenic. Toxicological sciences. 2012; 129:293-304.
56. Andrew AS, Mason RA, Memoli V and Duell EJ. Arsenic activates EGFR pathway signaling in the lung. Toxicological sciences. 2009; 109:350-357.
57. Wu W, Graves LM, Jaspers I, Devlin RB, Reed W and Samet JM. Activation of the EGF receptor signaling pathway in human airway epithelial cells exposed to metals. The American journal of physiology. 1999; 277:L924-931.
58. Eblin KE, Bredfeldt TG, Buffington S and Gandolfi AJ. Mitogenic signal transduction caused by monomethylarsonous acid in human bladder cells: role in arsenic-induced carcinogenesis. Toxicological sciences. 2007; 95:321-330.
59. Simeonova PP, Wang S, Hulderman T and Luster MI. c-Src-dependent activation of the epidermal growth factor receptor and mitogen-activated protein kinase pathway by arsenic. Role in carcinogenesis. The Journal of biological chemistry. 2002; 277:2945-2950.
60. Tanaka-Kagawa T, Hanioka N, Yoshida H, Jinno H and Ando M. Arsenite and arsenate activate extracellular signal-regulated kinases 1/2 by an epidermal growth factor receptor-mediated pathway in normal human keratinocytes. The British journal of dermatology. 2003; 149:1116-1127.
61. Herbert KJ and Snow ET. Modulation of arsenic-induced epidermal growth factor receptor pathway signalling by resveratrol. Chemico-biological interactions. 2012; 198:38-48.
62. Lee CH, Wu SB, Hong CH, Chen GS, Wei YH and Yu HS. Involvement of mtDNA damage elicited by oxidative stress in the arsenical skin cancers. The Journal of investigative dermatology. 2013; 133:1890-1900.
63. Modica-Napolitano JS and Singh KK. Mitochondria as targets for detection and treatment of cancer. Expert reviews in molecular medicine. 2002; 4:1-19.
64. Banerjee N, Banerjee M, Ganguly S, Bandyopadhyay S, Das JK, Bandyopadhay A, Chatterjee M and Giri AK. Arsenic-induced mitochondrial instability leading to programmed cell death in the exposed individuals. Toxicology. 2008; 246:101-111.
65. Miller WH, Jr., Schipper HM, Lee JS, Singer J and Waxman S. Mechanisms of action of arsenic trioxide. Cancer research. 2002; 62:3893-3903.
66. Wang XJ, Sun Z, Chen W, Eblin KE, Gandolfi JA and Zhang DD. Nrf2 protects human bladder urothelial cells from arsenite and monomethylarsonous acid toxicity. Toxicology and applied pharmacology. 2007; 225:206-213.
67. Bhattacharjee P, Banerjee M and Giri AK. Role of genomic instability in arsenic-induced carcinogenicity. A review. Environment international. 2013; 53:29-40.
68. Srivastava R, Bhattacharya S, Chakraborty A and Chattopadhyay A. Differential in vivo genotoxicity of arsenic trioxide in glutathione depleted mouse bone marrow cells: expressions of Nrf2/Keap1/P62. Toxicology mechanisms and methods. 2015; 25:223-228.
69. Kesari VP, Kumar A and Khan PK. Genotoxic potential of arsenic at its reference dose. Ecotoxicology and environmental safety. 2012; 80:126-131.
70. Rossner P, Boffetta P, Ceppi M, Bonassi S, Smerhovsky Z, Landa K, Juzova D and Sram RJ. Chromosomal aberrations in lymphocytes of healthy subjects and risk of cancer. Environmental health perspectives. 2005; 113:517-520.
71. Zhao Y, Toselli P and Li W. Microtubules as a critical target for arsenic toxicity in lung cells in vitro and in vivo. International journal of environmental research and public health. 2012; 9:474-495.
72. Chang P, Li Y and Li D. Micronuclei levels in peripheral blood lymphocytes as a potential biomarker for pancreatic cancer risk. Carcinogenesis. 2011; 32:210-215.
73. Khan PK, Kesari VP and Kumar A. Mouse micronucleus assay as a surrogate to assess genotoxic potential of arsenic at its human reference dose. Chemosphere. 2013; 90:993-997.
74. Huang Y, Zhang J, McHenry KT, Kim MM, Zeng W, Lopez-Pajares V, Dibble CC, Mizgerd JP and Yuan ZM. Induction of cytoplasmic accumulation of p53: a mechanism for low levels of arsenic exposure to predispose cells for malignant transformation. Cancer research. 2008; 68:9131-9136.
75. Hanahan D and Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57-70.
76. Martinez VD, Buys TP, Adonis M, Benitez H, Gallegos I, Lam S, Lam WL and Gil L. Arsenic-related DNA copy-number alterations in lung squamous cell carcinomas. British journal of cancer. 2010; 103:1277-1283.
77. Thu KL, Vucic EA, Chari R, Zhang W, Lockwood WW, English JC, Fu R, Wang P, Feng Z, MacAulay CE, Gazdar AF, Lam S and Lam WL. Lung adenocarcinoma of never smokers and smokers harbor differential regions of genetic alteration and exhibit different levels of genomic instability. PloS one. 2012; 7:e33003.
78. Kim IM, Ackerson T, Ramakrishna S, Tretiakova M, Wang IC, Kalin TV, Major ML, Gusarova GA, Yoder HM, Costa RH and Kalinichenko VV. The Forkhead Box m1 transcription factor stimulates the proliferation of tumor cells during development of lung cancer. Cancer research. 2006; 66:2153-2161.
79. Katoh M, Igarashi M, Fukuda H, Nakagama H and Katoh M. Cancer genetics and genomics of human FOX family genes. Cancer letters. 2013; 328:198-206.
80. Yaghmaie M, Mozdarani H, Alimoghaddam K, Ghaffari SH, Ghavamzadeh A and Hajhashemi M. Characterization of arsenic-induced cytogenetic alterations in acute promyelocytic leukemia cell line, NB4. Medical oncology. 2012; 29:1209-1216.
81. Lou Y, Ma Y, Suo S, Ni W, Wang Y, Pan H, Tong H, Qian W, Meng H, Mai W, Huang J, Yu W, Wei J, Mao L and Jin J. Prognostic factors of patients with newly diagnosed acute promyelocytic leukemia treated with arsenic trioxide-based frontline therapy. Leukemia research. 2015; 39:938-944.
82. Kala SV, Neely MW, Kala G, Prater CI, Atwood DW, Rice JS and Lieberman MW. The MRP2/cMOAT transporter and arsenic-glutathione complex formation are required for biliary excretion of arsenic. The Journal of biological chemistry. 2000; 275:33404-33408.
83. Marapakala K, Packianathan C, Ajees AA, Dheeman DS, Sankaran B, Kandavelu P and Rosen BP. A disulfide-bond cascade mechanism for arsenic(III) S-adenosylmethionine methyltransferase. Acta crystallographica Section D, Biological crystallography. 2015; 71:505-515.
84. Hughes MF. Arsenic toxicity and potential mechanisms of action. Toxicology letters. 2002; 133:1-16.
85. Martinez VD, Vucic EA, Adonis M, Gil L and Lam WL. Arsenic biotransformation as a cancer promoting factor by inducing DNA damage and disruption of repair mechanisms. Molecular biology international. 2011; 2011:718974.
86. Joyce BT, Gao T, Zheng Y, Liu L, Zhang W, Dai Q, Shrubsole MJ, Hibler EA, Cristofanilli M, Zhang H, Yang H, Vokonas P, Cantone L, Schwartz J, Baccarelli A and Hou L. Prospective changes in global DNA methylation and cancer incidence and mortality. British journal of cancer. 2016; 115:465-472.
87. Kuchiba A, Iwasaki M, Ono H, Kasuga Y, Yokoyama S, Onuma H, Nishimura H, Kusama R, Tsugane S and Yoshida T. Global methylation levels in peripheral blood leukocyte DNA by LUMA and breast cancer: a case-control study in Japanese women. British journal of cancer. 2014; 110:2765-2771.
88. Sarabi MM and Naghibalhossaini F. Association of DNA methyltransferases expression with global and gene-specific DNA methylation in colorectal cancer cells. Cell biochemistry and function. 2015; 33:427-433.
89. Bailey KA, Wu MC, Ward WO, Smeester L, Rager JE, Garcia-Vargas G, Del Razo LM, Drobna Z, Styblo M and Fry RC. Arsenic and the epigenome: interindividual differences in arsenic metabolism related to distinct patterns of DNA methylation. Journal of biochemical and molecular toxicology. 2013; 27:106-115.
90. Mauro M, Caradonna F and Klein CB. Dysregulation of DNA methylation induced by past arsenic treatment causes persistent genomic instability in mammalian cells. Environmental and molecular mutagenesis. 2016; 57:137-150.
91. Struck AW, Thompson ML, Wong LS and Micklefield J. S-adenosyl-methionine-dependent methyltransferases: highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. Chembiochem. 2012; 13:2642-2655.
92. Bustaffa E, Stoccoro A, Bianchi F and Migliore L. Genotoxic and epigenetic mechanisms in arsenic carcinogenicity. Archives of toxicology. 2014; 88:1043-1067.
93. Pelch KE, Tokar EJ, Merrick BA and Waalkes MP. Differential DNA methylation profile of key genes in malignant prostate epithelial cells transformed by inorganic arsenic or cadmium. Toxicology and applied pharmacology. 2015; 286:159-167.
94. Benbrahim-Tallaa L, Waterland RA, Styblo M, Achanzar WE, Webber MM and Waalkes MP. Molecular events associated with arsenic-induced malignant transformation of human prostatic epithelial cells: aberrant genomic DNA methylation and K-ras oncogene activation. Toxicology and applied pharmacology. 2005; 206:288-298.
95. Du J, Zhou N, Liu H, Jiang F, Wang Y, Hu C, Qi H, Zhong C, Wang X and Li Z. Arsenic induces functional re-expression of estrogen receptor alpha by demethylation of DNA in estrogen receptor-negative human breast cancer. PloS one. 2012; 7:e35957.
96. Chen H, Li S, Liu J, Diwan BA, Barrett JC and Waalkes MP. Chronic inorganic arsenic exposure induces hepatic global and individual gene hypomethylation: implications for arsenic hepatocarcinogenesis. Carcinogenesis. 2004; 25:1779-1786.
97. Niedzwiecki MM, Hall MN, Liu X, Oka J, Harper KN, Slavkovich V, Ilievski V, Levy D, van Geen A, Mey JL, Alam S, Siddique AB, Parvez F, Graziano JH and Gamble MV. A dose-response study of arsenic exposure and global methylation of peripheral blood mononuclear cell DNA in Bangladeshi adults. Environmental health perspectives. 2013; 121:1306-1312.
98. Rojas D, Rager JE, Smeester L, Bailey KA, Drobna Z, Rubio-Andrade M, Styblo M, Garcia-Vargas G and Fry RC. Prenatal arsenic exposure and the epigenome: identifying sites of 5-methylcytosine alterations that predict functional changes in gene expression in newborn cord blood and subsequent birth outcomes. Toxicological sciences. 2015; 143:97-106.
99. Majumdar S, Chanda S, Ganguli B, Mazumder DN, Lahiri S and Dasgupta UB. Arsenic exposure induces genomic hypermethylation. Environmental toxicology. 2010; 25:315-318.
100. Chanda S, Dasgupta UB, Guhamazumder D, Gupta M, Chaudhuri U, Lahiri S, Das S, Ghosh N and Chatterjee D. DNA hypermethylation of promoter of gene p53 and p16 in arsenic-exposed people with and without malignancy. Toxicological sciences. 2006; 89:431-437.
101. Treas J, Tyagi T and Singh KP. Chronic exposure to arsenic, estrogen, and their combination causes increased growth and transformation in human prostate epithelial cells potentially by hypermethylation-mediated silencing of MLH1. The Prostate. 2013; 73:1660-1672.
102. Banerjee N, Paul S, Sau TJ, Das JK, Bandyopadhyay A, Banerjee S and Giri AK. Epigenetic modifications of DAPK and p16 genes contribute to arsenic-induced skin lesions and nondermatological health effects. Toxicological sciences. 2013; 135:300-308.
103. Marsit CJ, Karagas MR, Danaee H, Liu M, Andrew A, Schned A, Nelson HH and Kelsey KT. Carcinogen exposure and gene promoter hypermethylation in bladder cancer. Carcinogenesis. 2006; 27:112-116.
104. Khaleghian A, Ghaffari SH, Ahmadian S, Alimoghaddam K and Ghavamzadeh A. Metabolism of arsenic trioxide in acute promyelocytic leukemia cells. Journal of cellular biochemistry. 2014; 115:1729-1739.
105. Jadhav V, Ray P, Sachdeva G and Bhatt P. Biocompatible arsenic trioxide nanoparticles induce cell cycle arrest by p21(WAF1/CIP1) expression via epigenetic remodeling in LNCaP and PC3 cell lines. Life sciences. 2016; 148:41-52.
106. Miao Z, Wu L, Lu M, Meng X, Gao B, Qiao X, Zhang W and Xue D. Analysis of the transcriptional regulation of cancer-related genes by aberrant DNA methylation of the cis-regulation sites in the promoter region during hepatocyte carcinogenesis caused by arsenic. Oncotarget. 2015; 6:21493-21506. doi: 10.18632/oncotarget.4085.
107. Hartwig A, Groblinghoff UD, Beyersmann D, Natarajan AT, Filon R and Mullenders LH. Interaction of arsenic(III) with nucleotide excision repair in UV-irradiated human fibroblasts. Carcinogenesis. 1997; 18:399-405.
108. Rossman TG, Uddin AN and Burns FJ. Evidence that arsenite acts as a cocarcinogen in skin cancer. Toxicology and applied pharmacology. 2004; 198:394-404.
109. Lu T, Liu J, LeCluyse EL, Zhou YS, Cheng ML and Waalkes MP. Application of cDNA microarray to the study of arsenic-induced liver diseases in the population of Guizhou, China. Toxicological sciences. 2001; 59:185-192.
110. Wu MM, Chiou HY, Ho IC, Chen CJ and Lee TC. Gene expression of inflammatory molecules in circulating lymphocytes from arsenic-exposed human subjects. Environmental health perspectives. 2003; 111:1429-1438.
111. Osmond MJ, Kunz BA and Snow ET. Age and exposure to arsenic alter base excision repair transcript levels in mice. Mutagenesis. 2010; 25:517-522.
112. Komissarova EV and Rossman TG. Arsenite induced poly(ADP-ribosyl)ation of tumor suppressor P53 in human skin keratinocytes as a possible mechanism for carcinogenesis associated with arsenic exposure. Toxicology and applied pharmacology. 2010; 243:399-404.
113. Miwa M and Masutani M. PolyADP-ribosylation and cancer. Cancer science. 2007; 98:1528-1535.
114. Person RJ, Ngalame NN, Makia NL, Bell MW, Waalkes MP and Tokar EJ. Chronic inorganic arsenic exposure in vitro induces a cancer cell phenotype in human peripheral lung epithelial cells. Toxicology and applied pharmacology. 2015; 286:36-43.
115. Faes S and Dormond O. PI3K and AKT: Unfaithful Partners in Cancer. International journal of molecular sciences. 2015; 16:21138-21152.
116. Liu J, Chen B, Lu Y, Guan Y and Chen F. JNK-dependent Stat3 phosphorylation contributes to Akt activation in response to arsenic exposure. Toxicological sciences. 2012; 129:363-371.
117. Tokar EJ, Diwan BA and Waalkes MP. Arsenic exposure transforms human epithelial stem/progenitor cells into a cancer stem-like phenotype. Environmental health perspectives. 2010; 118:108-115.
118. Gao N, Shen L, Zhang Z, Leonard SS, He H, Zhang XG, Shi X and Jiang BH. Arsenite induces HIF-1alpha and VEGF through PI3K, Akt and reactive oxygen species in DU145 human prostate carcinoma cells. Molecular and cellular biochemistry. 2004; 255:33-45.
119. Dong Z. The molecular mechanisms of arsenic-induced cell transformation and apoptosis. Environmental health perspectives. 2002; 110:757-759.
120. Evens AM, Tallman MS and Gartenhaus RB. The potential of arsenic trioxide in the treatment of malignant disease: past, present, and future. Leukemia research. 2004; 28:891-900.
121. Kumar S, Yedjou CG and Tchounwou PB. Arsenic trioxide induces oxidative stress, DNA damage, and mitochondrial pathway of apoptosis in human leukemia (HL-60) cells. Journal of experimental & clinical cancer research. 2014; 33:42.
122. Akao Y, Mizoguchi H, Kojima S, Naoe T, Ohishi N and Yagi K. Arsenic induces apoptosis in B-cell leukaemic cell lines in vitro: activation of caspases and down-regulation of Bcl-2 protein. British journal of haematology. 1998; 102:1055-1060.
123. Rousselot P, Labaume S, Marolleau JP, Larghero J, Noguera MH, Brouet JC and Fermand JP. Arsenic trioxide and melarsoprol induce apoptosis in plasma cell lines and in plasma cells from myeloma patients. Cancer research. 1999; 59:1041-1048.
124. Shen ZY, Tan LJ, Cai WJ, Shen J, Chen C, Tang XM and Zheng MH. Arsenic trioxide induces apoptosis of oesophageal carcinoma in vitro. International journal of molecular medicine. 1999; 4:33-37.
125. Cai X, Shen YL, Zhu Q, Jia PM, Yu Y, Zhou L, Huang Y, Zhang JW, Xiong SM, Chen SJ, Wang ZY, Chen Z and Chen GQ. Arsenic trioxide-induced apoptosis and differentiation are associated respectively with mitochondrial transmembrane potential collapse and retinoic acid signaling pathways in acute promyelocytic leukemia. Leukemia. 2000; 14:262-270.
126. Esteller M. Non-coding RNAs in human disease. Nature reviews Genetics. 2011; 12:861-874.
127. Gibb EA, Vucic EA, Enfield KS, Stewart GL, Lonergan KM, Kennett JY, Becker-Santos DD, MacAulay CE, Lam S, Brown CJ and Lam WL. Human cancer long non-coding RNA transcriptomes. PloS one. 2011; 6:e25915.
128. Gibb EA, Enfield KS, Stewart GL, Lonergan KM, Chari R, Ng RT, Zhang L, MacAulay CE, Rosin MP and Lam WL. Long non-coding RNAs are expressed in oral mucosa and altered in oral premalignant lesions. Oral oncology. 2011; 47:1055-1061.
129. Gibb EA, Brown CJ and Lam WL. The functional role of long non-coding RNA in human carcinomas. Molecular cancer. 2011; 10:38.
130. Stefani G and Slack FJ. Small non-coding RNAs in animal development. Nature reviews Molecular cell biology. 2008; 9:219-230.
131. Le Thomas A, Toth KF and Aravin AA. To be or not to be a piRNA: genomic origin and processing of piRNAs. Genome biology. 2014; 15:204.
132. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009; 136:215-233.
133. Wen W, Lu L, He Y, Cheng H, He F, Cao S, Li L, Xiong L and Wu T. LincRNAs and base modifications of p53 induced by arsenic methylation in workers. Chemico-biological interactions. 2016; 246:1-10.
134. Tani H, Onuma Y, Ito Y and Torimura M. Long non-coding RNAs as surrogate indicators for chemical stress responses in human-induced pluripotent stem cells. PloS one. 2014; 9:e106282.
135. Luo F, Sun B, Li H, Xu Y, Liu Y, Liu X, Lu L, Li J, Wang Q, Wei S, Shi L, Lu X, Liu Q and Zhang A. A MALAT1/HIF-2α feedback loop contributes to arsenite carcinogenesis. Oncotarget. 2016; 7:5769-87. doi: 10.18632/oncotarget.6806.
136. Esquela-Kerscher A and Slack FJ. Oncomirs - microRNAs with a role in cancer. Nature reviews Cancer. 2006; 6:259-269.
137. Sturchio E, Colombo T, Boccia P, Carucci N, Meconi C, Minoia C and Macino G. Arsenic exposure triggers a shift in microRNA expression. The Science of the total environment. 2014; 472:672-680.
138. Liu DZ, Zhang HY, Long XL, Zou SL, Zhang XY, Han GY and Cui ZG. MIR-150 promotes prostate cancer stem cell development via suppressing p27Kip1. European review for medical and pharmacological sciences. 2015; 19:4344-4352.
139. Aherne ST, Madden SF, Hughes DJ, Pardini B, Naccarati A, Levy M, Vodicka P, Neary P, Dowling P and Clynes M. Circulating miRNAs miR-34a and miR-150 associated with colorectal cancer progression. BMC cancer. 2015; 15:329.
140. Dinh TK, Fendler W, Chalubinska-Fendler J, Acharya SS, O’Leary C, Deraska PV, D’Andrea AD, Chowdhury D and Kozono D. Circulating miR-29a and miR-150 correlate with delivered dose during thoracic radiation therapy for non-small cell lung cancer. Radiation oncology. 2016; 11:61.
141. Yang K, He M, Cai Z, Ni C, Deng J, Ta N, Xu J and Zheng J. A decrease in miR-150 regulates the malignancy of pancreatic cancer by targeting c-Myb and MUC4. Pancreas. 2015; 44:370-379.
142. Dezhong L, Xiaoyi Z, Xianlian L, Hongyan Z, Guohua Z, Bo S, Shenglei Z and Lian Z. miR-150 is a factor of survival in prostate cancer patients. Journal of BUON. 2015; 20:173-179.
143. Wang Y, Wang L, Yin C, An B, Hao Y, Wei T, Li L and Song G. Arsenic trioxide inhibits breast cancer cell growth via microRNA-328/hERG pathway in MCF-7 cells. Molecular medicine reports. 2015; 12:1233-1238.
144. Martinez VD, Vucic EA, Thu KL, Hubaux R, Enfield KS, Pikor LA, Becker-Santos DD, Brown CJ, Lam S and Lam WL. Unique somatic and malignant expression patterns implicate PIWI-interacting RNAs in cancer-type specific biology. Scientific reports. 2015; 5:10423.
145. Gebert D, Ketting RF, Zischler H and Rosenkranz D. piRNAs from Pig Testis Provide Evidence for a Conserved Role of the Piwi Pathway in Post-Transcriptional Gene Regulation in Mammals. PloS one. 2015; 10:e0124860.
146. Ku HY and Lin H. PIWI proteins and their interactors in piRNA biogenesis, germline development and gene expression. National science review. 2014; 1:205-218.
147. Vagin VV, Sigova A, Li C, Seitz H, Gvozdev V and Zamore PD. A distinct small RNA pathway silences selfish genetic elements in the germline. Science. 2006; 313:320-324.
148. Yin H and Lin H. An epigenetic activation role of Piwi and a Piwi-associated piRNA in Drosophila melanogaster. Nature. 2007; 450:304-308.
149. Post C, Clark JP, Sytnikova YA, Chirn GW and Lau NC. The capacity of target silencing by Drosophila PIWI and piRNAs. Rna. 2014; 20:1977-1986.
150. Watanabe T and Lin H. Posttranscriptional regulation of gene expression by Piwi proteins and piRNAs. Molecular cell. 2014; 56:18-27.
151. Gou LT, Dai P, Yang JH, Xue Y, Hu YP, Zhou Y, Kang JY, Wang X, Li H, Hua MM, Zhao S, Hu SD, Wu LG, et al. Pachytene piRNAs instruct massive mRNA elimination during late spermiogenesis. Cell research. 2014; 24:680-700.
152. Kwon C, Tak H, Rho M, Chang HR, Kim YH, Kim KT, Balch C, Lee EK and Nam S. Detection of PIWI and piRNAs in the mitochondria of mammalian cancer cells. Biochemical and biophysical research communications. 2014; 446:218-223.
153. Huang XA, Yin H, Sweeney S, Raha D, Snyder M and Lin H. A major epigenetic programming mechanism guided by piRNAs. Developmental cell. 2013; 24:502-516.
154. Putila JJ and Guo NL. Association of arsenic exposure with lung cancer incidence rates in the United States. PloS one. 2011; 6:e25886.
155. Mohan D and Pittman CU, Jr. Arsenic removal from water/wastewater using adsorbents--A critical review. Journal of hazardous materials. 2007; 142:1-53.
156. Andrew AS, Burgess JL, Meza MM, Demidenko E, Waugh MG, Hamilton JW and Karagas MR. Arsenic exposure is associated with decreased DNA repair in vitro and in individuals exposed to drinking water arsenic. Environmental health perspectives. 2006; 114:1193-1198.
157. Walter I, Schwerdtle T, Thuy C, Parsons JL, Dianov GL and Hartwig A. Impact of arsenite and its methylated metabolites on PARP-1 activity, PARP-1 gene expression and poly(ADP-ribosyl)ation in cultured human cells. DNA repair. 2007; 6:61-70.
158. Hamadeh HK, Trouba KJ, Amin RP, Afshari CA and Germolec D. Coordination of altered DNA repair and damage pathways in arsenite-exposed keratinocytes. Toxicological sciences. 2002; 69:306-316.
159. He X, Chen MG, Lin GX and Ma Q. Arsenic induces NAD(P)H-quinone oxidoreductase I by disrupting the Nrf2 x Keap1 x Cul3 complex and recruiting Nrf2 x Maf to the antioxidant response element enhancer. The Journal of biological chemistry. 2006; 281:23620-23631.
160. Lu TH, Tseng TJ, Su CC, Tang FC, Yen CC, Liu YY, Yang CY, Wu CC, Chen KL, Hung DZ and Chen YW. Arsenic induces reactive oxygen species-caused neuronal cell apoptosis through JNK/ERK-mediated mitochondria-dependent and GRP 78/CHOP-regulated pathways. Toxicology letters. 2014; 224:130-140.
161. Pozo-Molina G, Ponciano-Gomez A, Rivera-Gonzalez GC, Hernandez-Zavala A and Garrido E. Arsenic-induced S phase cell cycle lengthening is associated with ROS generation, p53 signaling and CDC25A expression. Chemico-biological interactions. 2015; 238:170-179.
162. Liu Y, Hock JM, Van Beneden RJ and Li X. Aberrant overexpression of FOXM1 transcription factor plays a critical role in lung carcinogenesis induced by low doses of arsenic. Molecular carcinogenesis. 2014; 53:380-391.
163. Germolec DR, Yoshida T, Gaido K, Wilmer JL, Simeonova PP, Kayama F, Burleson F, Dong W, Lange RW and Luster MI. Arsenic induces overexpression of growth factors in human keratinocytes. Toxicology and applied pharmacology. 1996; 141:308-318.
164. Chen H, Liu J, Merrick BA and Waalkes MP. Genetic events associated with arsenic-induced malignant transformation: applications of cDNA microarray technology. Molecular carcinogenesis. 2001; 30:79-87.
165. Michailidi C, Hayashi M, Datta S, Sen T, Zenner K, Oladeru O, Brait M, Izumchenko E, Baras A, VandenBussche C, Argos M, Bivalacqua TJ, Ahsan H, et al. Involvement of epigenetics and EMT-related miRNA in arsenic-induced neoplastic transformation and their potential clinical use. Cancer prevention research. 2015; 8:208-221.
166. Cavigelli M, Li WW, Lin A, Su B, Yoshioka K and Karin M. The tumor promoter arsenite stimulates AP-1 activity by inhibiting a JNK phosphatase. The EMBO journal. 1996; 15:6269-6279.
167. Yih LH, Peck K and Lee TC. Changes in gene expression profiles of human fibroblasts in response to sodium arsenite treatment. Carcinogenesis. 2002; 23:867-876.
168. Zhao S, Zhang J, Zhang X, Dong X and Sun X. Arsenic trioxide induces different gene expression profiles of genes related to growth and apoptosis in glioma cells dependent on the p53 status. Molecular biology reports. 2008; 35:421-429.
169. Wang F, Liu S, Xi S, Yan L, Wang H, Song Y and Sun G. Arsenic induces the expressions of angiogenesis-related factors through PI3K and MAPK pathways in SV-HUC-1 human uroepithelial cells. Toxicology letters. 2013; 222:303-311.
170. Zhang S, Ma C, Pang H, Zeng F, Cheng L, Fang B, Ma J, Shi Y, Hong H, Chen J, Wang Z and Xia J. Arsenic trioxide suppresses cell growth and migration via inhibition of miR-27a in breast cancer cells. Biochemical and biophysical research communications. 2016; 469:55-61.
171. Wang Z, Humphries B, Xiao H, Jiang Y and Yang C. MicroRNA-200b suppresses arsenic-transformed cell migration by targeting protein kinase Calpha and Wnt5b-protein kinase Calpha positive feedback loop and inhibiting Rac1 activation. The Journal of biological chemistry. 2014; 289:18373-18386.
172. Gonzalez H, Lema C, Kirken RA, Maldonado RA, Varela-Ramirez A and Aguilera RJ. Arsenic-exposed Keratinocytes Exhibit Differential microRNAs Expression Profile; Potential Implication of miR-21, miR-200a and miR-141 in Melanoma Pathway. Clinical cancer drugs. 2015; 2:138-147.
173. Pfeffer SR, Yang CH and Pfeffer LM. The Role of miR-21 in Cancer. Drug development research. 2015; 76:270-277.
174. Yang CH, Yue J, Pfeffer SR, Fan M, Paulus E, Hosni-Ahmed A, Sims M, Qayyum S, Davidoff AM, Handorf CR and Pfeffer LM. MicroRNA-21 promotes glioblastoma tumorigenesis by down-regulating insulin-like growth factor-binding protein-3 (IGFBP3). The Journal of biological chemistry. 2014; 289:25079-25087.
175. Yang CH, Yue J, Fan M and Pfeffer LM. IFN induces miR-21 through a signal transducer and activator of transcription 3-dependent pathway as a suppressive negative feedback on IFN-induced apoptosis. Cancer research. 2010; 70:8108-8116.
176. Chang YW, Chen MW, Chiu CF, Hong CC, Cheng CC, Hsiao M, Chen CA, Wei LH and Su JL. Arsenic trioxide inhibits CXCR4-mediated metastasis by interfering miR-520h/PP2A/NF-kappaB signaling in cervical cancer. Annals of surgical oncology. 2014; 21:S687-695.
177. Wang M, Ge X, Zheng J, Li D, Liu X, Wang L, Jiang C, Shi Z, Qin L, Liu J, Yang H, Liu LZ, He J, Zhen L and Jiang BH. Role and mechanism of miR-222 in arsenic-transformed cells for inducing tumor growth. Oncotarget. 2016; 7:17805-17814. doi: 10.18632/oncotarget.7525.
178. Brown KR, Otasek D, Ali M, McGuffin MJ, Xie W, Devani B, Toch IL and Jurisica I. NAViGaTOR: Network Analysis, Visualization and Graphing Toronto. Bioinformatics (Oxford, England). 2009; 25:3327-3329.