
Introduction
Abdominal aortic aneurysms (AAAs) are permanent dilations of the abdominal aorta with over 85% mortality after rupture. There are no therapeutic strategies but surgery proven to blunt AAA progression and rapture. Pathologically, AAA is characterized by dilatation of all layers of the arterial wall as a result of loss of elastin, vascular smooth muscle cell (VSMC) apoptosis, and compensatory collagen deposition [1-3]. For example, angiotensin II (AngII) increases matrix metalloproteinases (MMPs) to induce AAA formation in animal experiments by upregulating oxidative stress [4, 5].
Statins are cholesterol-lowering drugs and widely used in the treatment of hypercholesterolemia to prevent the development of atherosclerosis through improving endothelial function [6-8]. However, statins produce several adverse effects, such as insulin resistance [9], skeletal muscle toxicity and myocardial atrophy [10]. The AMP-activated protein kinase (AMPK), consisting of catalytic α subunit and regulatory subunits β and γ, has a pivotal function in energy homoeostasis in eukaryotes. AMPK is activated by pravastatin in skeletal muscle and endothelial cells [11, 12]. We reported that AMPK activation mediated AngII-induced AAA formation in Apoe-/- mice [13]. This alludes that pravastatin is potentially to promote AAA incidences.
Activator protein 2α (AP-2α) is a member of the AP-2 transcription factor family consisting of α, β, γ, δ, and ε subunits [14]. Mice deficient in AP-2 die after birth due to the abnormal development, suggesting an important role of AP-2 in mammals [15, 16]. Park et al reported that nerve growth factor stimulated endothelial cell invasion by augmenting MMP-2 via AP-2α-dependent gene transcription, which may be responsible for triggering angiogenesis [17]. We found that aspirin activates AMPK to increase AP-2α in accelerated atherosclerotic plaque [18]. This study is aimed to investigate the effects of pravastatin on AAA formations. Our results demonstrate that pravastatin augments AngII-induced AAA incidence through AMPKα2/AP-2α signaling.
Results
Pravastatin promotes the incidence and severity of AAA formation in AngII-infused Apoe−/− mice
As reported [19, 20], we found that treatment with AngII (1.44 mg/kg/day) for 28 consecutive days promotes AAA formation in Apoe-/- mice as evidenced by enlarged abdominal aortas morphologically (Figure 1A). To determine the effects of statin on AAA formation, we treated AngII-infused Apoe-/- mice with pravastatin (50 mg/kg/day) for 8 weeks. Compared with mice treated with vehicle, pravastatin significantly increased the AAA incidence (60.87% VS 91.30%, P<0.05), the mortality and the severity of AAA in AngII-infused mice (Figure 1B and 1C). Histological analysis also indicated that the maximal aortic diameter and the degradation of elastin were increased by pravastatin in Apoe−/− mice with AAA (Figure 1D-1F). These results suggest that pravastatin promotes the AngII-induced AAA formation in Apoe-/- mice.
We also detected whether pravastatin affected hemodynamic parameters or metabolic indexes in AngII-infused Apoe-/- mice. As indicated in Supplementary Table S1 and S2, pravastatin treatment had no effects on heart rates, blood pressures, plasma triglyceride and glucose levels in AngII-infused Apoe-/- mice. Interestingly, the levels of total cholesterol and LDL cholesterol, but not HDL cholesterol, were increased by pravastatin in Apoe-/- mice, demonstrating that the effect of pravastatin on AAA formation is not due to the lipid-lowering effects.
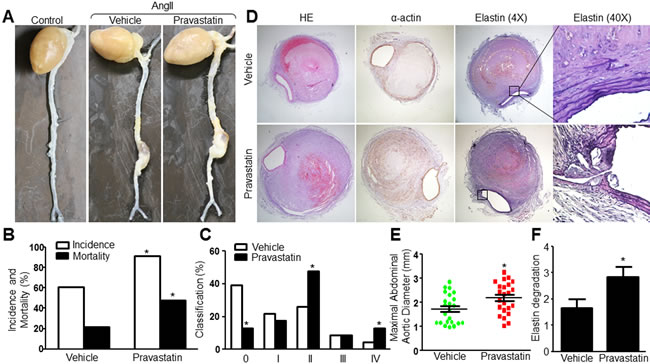
Figure 1: Pravastatin promotes AngII-induced AAA formation in Apoe−/− mice. Apoe-/- mice were received pravastatin administration (50 mg/kg) in drinking water for 4 weeks followed by AngII infusion (1.44 mg/kg/day) for another 4 weeks. A. Representative images showing macroscopic features of abdominal aortic specimens in mice. B. The incidence and the mortality, and C. the severity of AAA in AngII-infused mice. D. Histological analysis of HE staining, immunohistochemistry of α-actin, and Verhoff-Van Gieson staining in abdominal aortic cross-section. E. Maximal abdominal aortic diameters and F. grades of elastin degradation in aneurysmal tissues. N is 15-23 in each group. Chi-Square test was used for statistical comparisons in B. and C. Unpaired student’s t-test was used for statistical comparisons in E and F. *P < 0.05 vs. Vehicle.
Pravastatin via ROS increases AMPK phosphorylation in VSMCs
We previously reported that AMPK is activated by oxidants via threonine 172 phosphorylation (pAMPK-T172) and AMPK activation instigates AAA formation [13, 21]. Then we investigated whether pravastatin via oxidant activated AMPKα2. As indicated in Figure 2A-2C, the levels of pAMPK-T172 were gradually increased by pravastatin in concentration-dependent manner in cultured murine VSMCs. Tempol, as a SOD mimic, abolished the augments of Nox4, p47, 4-HNE, and pAMPK-T172 induced by pravastatin. Similarly, the ROS productions, assayed by determining the intensity of DHE fluorescence, were increased by pravastatin, which were abolished by tempol (Figure 2D and 2E). Taking all data together, it indicates that pravastatin via induction of oxidative stress activates AMPKα2 in VSMCs.
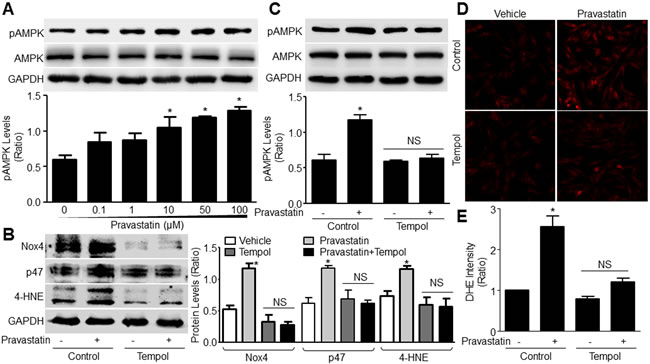
Figure 2: Pravastatin via ROS activates AMPK in murine VSMCs. A. Murine VSMCs were treated with pravastatin for 2 hours as indicated concentrations. Total cell lysates were subjected to perform western blot analysis of the levels of pAMPK-T172. The blot is a representative of three blots from three independent experiments. B.-E. Cultured VSMCs were pretreated with tempol (10 μM, 30 minutes) followed by co-incubation of pravastatin (50 μM) for 2 hours. Total cell lysates were subjected to perform western blot analysis of the levels of B. oxidative stress biomarkers of Nox4, p47 and 4-HNE and C. pAMPK-T172. ROS productions were determined by measuring DHE fluorescence in D. and performed quantitative analysis in E. N is 3 in each group. One-way ANOVA followed by Tukey post-hoc tests was used for multiple comparisons. In A, *P < 0.05 vs. point 0. In B., C. and E., *P < 0.05 vs. Control. NS indicates no significance.
Pravastatin via ROS enhances AP-2α phosphorylation in VSMCs
We have identified AP-2α as a downstream of AMPKα2 via phosphorylation at serine 219 (S219), which represents its transcriptional activity [13]. Thus, we detected whether pravastatin increases AP-2α S219 phosphorylation. As depicted in Figure 3A, pravastatin did not increase the level of pAP-2α-S219 at a concentration of 0.1-1 μM; however, at 10 μM, pravastatin significantly enhanced AP-2α S219 phosphorylation. Increasing concentrations of pravastatin (50-100 μM) further enhanced AP-2α phosphorylation. Further, reduction of ROS by tempol abolished pravastatin-induced AP-2α S219 phosphorylation (Figure 3B), revealing that pravastatin is a potential activator of AP-2α through ROS.
AMPKα2 is involved in pravastatin-increased AP-2α phosphorylation
To determine the role of AMPKα2 in pravastatin-activated AP-2α, we pretreated VSMCs with compound C (20 μM) which functions as an AMPK inhibitor (Supplementary Figure S1A). As presented in Figure 3B, pravastatin (50 μM) for 2 hours enhanced AP-2α phosphorylation in vehicle-treated cells, but not in VSMCs pretreated with compound C.
The effects of compound C on AP-2α phosphorylation were further confirmed by transfecting VSMCs with AMPKα2 siRNA to inhibit AMPKα2 expression (Supplementary Figure S1B). AMPKα2 siRNA, but not control siRNA, blocked the effects of pravastatin on AP-2α phosphorylation (Figure 3C), suggesting that the pravastatin-increased AP-2α phosphorylation is AMPKα2 dependent.
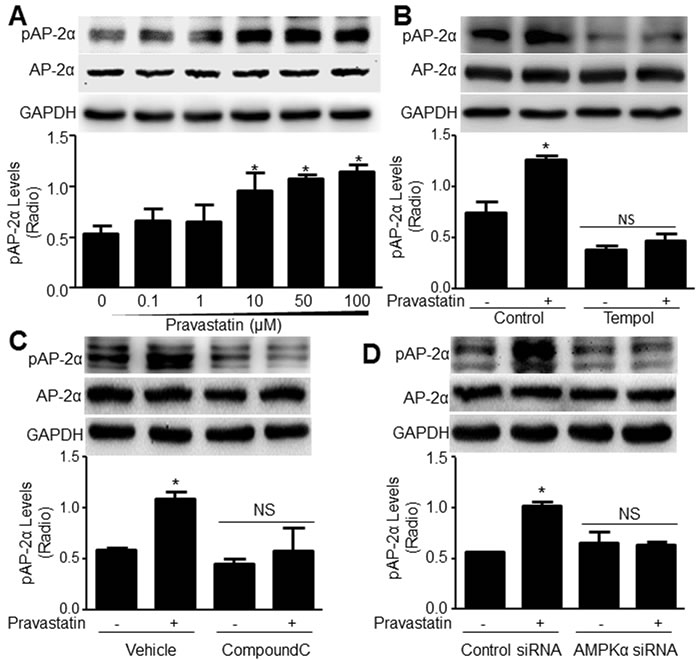
Figure 3: Pravastatin via AMPK activation increases AP-2α serine 219 phosphorylation in murine VSMCs. A. Mouse VSMCs were treated with pravastatin for 2 hours as indicated concentrations. B. Cultured VSMCs were pretreated with tempol (10 μM, 30 minutes) followed by co-incubation of pravastatin (50 μM) for 2 hours. C. VSMCs were pretreated with compound C (20 μM, 30 minutes) followed by co-incubation of pravastatin (50 μM) for 2 hours. D. VSMCs transfected with AMPKα2 siRNA for 48 hours were incubated with pravastatin (50 μM) for 2 hours. Total cell lysates in A.-D. were subjected to perform western blot analysis of the level of pAP-2α-S219. N is 3 in each group. One-way ANOVA followed by Tukey post-hoc tests was used for multiple comparisons. In A, *P < 0.05 vs. point 0. In B.-D., *P < 0.05 vs. Control (Vehicle) or control siRNA alone. NS indicates no significance.
Pravastatin via AMPKα2/AP-2α signaling increases MMP2 gene expressions in VSMCs
To validate how activation of AP-2α by pravastatin increases the AAA formation, we detected the levels of MMP2, which is a key contributor for elastin degradation in the process of AAA and a target of AP-2α in endothelial cells [17]. In Figure 4A, pravastatin concentration-dependently increased the protein levels of MMP2 in VSMCs. As expected, either tempol (Figure 4B) or compound C (Figure 4C) significantly abolished the pravastatin-enhanced the levels of MMP2 protein and activity, and mRNA (Supplementary Figure S1C).
To further confirm the essential roles of AMPKα2 and AP-2α in pravastatin-induced upregulations of MMP2, endogenous expressions of AMPKα2 and AP-2α in VSMCs were suppressed by transfection of AMPKα2 and AP-2α siRNA (Supplementary Figure S1B and S1D). Deficiency of AP-2α had no effects on pravastatin-increased AMPK phosphorylation, indicating that AP-2α is a downstream of AMPK (Supplementary Figure S1D). As shown in Figure 4D, 4E, S1E, S1F, pravastatin increased the levels of MMP2 protein, mRNA and activity in cells transfected with control siRNA. All these effects produced by pravastatin were bypassed by knockdown of AMPKα2 and AP-2α, demonstrating that both AMPKα2 and AP-2α are required for pravastatin-induced MMP2 upregulation in VSMCs.
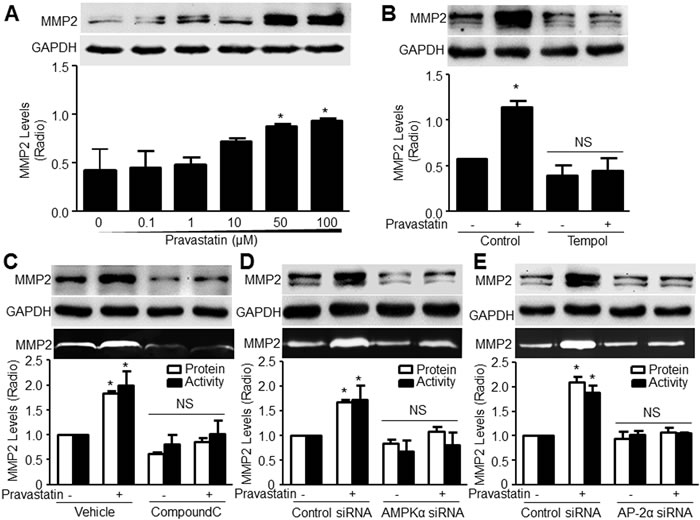
Figure 4: Pravastatin increases the levels of MMP2 protein and activity in murine VSMCs, which depends on AMPK and AP-2α. A. Mouse VSMCs were treated with pravastatin for 24 hours as indicated concentrations. B. Cultured VSMCs were pretreated with tempol (10 μM, 30 minutes) followed by co-incubation of pravastatin (50 μM) for 24 hours. C. VSMCs were pretreated with compound C. (20 μM, 30 minutes) followed by co-incubation of pravastatin (50 μM) for 24 hours. D. and E. Cultured VSMCs transfected with D. AMPKα2 siRNA or E. AP-2α siRNA for 48 hours were incubated with pravastatin (50 μM) for 24 hours. Total cell lysates in A.-E. were subjected to perform western blot analysis of the level of MMP2 proteins. The activity of MMP2 in culture medium in C.-E. was assayed by zymography. The N is 3 in each group. One-way ANOVA followed by Tukey post-hoc tests was used for multiple comparisons. In A., *P < 0.05 vs. point 0. In B.-E., *P < 0.05 vs. Vehicle or control siRNA alone. NS indicates no significance.
Pravastatin activates AMPKα2/AP-2α/MMP2 signaling in AngII-treated murine VSMCs
Knowing that pravastatin activated the AMPKα2/AP-2α/MMP2 signaling in VSMCs under resting condition, we next examined whether pravastatin activates the AMPKα2/AP-2α/MMP2 signaling in VSMCs with AngII. As indicated in Figure 5A-5C, both AngII and pravastatin alone increased the levels of pAMPK, pAP-2α, and MMP2 protein and mRNA expressions in VSMCs. Importantly, co-incubation of pravastatin further increased the levels of pAMPK, pAP-2α, and MMP2 protein and mRNA expressions in AngII-treated VSMCs, suggesting that pravastatin activates AMPKα2/AP-2α signaling in AngII-stressed cells.
Pravastatin activates AMPKα2/AP-2α/MMP2 signaling in AngII-treated human VSMCs
To show the translational applicability of these observations from mice, we also tested whether they can be replicated in human VSMCs. Thus, primary human aortic VSMCs were incubated with AngII plus pravastatin. Similarly, pravastatin increased the levels of pAMPK, pAP-2α, and MMP2 protein and mRNA expressions in human VSMCs with or without AngII (Figure 5D-5F), implying that pravastatin activates AMPKα2/AP-2α/MMP2 signaling in humans.
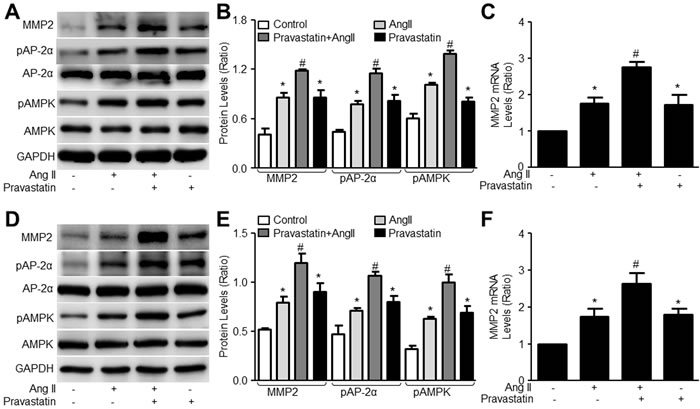
Figure 5: Pravastatin activates AMPK/AP-2α/MMP2 signaling in AngII-stimulated murine VSMCs. A.-C. Murine VSMCs were treated with pravastatin (50 μM) for 30 minutes followed by AngII (1 μM) for 24 hours. D.-F. Human VSMCs were treated with pravastatin (50 μM) for 30 minutes followed by AngII (1 μM) for 24 hours. Cells were harvested to measure the levels of MMP2, pAMPK and pAP-2α in total cell lysates by western blot in A. and D. The quantitative analysis was shown in B. and E. The level of MMP2 was determined in C. and F. by RT-qPCR and beta-actin was used as the housekeeping gene. N is 3 in each group. One-way ANOVA followed by Tukey post-hoc tests was used for multiple comparisons. *P < 0.05 vs. Control. #P < 0.05 vs. AngII alone.
Pravastatin induces oxidative stress, activates AMPKα2/AP-2α signaling, and upregulates MMP2 gene expression in vivo
The effects of pravastatin on oxidative stress, AMPKα2 and AP-2α phosphorylations, and MMP2 gene expression were further investigated in vivo. As indicated in Supplementary Figure S2A-S2C, in AngII-infused Apoe-/- mice, pravastatin administration dramatically increased the levels of 4-HNE, p47, Nox4, pAMPK-T172, pAP-2α-S219, and MMP2 protein and activity, compared to vehicle-treated mice, suggesting that pravastatin induces oxidative stress and activates AMPKα2/AP-2α/MMP2 signaling in vivo.
Knockdown of AMPKα2 ablates pravastatin-enhanced AAA formation in Apoe-/- mice
The role of AMPKα2 in pravastatin-increased AngII-induced AAA formation was also determined in Apoe-/- mice. Lentivirus-mediated RNA interference significantly inhibited AMPKα2 protein expression in abdominal aortic artery (Supplementary Figure S3B-S3E), but not affected hemodynamic parameters or metabolic indexes in AngII-infused Apoe-/- mice (Supplementary Table S3 and S4). Similarly, pravastatin significantly promoted the AngII-induced AAA formation in Apoe-/- mice infected with lentivirus expressing negative control shRNA, but not in Apoe-/- mice infected with lentivirus containing AMPKα2 shRNA (Figure 6A-6E). Collectively, it indicates that pravastatin via AMPKα2 activation promotes AAA formation in mice.
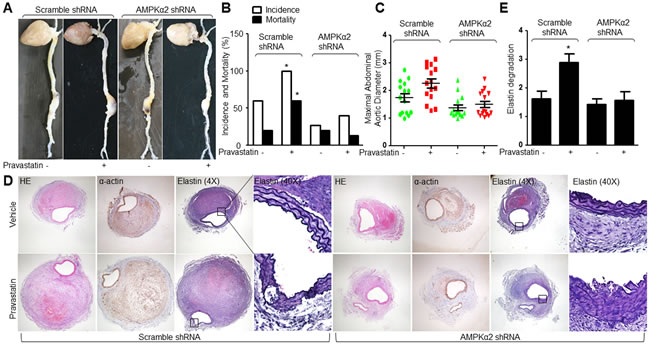
Figure 6: Deficiency of AMPKα2 blunts the effects of pravastatin on AAA formation in Apoe−/− mice. The protocol and experimental designs were described in Supplementary Methods and Figure S3A. A. Morphological analysis of abdominal aortic aneurysm, B. the incidence and the mortality, C. maximal abdominal aortic diameter, and D. histological analysis of HE staining, α-actin, and Verhoff-Van Gieson staining for elastin. E. Grades of elastin degradation in aneurysm tissues. 15 mice in each group. Chi-Square test was used for statistical comparisons in B. One-way ANOVA followed by Tukey post-hoc tests was used for multiple comparisons in C. and E. *P < 0.05 vs. Scramble shRNA alone.
Lentivirus-mediated gene knockdown of AP-2α abolishes the effects of pravastatin on AAA formation in Apoe-/- mice
We next examined the roles of AP-2α in the effects of pravastatin on promoting AAA formation in Apoe-/- mice. Due to the lethal phenotype of mice deficient in AP-2α [16], we generated AP-2α-knockdown mice by infecting lentivirus containing AP-2α short hairpin RNA (shRNA) to downregulate AP-2α protein expression in Apoe-/- mice (Supplementary Figure S3B-S3E), which did not affect hemodynamic parameters or metabolic indexes in AngII-infused Apoe-/- mice (Supplementary Table S5 and S6). As depicted in Figure 7A, pravastatin treatment significantly accelerated the AngII-induced AAA formation in Apoe-/- mice infected with lentivirus expressing scramble shRNA. The incidence and mortality of AAA (Figure 7B), maximal diameter (Figure 7C), and elastin degradation (Figure 7D and 7E) in abdominal aortic artery were increased, compared to vehicle-treated mice. However, all these effects induced by pravastatin were limited in Apoe-/- mice if they were infected with lentivirus expressing AP-2α shRNA. Together, these data demonstrate that AP-2α is required for pravastatin-enhanced AAA formation in mice.
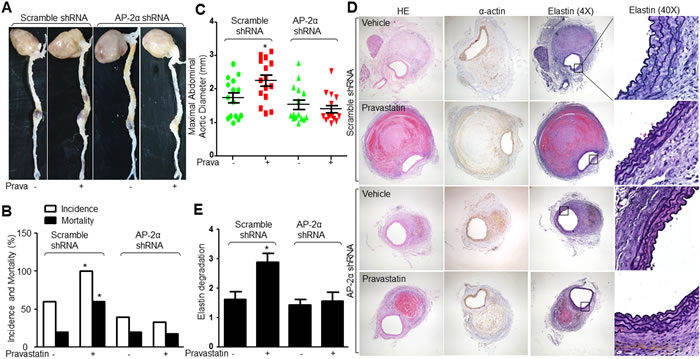
Figure 7: Lentivirus-mediated gene knockdown of AP-2α abolishes the effects of pravastatin on AAA formation in Apoe−/− mice. The protocol and experimental designs were described in Supplementary Methods and Figure S3A. A. Representative images showing macroscopic features of abdominal aortic specimens. B. The incidence and the mortality and C. maximal abdominal aortic diameter in all AngII-infused mice. D. Representative histological analysis of HE staining, IHC analysis of α-actin, Verhoff-Van Gieson staining for elastin in abdominal aortic cross-section. E. Grades of elastin degradation in aneurysm tissues. 15 mice in each group. Chi-Square test was used for statistical comparisons in B. One-way ANOVA followed by Tukey post-hoc tests was used for multiple comparisons in C and E. *P < 0.05 vs. Scramble shRNA alone.
AMPKα2 and AP-2α are crucial for pravastatin-induced MMP2 gene expression in vivo
The pathophysiological feather of AAA is the elastin degradation in vascular medium, which is mediated by MMPs (MMP2 and MMP9) [22]. We then examined the effects of AMPKα2 or AP-2α shRNA on MMP2/9 in vivo. As shown in Supplementary Figure S4A-S4B and S5A-S5D, the levels of MMP2 mRNA, protein, and activity were remarkably increased in Apoe-/- mice expressing scramble shRNA by pravastatin administration, compared to vehicle treated mice. However, either AP-2α or AMPKα2 shRNA inhibited the enchantment of MMP2 induced by pravastatin in AngII-infused Apoe-/- mice. The levels of MMP9 protein in cross-sections of the abdominal aortas were comparable among each group, indicating that MMP2 plays a dominant role in pravastatin-induced elastin degradation, similar to the report from Bradford C Berk’s lab [23]. Taken these data together, it reveals that AMPKα2 or AP-2α is crucial for pravastatin-enhanced MMP2 activation in vivo.
Pravastatin increases AP-2α and AMPKα2 phosphorylations in human subjects
To address if pravastatin at conventional dose (20 mg/day) activates AMPKα2/AP-2α signaling, we assayed the phosphorylated levels of AP-2α and AMPKα2 in peripheral blood leucocytes from human individuals taking pravastatin (Supplementary Table S7). In Figure 8A, the levels of pAP-2α-S219 and pAMPK-T172 were remarkably increased by taking pravastatin in humans, accompanied with the enhancement of serum MMP2 levels (Figure 8B). These data indicate that pravastatin is able to activate AMPKα2/AP-2α/MMP2 signaling in humans at regular dose.
Increased AP-2α and AMPKα2 phosphorylations in AAA patients
Finally, to establish human relevance, we conducted a pilot experiment by collecting samples of AAA from human subjects (Supplementary Table S8). As shown in Figure 8C-8E, human AAA samples exhibited higher levels of AMPKα2 and AP-2α phosphorylations in aortic tissues from AAA than those from non-AAA control patients. Although the pilot experiment did not establish the cause-effect relation among AP-2α and AAA in clinical investigations, it still implies the important role of AP-2α in the process of AAA.
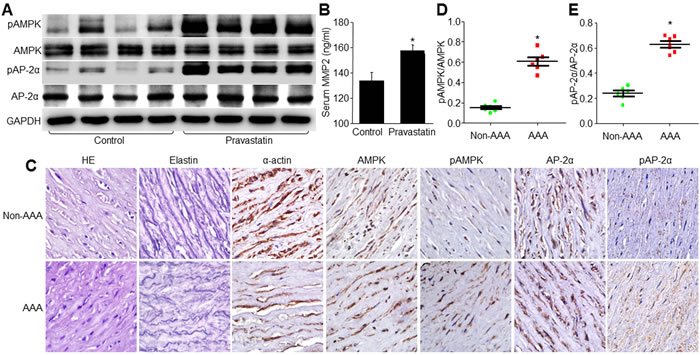
Figure 8: The levels of pAMPK and pAP-2α are increased in human individuals taking pravastatin and in AAA patients. A. and B. The profiles of these patients were shown in Supplementary Table S9. The peripheral blood cells were collected from human subjects with or without taking pravastatin (20 mg per day) for 0.5-3 years. The levels of pAMPK-T172 and pAP-2α-S219 in total cell lysates were detected by Western blot A. and serum levels of MMP2 were determined by ELISA B.. C.-E. The demographic data were presented in Supplementary Table S10. The levels of pAMPK and pAP-2α were assayed by IHC C.. The quantitative analyses of pAMPK D. and pAP-2α E. from C were shown. 8 human subjects in each group in A and B. 6 human subjects in each group in C.-E. Unpaired student’s t-test was used for all statistical comparisons. *P < 0.05 vs. Control patients B. or Non-AAA patients D., E.
Discussion
The major finding of this study is that pravastatin promotes the AAA formation. To date, the effects of statins on AAA remain controversial. Simvastatin has been reported to inhibit AAA formation induced by angiotensin II (AngII) in apolipoprotein E knockout (Apoe-/-) mice [24-26], while rosuvastatin and atorvastatin had no benefit for abdominal aortic aneurysms [27]. Pravastatin produces bidirectional effects on cerebral aneurysm in estrogen-deficient rats, which is associated with the dose [23]. Karrowni et al retrospectively found that statin therapy led to decreased AAA growth rate in humans, and while meta-analyses and individual studies have failed to show benefit or shown mildly reduced growth [28]. Hurks et al reported that there was a trend towards increased aortic MMPs and protease activity in AAA patients taking pravastatin, but not atorvastatin or simvastatin [29]. The distinct effects of statins on AAA may depend on the subtypes, doses, durations, and AAA models.
We also identified a novel molecular mechanism that AP-2α S219 phosphorylation is crucial for pravastatin-promoted AAA formation. As a transcription factor, AP-2α recognizes the consensus DNA sequence of 5′-GCCNNNGGC-3′, which regulates a number of genes including MMP2, p21, Bcl-2, and VEGF, which are involved in multiple cell functions. We previously identified AMPKα2 as an upstream kinase of AP-2α [13]. In this study, we further discovered that statins function as AP-2α activator through AMPKα2.
The results presented in this work have for the first time established a causal effect of statins on AAA in vivo and uncovered the molecular mechanism by which pravastatin promotes AAA formation (Supplementary Figure S6). We suggest that pravastatin administration would be cautiously reconsidered in patients with high serum level of AngII or low blood level of apolipoprotein E, and patients with AAA.
Materials and Methods
Detailed information is available in Supplementary Materials.
Animals and protocols of in vivo experiments
Male Apoe-/- mice were received pravastatin administration (50 mg/kg) in drinking water. The model of AAA was established by implanting an Alzet osmatic minipumps into Apoe-/- mice to deliver AngII (1.44 mg/kg/day, 28 days) as described previously [30]. Mice were injected with GFP-labeled lentivirus expressing AP-2α or AMPKα2 shRNA via tail vein as described previously [31]. At the end of experiment, all mice were sacrificed under anesthesia. The whole aortas including thoracic and abdominal aortas were collected for morphological and histological analysis of AAA.
Analysis and quantification of AAA
To quantify AAA incidence and size, the maximum width of the abdominal aorta was measured with Image Pro Plus software (Media Cybernetics Inc.). Aneurysm incidence was defined as an external width of the suprarenal aorta that was increased by 50% or greater as described previously [32].
Patients and sample processing
AAA tissues were obtained from human subjects under surgery. Patients were enrolled to take pravastatin (20 mg/kg/day) for 0.5-3 years. The blood was collected from human subjects before and after pravastatin treatment. Leucocytes were isolated from blood as described previously [33].
Generation of shRNA construct and lentivirus production
Based on the protocol from Signaling Gateway, the shRNA cassette containing target sequence of AP-2α (GGAGAGCGAAGTCTAAGAATG) or AMPKα2 (GGAGAGCUAUUUGAUUAUATT) was designed. The cassette was subcloned into pEN-hH1c vector as described previously [34].
Cell cultures
As described previously [35], mouse and human vascular smooth muscle cells (VSMCs) from ATCC were grown in basal medium (Clonetics Inc. Walkersville, MD) supplemented with 2% fetal bovine serum, penicillin (100 U/ml) and streptomycin (10 mg/ml). In all experiments, cells were used between passages 4 and 8. All cells were incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Cells were grown to 80% confluency before being treated with different agents.
Transfection of siRNA into cells
Transient transfection of siRNA was carried out according to Santa Cruz’s protocol [36].
Detection of ROS
ROS production in culture cells was detected using the fluorescent probe DHE as described previously [36, 37].
Gelatin Zymography
Zymography was completed by use of a MMP gelatin zymography kit (GenMed Scientific Inc., USA) as described previously [13].
Statistical analysis
For the relative quantitation of western blot, qPCR and zymography, the intensity of bands were calculated and the background was subtracted from the calculated area. We setup the ration of control group as 1. All quantitative data were reported as mean ± SEM. One-way ANOVA followed by Tukey post-hoc tests was used for multiple comparisons. Chi-Square test was used for statistical comparisons of AAA incidence, mortality, and severity. Two-sided P-value <0.05 was considered as significant.
Conflicts of Interest
There is no conflict of interest.
Grant Support
This work was supported by National 973 Basic Research Program of China (2013CB530700), National Natural Science Foundation of China (81320108004, 81370411, 81470591, 81570723, 81570729, 81673423), Natural Science Foundation of Shandong Province (ZR2016HM50), and Natural Science Foundation of Henan Province (162300410216, 121100910300, 132102310247). This project was also sponsored by Program for New Century Excellent Talents in University (NCET-13-0351), Program of Clinical Investigation (Nanshan Group), Qilu Hospital, Shandong University (2014QLKY15), and the Research Fund of Xinxiang Medical University (2016PN-KFKT-02, XYBSKYZZ201626, 2014QN153, ZD2011-30). Shuang-Xi Wang is a recipient of Qilu Professional Scholar of Shandong University and a Taihang Professional Scholar of Xinxiang Medical University (505067). Dr. Ya-Ling Yin is a recipient of Taihang Young Scholars of Xinxiang Medical University.
References
1. Satoh K, Nigro P, Matoba T, O’Dell MR, Cui Z, Shi X, Mohan A, Yan C, Abe J, Illig KA, Berk BC. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nat Med. 2009;15:649-656
2. Shah PK. Inflammation, metalloproteinases, and increased proteolysis: an emerging pathophysiological paradigm in aortic aneurysm. Circulation. 1997;96:2115-2117
3. Thompson RW, Geraghty PJ, Lee JK. Abdominal aortic aneurysms: basic mechanisms and clinical implications. Curr Probl Surg. 2002;39:110-230
4. Browatzki M, Larsen D, Pfeiffer CA, Gehrke SG, Schmidt J, Kranzhofer A, Katus HA, Kranzhofer R. Angiotensin II stimulates matrix metalloproteinase secretion in human vascular smooth muscle cells via nuclear factor-kappaB and activator protein 1 in a redox-sensitive manner. J Vasc Res. 2005;42:415-423
5. Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916-1923
6. Davignon J. Beneficial cardiovascular pleiotropic effects of statins. Circulation. 2004;109:III39-43
7. Li P, Yin YL, Guo T, Sun XY, Ma H, Zhu ML, Zhao FR, Xu P, Chen Y, Wan GR, Jiang F, Peng QS, Liu C, Liu LY, Wang SX. Inhibition of Aberrant MicroRNA-133a Expression in Endothelial Cells by Statin Prevents Endothelial Dysfunction by Targeting GTP Cyclohydrolase 1 in Vivo. Circulation. 2016;134:1752-1765
8. Wang F, Ma H, Liang WJ, Yang JJ, Wang XQ, Shan MR, Chen Y, Jia M, Yin YL, Sun XY, Zhang JN, Peng QS, Chen YG, et al. Lovastatin upregulates microRNA-29b to reduce oxidative stress in rats with multiple cardiovascular risk factors. Oncotarget. 2017;8:9021-9034. doi: 10.18632/oncotarget.14462.
9. Naci H, Brugts J, Ades T. Comparative tolerability and harms of individual statins: a study-level network meta-analysis of 246 955 participants from 135 randomized, controlled trials. Circ Cardiovasc Qual Outcomes. 2013;6:390-399
10. Bonifacio A, Mullen PJ, Mityko IS, Navegantes LC, Bouitbir J, Krahenbuhl S. Simvastatin induces mitochondrial dysfunction and increased atrogin-1 expression in H9c2 cardiomyocytes and mice in vivo. Arch Toxicol. 2016;90:203-215
11. Izumi Y, Shiota M, Kusakabe H, Hikita Y, Nakao T, Nakamura Y, Muro T, Miura K, Yoshiyama M, Iwao H. Pravastatin accelerates ischemia-induced angiogenesis through AMP-activated protein kinase. Hypertens Res. 2009;32:675-679
12. Ohira M, Endo K, Saiki A, Miyashita Y, Terai K, Murano T, Watanabe F, Tatsuno I, Shirai K. Atorvastatin and pitavastatin enhance lipoprotein lipase production in L6 skeletal muscle cells through activation of adenosine monophosphate-activated protein kinase. Metabolism. 2012;61:1452-1460
13. Wang S, Zhang C, Zhang M, Liang B, Zhu H, Lee J, Viollet B, Xia L, Zhang Y, Zou MH. Activation of AMP-activated protein kinase alpha2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. Nat Med. 2012;18:902-910
14. Zhao F, Satoda M, Licht JD, Hayashizaki Y, Gelb BD. Cloning and characterization of a novel mouse AP-2 transcription factor, AP-2delta, with unique DNA binding and transactivation properties. J Biol Chem. 2001;276:40755-40760
15. Schorle H, Meier P, Buchert M, Jaenisch R, Mitchell PJ. Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature. 1996;381:235-238
16. Zhang J, Hagopian-Donaldson S, Serbedzija G, Elsemore J, Plehn-Dujowich D, McMahon AP, Flavell RA, Williams T. Neural tube, skeletal and body wall defects in mice lacking transcription factor AP-2. Nature. 1996;381:238-241
17. Park MJ, Kwak HJ, Lee HC, Yoo DH, Park IC, Kim MS, Lee SH, Rhee CH, Hong SI. Nerve growth factor induces endothelial cell invasion and cord formation by promoting matrix metalloproteinase-2 expression through the phosphatidylinositol 3-kinase/Akt signaling pathway and AP-2 transcription factor. J Biol Chem. 2007;282:30485-30496
18. Yang JJ, Li P, Wang F, Liang WJ, Ma H, Chen Y, Ma ZM, Li QZ, Peng QS, Zhang Y, Wang SX. Activation of activator protein 2 alpha by aspirin alleviates atherosclerotic plaque growth and instability in vivo. Oncotarget. 2016;7:52729-52739. doi: 10.18632/oncotarget.10400.
19. Maegdefessel L, Azuma J, Toh R, Merk DR, Deng A, Chin JT, Raaz U, Schoelmerich AM, Raiesdana A, Leeper NJ, McConnell MV, Dalman RL, Spin JM, Tsao PS. Inhibition of microRNA-29b reduces murine abdominal aortic aneurysm development. J Clin Invest. 2012;122:497-506
20. Boon RA, Seeger T, Heydt S, Fischer A, Hergenreider E, Horrevoets AJ, Vinciguerra M, Rosenthal N, Sciacca S, Pilato M, van Heijningen P, Essers J, Brandes RP, Zeiher AM, Dimmeler S. MicroRNA-29 in aortic dilation: implications for aneurysm formation. Circ Res. 2011;109:1115-1119
21. Zhang J, Xie Z, Dong Y, Wang S, Liu C, Zou MH. Identification of nitric oxide as an endogenous activator of the AMP-activated protein kinase in vascular endothelial cells. J Biol Chem. 2008;283:27452-27461
22. Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625-632
23. Tada Y, Kitazato KT, Yagi K, Shimada K, Matsushita N, Kinouchi T, Kanematsu Y, Satomi J, Kageji T, Nagahiro S. Statins promote the growth of experimentally induced cerebral aneurysms in estrogen-deficient rats. Stroke. 2011;42:2286-2293
24. Zhang Y, Naggar JC, Welzig CM, Beasley D, Moulton KS, Park HJ, Galper JB. Simvastatin inhibits angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-knockout mice: possible role of ERK. Arterioscler Thromb Vasc Biol. 2009;29:1764-1771
25. Golledge J, Cullen B, Moran C, Rush C. Efficacy of simvastatin in reducing aortic dilatation in mouse models of abdominal aortic aneurysm. Cardiovasc Drugs Ther. 2010;24:373-378
26. Steinmetz EF, Buckley C, Shames ML, Ennis TL, Vanvickle-Chavez SJ, Mao D, Goeddel LA, Hawkins CJ, Thompson RW. Treatment with simvastatin suppresses the development of experimental abdominal aortic aneurysms in normal and hypercholesterolemic mice. Ann Surg. 2005;241:92-101
27. Wang JA, Chen WA, Wang Y, Zhang S, Bi H, Hong B, Luo Y, Daugherty A, Xie X. Statins exert differential effects on angiotensin II-induced atherosclerosis, but no benefit for abdominal aortic aneurysms. Atherosclerosis. 2011;217:90-96
28. Karrowni W, Dughman S, Hajj GP, Miller FJ, Jr. Statin therapy reduces growth of abdominal aortic aneurysms. J Investig Med. 2011;59:1239-1243
29. Hurks R, Hoefer IE, Vink A, Pasterkamp G, Schoneveld A, Kerver M, de Vries JP, Tangelder MJ, Moll FL. Different effects of commonly prescribed statins on abdominal aortic aneurysm wall biology. Eur J Vasc Endovasc Surg. 2010;39:569-576
30. Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605-1612
31. Maegdefessel L, Azuma J, Toh R, Deng A, Merk DR, Raiesdana A, Leeper NJ, Raaz U, Schoelmerich AM, McConnell MV, Dalman RL, Spin JM, Tsao PS. MicroRNA-21 blocks abdominal aortic aneurysm development and nicotine-augmented expansion. Sci Transl Med. 2012;4:122ra122
32. Sparks AR, Johnson PL, Meyer MC. Imaging of abdominal aortic aneurysms. Am Fam Physician. 2002;65:1565-1570
33. Nimrichter L, Burdick MM, Aoki K, Laroy W, Fierro MA, Hudson SA, Von Seggern CE, Cotter RJ, Bochner BS, Tiemeyer M, Konstantopoulos K, Schnaar RL. E-selectin receptors on human leukocytes. Blood. 2008;112:3744-3752
34. Godec J, Cowley GS, Barnitz RA, Root DE, Sharpe AH, Haining WN. Inducible RNAi in vivo reveals that the transcription factor BATF is required to initiate but not maintain CD8+ T-cell effector differentiation. Proc Natl Acad Sci U S A. 2015;112:512-517
35. Wang J, Guo T, Peng QS, Yue SW, Wang SX. Berberine via suppression of transient receptor potential vanilloid 4 channel improves vascular stiffness in mice. J Cell Mol Med. 2015;19:2607-2616
36. Wang S, Xu J, Song P, Wu Y, Zhang J, Chul Choi H, Zou MH. Acute inhibition of guanosine triphosphate cyclohydrolase 1 uncouples endothelial nitric oxide synthase and elevates blood pressure. Hypertension. 2008;52:484-490
37. Yang XH, Li P, Yin YL, Tu JH, Dai W, Liu LY, Wang SX. Rosiglitazone via PPARgamma-dependent suppression of oxidative stress attenuates endothelial dysfunction in rats fed homocysteine thiolactone. J Cell Mol Med. 2015;19:826-835