
INTRODUCTION
Glioblastoma (GBM) is the most lethal form of brain tumor and the most prevalent malignant primary brain tumor in adults. This highly aggressive tumor has not only a poor prognosis and treatment limitations, but also direct repercussions on quality of life and cognitive functions of the patient. With 3.19 in 100,000 people newly diagnosed each year GBM has the highest number of cases of all malignant brain tumors [1]. Until recently, the World Health Organization (WHO) classified gliomas into grades I to IV defined by increasing degrees of undifferentiated phenotype, grade of malignancy, proliferative index, response to treatment and survival time. While Grade I and II described benign and relatively non-malignant tumors, Grade III were tumors of low-grade malignancy, and Grade IV astrocytoma were designated the term glioblastoma (GBM) [2]. In 2016, CNS tumors were redefined by the WHO to incorporate both histological and molecular features including isocitrate dehydrogenase 1 (IDH)-wildtype, IDH-mutant, giant cell glioblastoma, gliosarcoma, epithelioid GBM and diffuse midline glioma, H3 K27 mutant [3]. Unfortunately however the median survival rate for GBM remains at 14.6 months [4], with a five year survival rate of only 4–5%. This dismal outcome motivates a search for novel and effective therapies to treat this incurable disease.
A combination of resection, radiotherapy (RT) and chemotherapy is the current standard of care for treating GBM. The degree of resection is of greatest importance as it can prolong survival significantly [5–7]; however, it is difficult to achieve complete resection without causing neurological deficits due to the highly invasive capacity of GBM cells in infiltrating adjacent brain tissue [8]. Ionizing radiation therapy remains the most clinically effective treatment strategy for most CNS tumors. First-line chemotherapy for GBM is the DNA alkylating agent temozolomide (TMZ). Despite the fact that several chemotherapies are available, patients treated with TMZ have the highest median survival rate compared to other chemotherapeutic agents [4, 9]. TMZ has much less toxicity than other chemotherapeutics and is administered orally.
Although the number of studies of GBM has grown and current therapy regimes have improved, only little improvement has been achieved in the last decade, with patients still waiting for a breakthrough which extends life beyond the median 14 months of survival post initial diagnosis. Unfortunately the majority of preclinical therapies which were shown to be effective have subsequently failed in the clinic. A promising approach, which holds real translation potential for the treatment of GBM, is drug repurposing [10–12].
Aromatase, or estrogen synthase, is a critical enzyme in the irreversible conversion of androgens to estrogen [13]. The aromatase enzyme is comprised of a microsomal cytochrome P450 complex which is a product of the CYP19A1 gene. Innate aromatase activity is found to be present in gonadal tissues, uterus, breast, prostate, epididymis, placenta, adrenal glands, liver, skin, muscle, vascular endothelium, bone and brain [14]. Moreover estrogen is associated with several cancers and protects against adverse symptoms during the peri- and postmenopausal intervals, stimulating cellular proliferation, migration and growth of reproductive tissues [15]. The inhibition of aromatase enzyme is currently used in the treatment of hormone-dependent breast cancer, alterations of ovarian and endometrial function and treatment of benign disorders like gynecomastia as uncontrolled proliferation is targeted. In the present study, we therefore investigated the effects of aromatase inhibition on GBM cells proliferation, migration and, ultimately when used as an adjunct therapy, chemoresistance.
Specifically targeting drugs to disease sites within targeted polymeric carriers offers great potential to eliminate adverse side effects. Poly lactic-co-glycolic acid (PLGA) is a well-established polymeric excipient (FDA approved) consisting of poly lactic acid (PLA) and poly glycolic acid (PGA). Disialogangliosidase2 (GD2) is a highly glycosylated sphingolipid which has been shown to be highly expressed in high-grade gliomas [16] in addition to several other cancer types [17]. It was also reported as a target for glioblastoma [18] but, to date, there has been no clinical development related to GBM. Clinical experience with anti-GD2 antibodies was generated in neuroblastoma, a paediatric malignancy characterized by high GD2 expression. Several anti-GD2 antibodies are under clinical development and dinutuximab (ch14.18 produced in SP2/0 cells) was approved for the treatment of neuroblastoma in combination with IL2 and GM-CSF [19]. In Europe, ch14.18 was remanufactured in CHO cells (anti-GD2-ch14.18/CHO) [20, 21], which is currently used in clinical trials [22, 23]. Based on these considerations, we investigated whether ch14.18/CHO may provide a novel approach to target nanoparticles to glioblastoma and thereby minimise off-target effects.
In this study, biodegradable PLGA nanoparticles containing the aromatase inhibitor, Letrozole, were synthesized and capable of selective target recognition in co-culture experiments. Furthermore it was determined that aromatase inhibition lead to improved drug response to clinically relevant chemotherapeutic treatment.
RESULTS
Assessment of aromatase expression
To evaluate the expression of aromatase as a target in patient-derived glioblastoma cells we investigated the protein levels of P450 (aromatase) in a panel of both patient-derived cell lines and xenograft tumor lysates. Glioblastoma cell lines derived from primary tumors, MZ-327 and RN1 and recurrent derived GBM tumors, MZ-256 and MZ-304, in addition to several patient derived xenograft tumour lysates provided by the Mayo Clinic Brain Tumor SPORE (G46, G59, G64, G75, G76, G79, G80, G84, G85 and G91, Supplementary Table 1) [24–27] were assessed by Western blot (Figure 1). Notably, all GBM samples assessed express P450 at the molecular weight of 50–55 kDa, corresponding to the apparent molecular weight of aromatase.
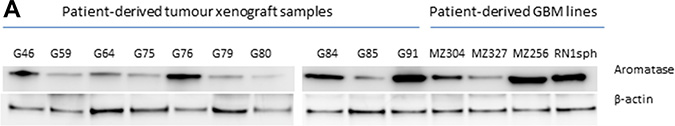
Figure 1: Evaluation of aromatase expression. Aromatase (Cytochrome P450) protein expression was assessed in a panel of patient-derived GBM tumor xenograft samples and cell lines. Isolated protein lysates were provided from patient-derived glioblastoma xenografts (G46, G59, G64, G76, G79, G80, G84, G85 and G91) by the Mayo Clinic Brain Tumor SPORE [24–27]. These lysates, in addition to primary (MZ-327 and RN1) and recurrent (MZ-256 and MZ-304) GBM patient-derived cell lines were assessed by Western blotting for aromatase expression (50–55 kDa, Abcam, 1:250) with β-actin (1:5000) as a loading control. All clinical data pertaining to patient-derived glioblastoma xenograft tumor samples is provided in Supplementary Table 1.
The effects of Letrozole treatment on GBM migration, proliferation and spheroid formation
To address the role of aromatase in GBM cells, the effects of P450-specific inhibitor, Letrozole, was further assessed. Primary (RN1) and recurrent (MZ-256 and MZ-304) GBM cell lines were incubated with Letrozole (0.1 μM) for 24–72 hours, to determine if the Letrozole has an impact on biological characteristics of GBM cell lines such as motility, proliferation and sphere-forming potential. Notably all cells treated with Letrozole showed a significant decrease in cell motility compared to vehicle-treated (DMSO) controls (Figure 2A and 2B). As shown in Figure 2C, cell number was significantly reduced in all three cell lines when treated with Letrozole (0.1 μM) for 72 hours and RN1 cells displayed reduced spheroid formation capabilities when exposed to Letrozole (0.1 μM) over a 72 hour period (Figure 2D and 2E *p < 0.05, **p < 0.01, ***p < 0.001).
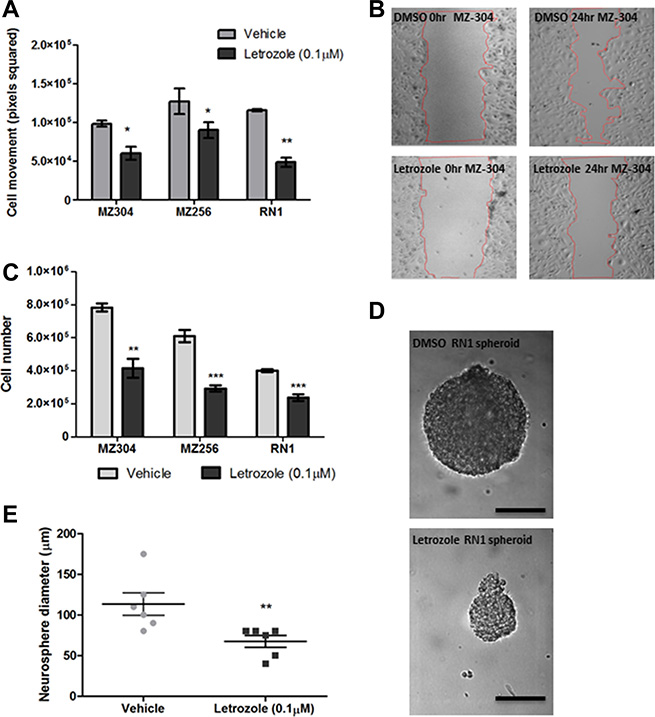
Figure 2: Assessment of the effects of Letrozole treatment on GBM migration, proliferation and spheroid formation. Primary (RN1) and recurrent (MZ-256 and MZ-304) GBM cell lines were incubated with Letrozole (0.1 μM) for 24–72 hours, to determine if the cellular function of GBM cell lines changes upon treatment with the aromatase inhibitor, Letrozole. Notably all cells treated with Letrozole showed a significant decrease of cell movement compared to vehicle-treated (DMSO) controls (A and B). As shown (C), cell number was significantly reduced in all three cell lines when treated with Letrozole (0.1 μM) for 72 hours and RN1luc cells displayed reduced spheroid formation capabilities when exposed to Letrozole (0.1 μM) over a 72 hour period (D and E), n = 3 mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001).
Assessment of GD2 expression in GBM cells
β1,4-N-acetylgalactosaminyltransferase (β4GANT1, GM2/GD2 synthase) is a key enzyme which is responsible for the synthesis of the glycosphingolipids GM2, GD2, and GA2. In a previous publication by Woo et al. [16], disialogangliosidase 2 (GD2) was identified as a GBM-specific antigen, in addition to a glycosphingolipid, CD90. As an initial screen, a variety of cell lines were assessed with respect to their relative expression of β4GANT1 mRNA. As a reference, we used cervical cancer HeLa cells, which have been shown to express β4GANT1 [16]. Cell lines assessed included the colorectal HT29 and lung cancer A549 lines, expressing less or equivalent amounts of β4GANT1 as the HeLa controls. The GBM commercial and primary lines, A172, MZ-327 and MZ18 expressed little or no GD2 synthase compared to HeLa cells, while U251 or patient derived primary RN1 spheroid culture and JK2, or recurrent MZ-256 and MZ-304 lines expressed significantly higher levels of GD2 synthase (Figure 3A). Although the commercially available GBM cell line U251 expressed β4GANT1, it was of interest to the authors to assess patient-derived lines in this study. Expression of GD2 synthase patient-derived lines MZ-256, MZ-304 and RN1-spheroid was further confirmed by flow cytometry analysis (Figure 3B). Based on their high GD2 synthase expression levels these lines were chosen for further investigations.
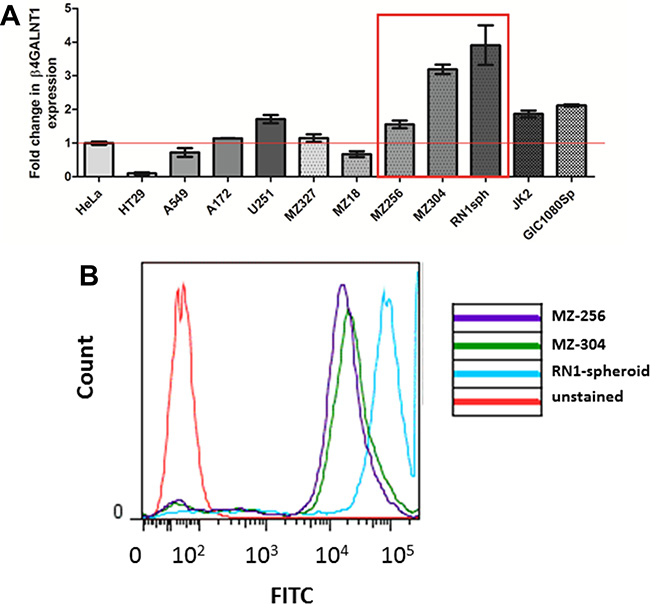
Figure 3: GD2 expression in GBM and non-GBM cell lines. In order to carry out a preliminary screen of a panel of both GBM and non-GBM cell lines, qualitative PCR (qPCR) was carried out to asses mRNA expression of the key enzyme responsible for the synthesis of disialogangliosidase 2 (GD2). The expression levels of β1,4-N-acetylgalactosaminyltransferase (β4GANT1) mRNA transcript was assessed in n = 2 non-GBM lines (HT29 and A549), n = 2 commercially available GBM lines (A152 and U251), n = 4 primary GBM-patient derived lines (MZ-327, MZ-18, RN1spheroid and JK2), n = 2 recurrent GBM lines (MZ-256 and MZ-304) and a glioma initiating cell line (GIC1080Sp) relative to the cervical cancer cell line, HeLa. As shown (A) RN1spheroid, MZ-256 and MZ-304 have greatest β4GANT1 expression. These highlighted lines were assessed by flow cytometry using a GD2-FITC tagged primary antibody (B) showing positive GD2 antigen presentation on the surface of these cell types (representative flow of n = 4).
Characterisation of PLGA-Let-NPs and visualisation of anti-GD2-ch14.18/CHO-PLGA-Alexafluor647-NPs in glioblastoma-colorectal cancer cell co-culture
PLGA is an FDA-approved biodegradable, physically strong and highly biocompatible polymer; especially suitable as a delivery vehicle for drugs, proteins and other macromolecules including DNA, RNA and peptides [28–30]. Reasons for its popularity among various available biodegradable polymers are its favourable degradation characteristics and its suitability for sustained drug delivery. Toxicological studies have designated PLGA as an extremely safe material for macroscopic and micro particle systems [31]. Nanoparticles were prepared from a PLGA polymer using a modified single emulsion evaporation method [32]. In order to characterize the synthesized nanoparticles, size and zeta potential of each nanoparticle batch was measured in a Malvern Zetasizer. Nanoparticles, which contained Letrozole, (PLGA-Let-NPs) were found to possess a mean size of 143.6 nm ± 27.84, with a mean zeta-potential of -21.58 mV ± 0.632 and an encapsulation efficiency of 82.22% + 5.77. Supplementary Figure 1 Nanoparticle imaging was carried out using scanning electron microscopy (SEM, Figure 4B). As evaluated through a content release study, there is an initial burst release of Letrozole within the first 12 hours, with 50% of content release noted after 50 hours of incubation in both cell culture media and phosphate buffered saline (PBS) at 37°C (Figure 4A). The anti-GD2-ch14.18/CHO antibody-NP conjugation protocol, modified from Kocbek et al. [33] was optimised through positive conjugation verified by rhodamine fluorescence resulting from anti-mouse-Rhodamine labelled secondary binding (data not shown). The morphological effects of anti-GD2-ch14.18/CHO-Let-NP treatment of MZ-304 GBM cells was evaluated by SEM imaging (Dr. Clodagh Dooley, AML, CRANN-TCD). NPs can be visualised 15 minutes post-NP addition in 4% paraformaldehyde fixed cells. These NPs are no longer evident on the cell surface after 3 hours due to internalisaton with the effects of Let-release within the GBM cells evident in Figure 4C. Data of control-NP treatment of GBM cells is displayed in Supplementary Figure 1.
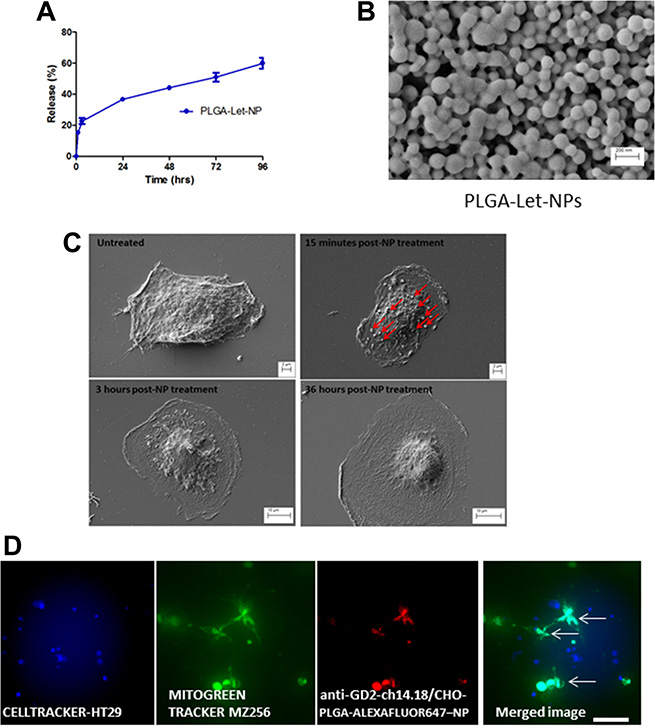
Figure 4: Characterisation of PLGA-Letrozole-NPs and anti-GD2-ch14.18/CHO-PLGA-NP mediated delivery of Letrozole exclusively to GBM cells in co-culture. Nanoparticles were prepared from a PLGA polymer using a modified single emulsion evaporation method [32]. Size and zeta potential of each nanoparticle batch was measured in a Malvern Zetasizer. As evaluated through a content release study, 50% content release of PLGA-Let-NPs occurs 72 hours post-incubation in phosphate buffered saline (PBS) at 37°C (A). This was also the case in cell culture media (data not shown). SEM imaging displays PLGA-NPs at 50Kx maginification (B). In addition, SEM imaging (C) display the morphological changes which occur 15 minutes post-NP treatment with NPs evident on the cell surface (denoted by red arrows), 3 hours post-NP treatment and 36 hours later. (D) HT29 colorectal cancer cells were pre-incubated with Cell tracker Blue reagent while MZ-256 GBM cells were incubated with MitoGreen tracker prior to co-culture on glass bottom dishes. As shown, NP positive cells are represented as Cyan in the merged image of Blue Cell-tracker-HT29, Green Mitogreen-MZ-256 and Red-NP. Notably, NP encapsulation is noted exclusively in those cells who are also MitoGreen positive, that is, MZ-256 cells and not in HT29 (Blue) cells (Scale 50 μm).
Specific delivery to GBM cells using anti-GD2-ch14.18/CHO conjugated PLGA nanoparticles
Specific cell delivery of potential therapeutics is a highly relevant issue, especially for diseases such as brain cancer where off-target drug delivery holds the potential to cause deleterious adverse effects and toxicity. To evaluate the potential of anti-GD2-ch14.18/CHO antibody to direct targeted delivery in GBM cells, PLGA-Alexafluor647-tagged-NPs were surface activated and coupled to anti-GD2-ch14.18/CHO antibody. HT29 colorectal cancer cells were pre-incubated with Cell tracker Blue reagent while MZ-256 GBM cells were incubated with MitoGreen tracker prior to co-culture on glass bottom dishes. As shown in Figure 4D, NP positive cells appear Cyan in the merged image of Blue Cell-tracker-HT29, Green Mitogreen-MZ-256 and Red-NP. Notably, NP encapsulation occurs exclusively in mitotically active MZ-256 cells as evidenced by their positivity for MitoGreen.
Treatment of GBM cells with anti-GD2-ch14.18/CHO -PLGA-Let-NPs ± TMZ
The standard treatment regimen for newly diagnosed glioblastoma patients is based on the so-called Stupp protocol [4], involving fractionated radiotherapy (RT) in combination with temozolomide (TMZ). The addition of TMZ to RT results in a clinically significant survival benefit [9]. TMZ is administered with fractionated radiotherapy and consists of a dose of 75 mg/m2 per day, given from the first until the last day of RT, but not longer than 49 days. After a four week break up to six cycles of adjuvant TMZ at a dose of 150 mg/m2 follow. Based on the findings of Stupp et al. median survival rate was increased from 12.1 months with RT alone to 14.6 months with TMZ and RT treatment combination. The contribution of aromatase to chemoresistance has been noted for several other cancers [34]. In order to assess if this may also be the case for GBMs, glioblastoma cell lines MZ-304 and RN1 were treated with anti-GD2-ch14.18/CHO-PLGA-Let-NPs or the corresponding control NP containing DMSO vehicle (anti-GD2-ch14.18/CHO-PLGA-empty-NPs) with or without temozolomide (150 μM)-supplemented cell media. Notably, treatment of both cell lines with anti-GD2-ch14.18/CHO-PLGA-Let-NPs in combination with TMZ (Figure 5A and 5B) significantly reduced cell numbers when compared to anti-GD2-ch14.18/CHO-PLGA-empty-NPs+TMZ or anti-GD2-ch14.18/CHO-PLGA-Let-NPs+DMSO treatments alone (*p < 0.05, **p < 0.01, ***p < 0.001).
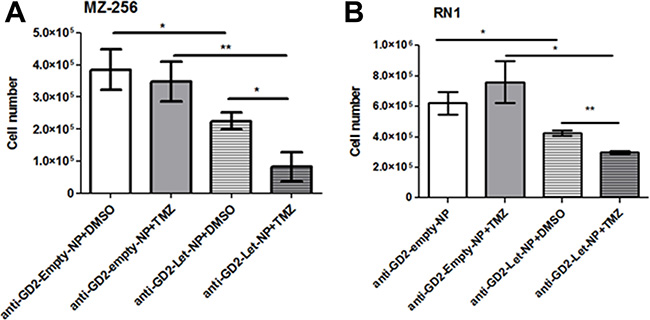
Figure 5: Treatment of GBM cells with anti-GD2-ch14.18/CHO-PLGA-Let-NPs ± TMZ. Glioblastoma cell lines MZ-304 (A) and RN1 (B) were treated with anti-GD2-ch14.18/CHO-PLGA-Let-NPs or the corresponding control anti-GD2-ch14.18/CHO-PLGA-Veh-NPs with or without temozolomide (150 μM)-supplemented cell media. Treatment of both cell lines with anti-GD2-ch14.18/CHO-PLGA-Let-NPs in combination with TMZ significantly reduced cell numbers when compared to anti-GD2-ch14.18/CHO-PLGA-Veh-NPs+TMZ or anti-GD2-ch14.18/CHO-PLGA-Let-NPs+DMSO treatments alone (n = 3, mean ±SEM, *p < 0.05, **p < 0.01, ***p < 0.001). Notably, for ease of labelling in the above figure anti-GD2-ch14.18/CHO-PLGA-NPs are referred to as anti-GD2-NPs.
Assessment of the role of miR-191 in aromatase inhibitor-induced reduction of GBM cell proliferation
The microRNA-191 (miR-191) has been implicated in several cancer types [35–38] showing an active role in cell proliferation, migration, chemoresistance and ultimately disease progression. As miR-191 is an estrogen-responsive microRNA [39], and its expression has been correlated to the aggressive mesenchymal subtype of GBM [40], it was of interest to evaluate if the treatment with Letrozole (0.1 μM) (Figure 2) alters miR-191 expression in glioma cells. Therefore, qPCR was carried out to assess the effect of aromatase inhibition on levels of miR-191. The results showed that Letrozole treatment results in a 34.46% ± 4.99 reduction in miR-191 expression relative to vehicle-treated controls (Figure 6A *p < 0.05). It was of interest, therefore, to assess if the reduction of GBM cell proliferation after Letrozole-treatment could be reversed by reintroduction of miR-191. As shown in Figure 6B, transient transfection of synthetic miR-191 into MZ-304, whose growth rate had been significant reduced by Letrozole treatment, led to a restoration of cell proliferation rates back to basal levels of untreated cells. Treatment with premiR-191 returned Letrozole-treated MZ-304 cells growth rate to that similar to untreated and DMSO+premiRneg-treated cells over a 120 hr period (***p < 0.001, n = 3, mean ± SEM, Two way ANOVA).
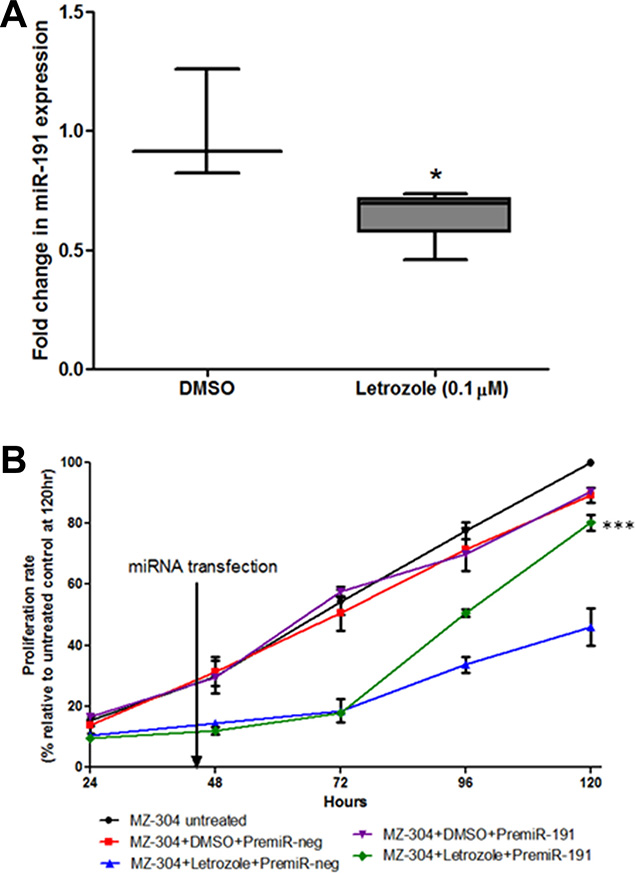
Figure 6: Assessment of miR-191contribution to GBM-letrozole treatment. GBM cells (MZ-304) were treated with Letrozole (0.1 μM) and qPCR was carried out to assess the effect of aromatase inhibition on the oestrogen response microRNA, miR-191. Notably, Letrozole treatment resulted in a 34.46% ± 4.99 reduction in miR-191 expression relative to vehicle-treated controls (A), n = 3, mean ± SEM *p < 0.05). Therefore, the ability of reintroduced synthetics premiR-191 into Letrozole-treated GBM cells to rescue the reduced-proliferation phenotype was assessed over 120 hours (B). It was found that transient transfection of miR-191 Let-treated MZ-304 cells led to a restoration of ‘normal’ cell proliferation rates, similar to those noted in untreated and DMSO+premiRneg-treated MZ-304 cells (***p < 0.001, n = 3, mean ± SEM, Two way ANOVA).
DISCUSSION
Previous studies carried out by Dave et al. [41] investigated the efficacy of the aromatase inhibitor, Letrozole, on GBM cell growth in vitro and in vivo exclusively in the conventional human glioma cell lines and the rat glioma C6 cells. Several studies have shown that treatment response in established glioma cells differs greatly to that noted in patient-derived primary cultures [42, 43]. Therefore, it was of great interest to the authors to ascertain if aromatase inhibitors manifest an anti-tumor effect also in primary cultures derived from both primary and recurrent GBMs, and on patient-derived stem-like glioma cells. In addition, the possibility of selective targeting of glioma cells through anti-GD2-ch14.18/CHO conjugated PLGA nanoparticles as an approach to limiting off target effects was addressed.
Aromatase is a microsomal cytochrome P450 complex coded by the CYP19A1 gene [44]. Aromatase catalyses the irreversible conversion of androgens to oestrogens [13] with activity in several normal and cancer tissues, the latter including brain cancers [14, 45, 46] and this study. Inhibition of aromatase is promising for the treatment of hormone-dependent breast cancer, pathological alterations of ovarian and endometrial function and some benign disorders like gynecomastia associated with uncontrolled cell proliferation. As found in this study, the addition of the aromatase inhibitor, Letrozole, to patient-derived GBM cultures led to a significant reduction in proliferation, migration and gliomasphere forming capacity in vitro. Our findings are not only concordant with those reported in conventional glioma cell lines by Dave et al. [41] but also extend these previous findings to a more clinically relevant experimental models such as glioma stem cells by integrating cell type-specific targeting approach based on the utilization of surface biomarkers expressed in GBM cells.
In the context of breast cancer, an estrogen-dependent cancer, where the use of aromatase inhibitors has been highly researched [47–51], deleterious effects of non-specific delivery of Letrozole includes osteoporosis leading to increased bone fractures, joint and muscle pain, menopausal symptoms, depression, cardiac problems, increased cholesterol and increased blood pressure were shown. In this regard, specific delivery of aromatase-inhibitors exclusively to target cancer cells using antibody conjugated biodegradable nanoparticles provides a promising approach for increasing drug efficacy and reducing therapeutic side effects. With respect to GBM, recent studies have shown that several antigens including cluster of differentiation 90 (CD90, Thy-1) and GD2 disialoganglioside are frequently overexpressed in glioblastoma cells [52]. GD2 disialoganglioside is a sialic-containing glycosphingolipid, also shown to be overexpressed in neuroblastoma with the GD2 specific antibody anti-GD2-ch14.18/CHO being used in several neuroblastoma clinical trials [20, 21]. We also reported previously that the anti-GD2-ch14.18/CHO antibody was successfully used for nanoparticle-mediated specific delivery of microRNAs to neuroblastoma tumors in vivo [53].
β-1,4 N-acetylgalactosaminyltransferase 1 encoded by the β4GALNT1 gene is an enzyme responsible for the biosynthesis of G(M2) and GD2 glycosphingolipids [54]. As depicted in Figure 3A, β4GALNT1 transcript expression was assessed as an indicator of GD2 expression across a panel of human cancer cell lines including conventional cell lines derived from GBM and patient-derived primary cultures of first diagnosed or recurrent GBM. Our results reveal the benefits of a combinatorial approach integrating nanoparticles-based delivery of the aromatase inhibitor Letrozole with the selective antibody-mediated cell targeting cell-specific recognition.
Although Letrozole has shown to be capable of blood-brain barrier (B-BB) transition in rat models of glioblastoma with high uptake rates into the tumor itself [55], specific delivery through encapsulation within biodegradable nanoparticles is a promising avenue of research and may provide the means to minimize the adverse effects of current chemotherapeutics in use for GBM treatment. The intricate composition of the B-BB ensures the highly regulated and restricted transport of molecules with limitations further imposed by electrical resistance across B-BB endothelia [56]. Differences in nanoparticle material, size and shape and receptor (over)expression makes it difficult to define optimal criteria for B-BB diffusions; however, passive transport typically occurs for lipophilic molecules below 400Da and through several studies it has been shown that 80–200 nm nanoparticle size range is optimal for tumour inclusion and effect [57]. In addition, conjugation of a GD2 specific antibody to the surface of activated Letrozole-encapsulated PLGA-nanoparticles renders GBM cell specificity. It should be noted that antibody conjugation significantly alters the cell surface of the nanoparticles and nanoparticle size as shown in Supplementary Table 2, presumably due to anti-GD2-ch14.18/CHO antibody-antibody aggregation. Such an aggregation causes the nanoparticles to behave like larger particles [58] and may affect systemic delivery for therapeutic application in the brain. However, evaluation of potential therapeutics through direct intratumoral injection including nanoparticle delivery to in vivo models of GBM [59, 60] or at the point of patient tumor resection or biopsy [61–65] in several studies indicates that such systemic transport challenges can be circumvented to facilitate therapeutic delivery in a clinical setting.
Nanoparticle formulation for this study was based on the protocol by Jana et al. (2014) which used a single emulsion and evaporation method for letrozole encapsulation and formulation of PLGA nanoparticles [32]. Similar to the aforementioned protocol, this study resulted in the formulation of PLGA-NPs less than 150 nm with an encapsulation efficiency of over 80%. Notably, however, anti-GD2-ch14.18/CHO antibody conjugation resulted in a dramatic increase in NP size (>360 nm) which although would affect B-BB penetrance and cellular diffusion; had no observable bearing on GBM cellular uptake as a result of direct administration in this study due to the pharmacokinetics of GD2 antigen-antibody endocytosis on the GBM cell surface. Although these sized NPs would have a significant effect on B-BB penetrance, treatment protocols for recurrent GBM tumour resection provides an opportunity for direct therapeutic administration to the brain tumour cavity; thereby circumventing the B-BB.
In order to assess whether Letrozole administration improves chemoresponse to TMZ in GBM cells, anti-GD2-ch14.18/CHO-PLGA-Let-NP in combination with temozolomide (TMZ, 150 μM) treatment in GBM cells in vitro was assessed. The findings show that the anti-cancer effects elicited by the Letrozole-nanoparticles in GBM cells attenuate TMZ-induced reduction in cell numbers over 72 hours. Although these findings need to be assessed in an orthotopic model of glioblastoma, this in vitro study highlights the successful encapsulation and delivery of functional Letrozole exclusively to GBM cells.
In an attempt to understand the mechanism through which Letrozole elicits its anti-tumor effect in GBM cells, the authors evaluated the expression levels of the estrogen-responsive microRNA-191 (miR-191) post-Letrozole treatments. MiR-191 has been shown to be a prognostic indicator of poor survival rates in GBM patients [40], indicating a potential role for hormonal control of GBM pathogenesis. Oestrogen is synthesised, catalysed by aromatase, and subsequently binds to the estrogen receptor (ER) alpha/beta subunits which dynamically interact with the estrogen responsive elements (ERE) in the promoter region of miR-191 [39], increasing expression and driving proliferation, migration and chemoresistance in several cancer types [54, 66–68]. It is hypothesized that aromatase inhibition through Letrozole treatment reduces oestrogen-mediated binding of the ER to the ERE and therefore reduces miR-191 expression. In order to assess this hypothesis the authors investigated the ability of miR-191 to rescue the reduced proliferative phenotype induced by Letrozole in GBM cells and found that reintroduction of synthetic premicroRNA-191 led to significant restoration of ‘normal’ growth patterns of the GBM cell in vitro. In this regard, it may be of interest to evaluate the therapeutic merit of miR-191 as a potential anti-tumor molecule for future studies.
Glioblastoma is a highly aggressive brain cancer with an extremely poor prognostic outcome despite intensive treatment regimes. With an average five year survival rate of less than 3%, this dismal prognosis necessitates exploration of novel therapeutics and drug delivery systems such as those detailed in this study. The identification of new mechanism-based therapeutic targets through repurposing of currently approved clinical drugs, and the use of alternative drug-delivery strategies such as tumour-specific mediated delivery of effective anti-cancer drugs, improves upon this bleak outlook, and is an important agenda with respect to GBM research.
MATERIALS AND METHODS
Cell lines and patient-derived tumor samples
For all in vitro experiments glioblastoma cell lines derived from patient GBM tumor biopsies and from other cancer types such as colorectal (HT29), lung (A549) and cervical (HeLa) were used. A172 and U251 are commercially available GBM cell lines which, in addition to the MZ-304, MZ-327 and MZ-256 cell lines [69, 70] were grown as a monolayer in DMEM media (BioWhittaker®) with 10% heat-inactivated fetal calf serum (FCS; GIBCO), 100U/mL penicillin (Sigma-Aldrich®), and 100 μg/mL streptomycin (Sigma-Aldrich®). Stem-like glioma initiating cell lines (GICs) established from human GBMs were previously described [71, 72]. Low-passage primary GBM patient-derived lines, RN1 and JK2, were kindly donated by the Brain Cancer Research Unit, QIMR Berghofer Medical Research Institute, Australia. The cells were grown as a monolayer (adherent condition) in Serum-Free Human NSC Culture Medium, as previously described [73], supplemented with β-FGF and EGF (10 ng/ml and 20 ng/ml respectively) on 1% matrigel-coated flasks or plates [73] or as a gliomasphere culture in the absence of matrigel and presence of heparin (10 ng/μl, Sigma Aldrich). These RN1 cells stably express luciferase if required. All cells were maintained in a humidified incubator at 37°C with 5% CO2. Patient clinical data pertaining to the GBM cell lines used is presented in Supplementary Table 1.
Protein extraction
Glioblastoma cells were washed with 1x Phosphate buffered saline (PBS) (BioWhittaker®), trypsinised (Trypsin:EDTA, Sigma-Aldrich®) and centrifuged at 1,200 rpm for 4 minutes. The supernatant was removed and protein was extracted from the resulting pellet in Protease inhibitor (1:100, Sigma-Aldrich®) and RIPA buffer (1M Tis pH 7.4, 5M NaCl, 1% NP-40, 50 mM NaF, 0.5M EDTA, 0.1% SDS, 0.5% Na-deoxycholate). Resuspended cells in RIPA were left on ice for 20 minutes before centrifugation at 4°C at 12,000 rpm for 10 minutes. Supernatant was isolated, quantified using a BCA™ Protein Assay kit (Thermo Fisher Scientific) and stored at –20°C.
Western blot
Western blot analysis was performed on lysates prepared as outlined previously, or lysates provided from patient-derived glioblastoma xenografts (G46, G59, G64, G76, G79, G80, G84, G85 and G91) by Mayo Clinic Brain Tumor SPORE [24–27]. Clinical data related to these patient-derived samples are detailed in Supplementary Table 1. Western blots were carried out using 4–10% gradient pre-cast gels (Thermo Fisher Scientific) and Tris-HEPES-SDS running buffer (0.1M Tris, 0.1M HEPES, 3 mM SDS, pH 8) (Thermo Fisher Scientific). Protein samples were mixed with 1x Lamelli buffer containing 1,4-dithiothreitol (DTT) (31.5 mM Tris-HCL, pH 6.8; 10% glycerol, 1% SDS, 0.005% Bromophenol blue, 50 mM DTT). Gels were run at 80V for 45 minutes, followed by transfer onto nitrocellulose membranes (GE Healthcare Life Sciences) at 40V for 90 minutes, in transfer buffer (48 mM Tris, 30 mM Glycine, 0.037% SDS, 2.5% methanol pH8.3). Blocking buffer (10%-milk-TBST, Marvel, (Tris, Tween20®) Sigma-Aldrich®) was used to block nonspecific sites on the membrane at 4°C overnight. Membranes were incubated with primary anti-P450 antibody (Abcam 1:250 in TBST) at room temperature for 2 hours and washed with TBST. Secondary anti-rat IgG antibody (Abcam, 1:2000 in TBST) was then added for two hours, followed by further washes with TBST. For control loading, β-actin antibody (Abcam, 1:5000 in TBST) was added for one hour at room temperature. Membranes were washed with TBST and secondary anti-mouse IgG antibody (Abcam, 1:2000 in TBST) was added for one hour, followed by further washes with TBST. All membranes were developed using the Pierce™ ECL Plus Western Blotting Substrate and the images were assessed on a LAS Imager4000.
RNA extraction, cDNA synthesis and qPCR
RNeasy® Mini kit (QIAGEN) was used to extract RNA from all cell lines. cDNA synthesis was performed using the High capacity cDNA Reverse Transcription Kit (Applied Biosystems™) according to the manufacturer’s protocol. Aromatase mRNA transcript (CYP19A1) expression was assessed in several cell lines relative to β-actin by qPCR, using a CYP19A1-specific Taqman probe (Applied Biosciences) and β-actin as an endogenous gene control. GD2 expression was assessed using qPCR of the β1,4-N-acetylgalactosaminyltransferase (β4GALNT1, GM2/GD2 synthase) gene using a specific Taqman probe. Detection of mRNA transcript was carried out on a StepOne Plus qPCR machine under the following parameters 50°C for 2 minutes, 95°C for 10 minutes, followed by 40 cycles of denaturation at 95°C for 15 seconds and 60°C for 60 seconds. Expression levels were calculated based on the delta delta CT (ΔΔCT) method.
Fluorescence activated sorting analysis
Log phase GBM cells (2 × 106, MZ-304, MZ-256 and RN1-spheroid) were harvested and stained with GD2-FITC antibody (Santa Cruz, sc-53831). Cells were then analysed by a FACS Calibur (BD Bioscience).
Cell proliferation assay
GBM cells (MZ-304, MZ-256 and RN1) were seeded at 1 × 105cells/well in a 6-well plate (Sarstedt AG&Co). After 24 hours, media was removed and replaced by either vehicle control (DMSO)-media (Sigma-Aldrich®) or Aromatase inhibitor (AI) Letrozole (Sigma-Aldrich®) supplemented media (0.1 μM). Cells were then allowed to grow for a further 72 hours and cell numbers were determined through trypan-blue exclusion.
2-Dimensional migration assay (Wound healing/scratch assay)
In order to test the migratory potential of cells post-treatment with DMSO as a control or Letrozole (Let, 0.1 μM), GBM cells (MZ-304, MZ-256 and RN1) were seeded at 1x104cells per side of a 2D Migration chamber (Ibidi GmbH) and allowed to grow to confluency. After 24 hours, the separator was detached using sterile tweezers, DMSO or Letrozole (0.1 μM) supplemented media was added and initial ‘wound’ images were taken. Cells were allowed to grow for 24 hours and the extent of cell movement was determined by taking an image after the incubation time, relative to the DMSO-treated control.
PLGA-Let-Nanoparticle (PLGA-Let-NP) formulation
Letrozole (Let) loading of PLGA nanoparticles was carried out according to the modified single-emulsion evaporation method outlined by Jana et al. [32], modified from [74]. Briefly, PLGA 50:50 (Resomer 503H 200 mg/ml, Sigma Aldrich) polymer was dissolved in an organic phase of chloroform. Letrozole (10 mg) or control DMSO was dissolved in an acetone - dichloromethane (1.5 ml 1:2 v/v) mixture. The PLGA-letrozole mixture was then emulsified with an aqueous phase composed of 2% polyvinyl alcohol (PVA) solution in water (4.5 ml) by sonication using a micro tip probe sonicator at an output of 50W for 30s in an ice bath. The organic solvents were then rapidly removed by evaporation at 37ºC leaving behind colloidal suspension of PLGA NPs containing Letrozole (PLGA-Let-NPs) or a DMSO control (PLGA-empty-NPs) in water, which was then pelleted using an ultracentrifuge at 4°C at 30,000 rpm for 15 minutes. Each pellet was washed with 1.13% w/v NaCl solution to keep the particles small. This was carried out three times. The resulting pellet was resuspended in molecular grade water and placed at –80°C for at least 30 minutes. This was then lyophilized overnight and the resulting powder was weighed to determine yield.
Nanoparticle characterisation
Each nanoparticle preparation was weighed in a sterile Eppendorf tube and resuspended in molecular grade water. This suspension was then placed on a Zeta cell and measured in a Malvern Zetasizer to determine size and zeta potential of each nanoparticle batch. To determine encapsulation efficiency, an aliquot of PLGA-Let-NPs (1 mg) was resuspended in acetone and subjected to centrifugation at 20,000 rpm at 25°C for 30 min. The amount of drug present in the pellet was determined using a standard curve, constructed using varying Letrozole concentrations (0–10 μg/ml in acetone, R2 = 0.9778) versus absorbance at 238 nm [75]. Encapsulation efficiency was calculated using the following formula:
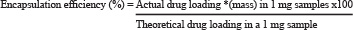
Letrozole release was calculated by resuspending lyophilized PLGA–Let-NPs (1 mg) in PBS (1.2 ml pH 7.4) and the solution was divided in 6 microfuge tubes (200 μl each), maintained at 37ºC in a thermo-stable water bath. After an appropriate time period (1 hr, 24 hr, 48 hr, 72 hr, 96 hr and 120 hr) the solution was centrifuged at 3000 rpm. Absorbance of this sample supernatant was measured at 238 nm. Concentration of the released Let was then calculated using the standard curve of the respective drug (0–10 μg/ml, R2 = 0.9966). The percentage of Let released was determined from the following equation:
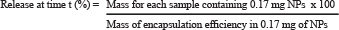
Note: For release, Starting mass of NPs was 1 mg, divided across 6 eppendorfs (therefore there was 0.17 mg in each tube containing 200 μl).
Conjugation of NPs to anti-GD2- ch14.18/CHO antibody
Based on the antibody conjugation to nanoparticle protocol used by Kocbek et al. [33], PLGA-NPs were resuspended in sterile filtered MES buffer (4-morpholinoethanesulfonic acid, pH 5.5) (Sigma-Aldrich®) and shaken at room temperature for 1 hour. After centrifugation at 4°C at 13,000 rpm for 20 minutes the MES buffer was discarded, giving a pellet of activated NPs. EDC-(N-[3-Dimethylaminopropyl]-N-ethylcarbodiimide hydrochloride) NHS-(N-Hydroxysuccinimide) (Sigma-Aldrich®) 1xPBS-mix at a 8:8:1 ratio was prepared. Both, the EDC-NHS-1xPBS-mix and anti-GD2-ch14.18/CHO antibody were added to the activated NPs, which were then shaken at room temperature overnight. The anti-GD2-ch14.18/CHO antibody was made available by a European funding effort by charities and commissioned by SIOPEN. This project is now further developed by APEIRON Biologics. A centrifugation at 4°C at 13,000 rpm for 20 minutes followed. The washing steps included a resuspension of the pellet in 1xPBS (0.01M; pH 7.15) and a further centrifugation as previously, which was repeated twice. The resulting pellet was resuspended in nuclease-free water and placed at -80°C for at least 30 minutes. NPs were then lyophilized overnight and the resulting powder was weighed to determine yield.
Assessment of GBM-specificity using anti-GD2-ch14.18/CHO-PLGA-Alexafluor647-NPs in a glioblastoma-colon cancer cell co-culture
In order to assess the ability of anti-GD2-ch14.18/CHO antibody to direct nanoparticle-mediated drug delivery to GBM cells, a co-culture system was generated involving the GBM cell line MZ-256 and the colorectal cell line (HT29). HT29 colorectal cancer cells (1 × 105) were pre-incubated with Cell tracker Blue reagent (25 μM) while MZ-256 GBM cells (1 × 105) were incubated with MitoGreen tracker (50 nM) prior to co-culture on glass bottom dishes in DMEM-10%FCS media. To enable visualisation of nanoparticle delivery PLGA-nanoparticles were generated which encapsulated an Alexafluor647 fluorophore. This fluorescent tag is only detectable after PLGA-NP degradation and content release. Anti-GD2-ch14.18/CHO-PLGA-Alexafluoro647-NPs were synthesized and added for a 6 hour period, removed and cells were assessed by fluorescent microscopy after 96 hours.
Treatment of GBM cells with anti-GD2-ch14.18/CHO-PLGA-Let-NPs and TMZ
Based upon encapsulation efficiency and release time calculations, MZ-304 and RN1 glioblastoma cells were treated with anti-GD2-ch14.18/CHO-PLGA-Let-NPs or the corresponding DMSO vehicle control nanoparticle (anti-GD2-ch14.18/CHO-PLGA-empty-NPs). GBM cells (MZ-256) were seeded at 1 × 104cells/well in a 6-well plate (Sarstedt AG&Co) and were allowed to grow. After 24 hours anti-GD2-ch14.18/CHO-PLGA-Let-NPs were resuspended in sterile media/PBS at a concentration of 1 mg/ml. Based on the determined average encapsulation efficiency of 82%, 8.2 mg (2.87 μM) of Letrozole was calculated to be present per mg of NP resuspended in 1 ml of media. Accordingly, an appropriate amount of resuspended NP was added to the cell culture media to ensure a final concentration of 0.1 μM of Letrozole. NPs were removed after 6 hours and temozolomide (TMZ) or DMSO vehicle control-supplemented (150 μM) cell media was used. Cells were then allowed to grow for a further 72 hours and cell numbers were assessed.
PremiR-191 rescue experiment of Letrozole-treated GBM (MZ-304) cells
GBM cells (MZ-304) were transiently transfected with premiR-191 (30 nM, Applied Biosystems) using siPORT NeoFx transfection reagent (Applied Biosystems). In brief, MZ-304 cells were seeded (1 × 104 cells) into a T25 flask in normal cell culture media. After 6 hours, to allow adherence, media was replaced with control (DMSO) or Letrozole (0.1 μM)-supplemented media and proliferation rates were determined using trypan blue exclusion at various time points. After 40 hours, MZ-304 ± Letrozole cells were transiently reverse transfected with control negative (premiR-neg) or synthetic precursor miR-191 (premiR-191) and returned to assess subsequent growth.
Statistical analysis
For comparison of multiple unpaired samples, OneWay ANOVA was used with Kruskal-Wallis H Test; while for direct comparison of two unpaired biological samples, Mann Whitney u test was used in Prism-Graph Pad. All assays were carried out with technical repeats of n = 2–3 and in biological triplicate and the values presented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
ACKNOWLEDGMENTS
All scanning electron microscopy (SEM) images were carried out by Dr. Clodagh Dooley, Centre for Research on adoptive nanostructures and nanodevices (CRANN) – Advanced Microscopy Laboratory (AML), Trinity College Dublin, College Green, Dublin 2, Ireland.
CONFLICTS OF INTEREST
The author(s) declare that they have no competing interests
FUNDING
This work was supported by the Irish Cancer Society Research Fellowship CRF13TIV, supported by Tesco Charity of the Year (AT). TH was funded by the ERASMUS+ program. JHMP receives support from Science Foundation Ireland (14/IA/2582) and Brain Tumor Ireland. BWS, BWD and AWB are members of the Brain Cancer Discovery Collaborative, which is supported by the Cure Brain Cancer Foundation. Work by JNS and JLP was supported by the Mayo SPORE in Brain Cancer (CA108961) and the Mayo Clinic.
REFERENCES
1. Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, Wolinsky Y, Kruchko C, Barnholtz-Sloan J. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014; 16:iv1–63.
2. Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, Cavenee WK. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002; 61:215–225; 226–219.
3. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta neuropathologica. 2016; 131:803–820.
4. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352:987–996.
5. Barker FG, 2nd, Prados MD, Chang SM, Gutin PH, Lamborn KR, Larson DA, Malec MK, McDermott MW, Sneed PK, Wara WM, Wilson CB. Radiation response and survival time in patients with glioblastoma multiforme. J Neurosurg. 1996; 84:442–448.
6. Lacroix M, Abi-Said D, Fourney DR, Gokaslan ZL, Shi W, DeMonte F, Lang FF, McCutcheon IE, Hassenbusch SJ, Holland E, Hess K, Michael C, Miller D, et al. A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. J Neurosurg. 2001; 95:190–198.
7. Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen HJ, Group AL-GS. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. The lancet oncology. 2006; 7:392–401.
8. Vecht CJ, Avezaat CJ, van Putten WL, Eijkenboom WM, Stefanko SZ. The influence of the extent of surgery on the neurological function and survival in malignant glioma. A retrospective analysis in 243 patients. J Neurol Neurosurg & Psych. 1990; 53:466–471.
9. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The lancet oncology. 2009; 10:459–466.
10. Hayashi K, Michiue H, Yamada H, Takata K, Nakayama H, Wei FY, Fujimura A, Tazawa H, Asai A, Ogo N, Miyachi H, Nishiki T, Tomizawa K, et al. Fluvoxamine, an anti-depressant, inhibits human glioblastoma invasion by disrupting actin polymerization. Scientific reports. 2016; 6:23372.
11. Sleire L, Skeie BS, Netland IA, Forde HE, Dodoo E, Selheim F, Leiss L, Heggdal JI, Pedersen PH, Wang J, Enger PO. Drug repurposing: sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system Xc-, leading to glutathione depletion. Oncogene. 2015; 34:5951–5959.
12. Triscott J, Lee C, Hu K, Fotovati A, Berns R, Pambid M, Luk M, Kast RE, Kong E, Toyota E, Yip S, Toyota B, Dunn SE. Disulfiram, a drug widely used to control alcoholism, suppresses the self-renewal of glioblastoma and over-rides resistance to temozolomide. Oncotarget. 2012; 3:1112–1123. doi: 10.18632/oncotarget.604.
13. Santen RJ, Brodie H, Simpson ER, Siiteri PK, Brodie A. History of aromatase: saga of an important biological mediator and therapeutic target. Endocr Rev. 2009; 30:343–375.
14. Kamat A, Hinshelwood MM, Murry BA, Mendelson CR. Mechanisms in tissue-specific regulation of estrogen biosynthesis in humans. Trends Endocrinol Metab. 2002; 13:122–128.
15. Lephart ED. Modulation of Aromatase by Phytoestrogens. Enzyme research. 2015; 2015:594656.
16. Woo SR, Oh YT, An JY, Kang BG, Nam DH, Joo KM. Glioblastoma specific antigens, GD2 and CD90, are not involved in cancer stemness. Anat & Cell Biol. 2015; 48:44–53.
17. Navid F, Santana VM, Barfield RC. Anti-GD2 antibody therapy for GD2-expressing tumors. Current cancer drug targets. 2010; 10:200–209.
18. Fleurence J, Cochonneau D, Fougeray S, Oliver L, Geraldo F, Terme M, Dorvillius M, Loussouarn D, Vallette F, Paris F, Birkle S. Targeting and killing glioblastoma with monoclonal antibody to O-acetyl GD2 ganglioside. Oncotarget. 2016; 7:41172–41185. doi: 10.18632/oncotarget.9226.
19. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, Shimada H, Grupp SA, Seeger R, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010; 363:1324–1334.
20. Zeng Y, Fest S, Kunert R, Katinger H, Pistoia V, Michon J, Lewis G, Ladenstein R, Lode HN. Anti-neuroblastoma effect of ch14.18 antibody produced in CHO cells is mediated by NK-cells in mice. Molecular immunology. 2005; 42:1311–1319.
21. Ladenstein R, Weixler S, Baykan B, Bleeke M, Kunert R, Katinger D, Pribill I, Glander P, Bauer S, Pistoia V, Michon J, Garaventa A, Lode HN. Ch14.18 antibody produced in CHO cells in relapsed or refractory Stage 4 neuroblastoma patients: a SIOPEN Phase 1 study. mAbs. 2013; 5:801–809.
22. Siebert N, Eger C, Seidel D, Juttner M, Zumpe M, Wegner D, Kietz S, Ehlert K, Veal GJ, Siegmund W, Weiss M, Loibner H, Ladenstein R, et al. Pharmacokinetics and pharmacodynamics of ch14.18/CHO in relapsed/refractory high-risk neuroblastoma patients treated by long-term infusion in combination with IL-2. mAbs. 2016; 8:604–616.
23. Mora J. Dinutuximab for the treatment of pediatric patients with high-risk neuroblastoma. Expert review of clinical pharmacology. 2016; 9:647–653.
24. Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, Schroeder MA, James CD. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro Oncol. 2005; 7:164–176.
25. Sarkaria JN, Carlson BL, Schroeder MA, Grogan P, Brown PD, Giannini C, Ballman KV, Kitange GJ, Guha A, Pandita A, James CD. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clinical cancer research. 2006; 12:2264–2271.
26. Sarkaria JN, Kitange GJ, James CD, Plummer R, Calvert H, Weller M, Wick W. Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clinical cancer research. 2008; 14:2900–2908.
27. Sarkaria JN, Yang L, Grogan PT, Kitange GJ, Carlson BL, Schroeder MA, Galanis E, Giannini C, Wu W, Dinca EB, James CD. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Molecular cancer therapeutics. 2007; 6:1167–1174.
28. Jeon O, Krebs M, Alsberg E. Controlled and sustained gene delivery from injectable, porous PLGA scaffolds. Journal of biomedical materials research Part A. 2011; 98:72–79.
29. Buske J, Konig C, Bassarab S, Lamprecht A, Muhlau S, Wagner KG. Influence of PEG in PEG-PLGA microspheres on particle properties and protein release. European journal of pharmaceutics and biopharmaceutics. 2012; 81:57–63.
30. Zhou J, Patel TR, Fu M, Bertram JP, Saltzman WM. Octa-functional PLGA nanoparticles for targeted and efficient siRNA delivery to tumors. Biomaterials. 2012; 33:583–591.
31. Dailey LA, Jekel N, Fink L, Gessler T, Schmehl T, Wittmar M, Kissel T, Seeger W. Investigation of the proinflammatory potential of biodegradable nanoparticle drug delivery systems in the lung. Toxicology and applied pharmacology. 2006; 215:100–108.
32. Jana SK, Chakravarty B, Chaudhury K. Letrozole and Curcumin Loaded-PLGA Nanoparticles: A Therapeutic Strategy for Endometriosis. Journal of Nanomedicine and Biotherapeutic Discovery. 2014; 4.
33. Kocbek P, Obermajer N, Cegnar M, Kos J, Kristl J. Targeting cancer cells using PLGA nanoparticles surface modified with monoclonal antibody. Journal of controlled release. 2007; 120:18–26.
34. Sui M, Zhang H, Fan W. The role of estrogen and estrogen receptors in chemoresistance. Current medicinal chemistry. 2011; 18:4674–4683.
35. Huang YZ, Zhang J, Shao HY, Chen JP, Zhao HY. MicroRNA-191 promotes osteosarcoma cells proliferation by targeting checkpoint kinase 2. Tumour biology. 2015; 36:6095–6101.
36. Sakurai M, Masuda M, Miki Y, Hirakawa H, Suzuki T, Sasano H. Correlation of miRNA expression profiling in surgical pathology materials, with Ki-67, HER2, ER and PR in breast cancer patients. The International journal of biological markers. 2015; 30:e190–199.
37. Ge YZ, Xin H, Lu TZ, Xu Z, Yu P, Zhao YC, Li MH, Zhao Y, Zhong B, Xu X, Zhou LH, Wu R, Xu LW, et al. MicroRNA expression profiles predict clinical phenotypes and prognosis in chromophobe renal cell carcinoma. Scientific reports. 2015; 5:10328.
38. Nagpal N, Ahmad HM, Chameettachal S, Sundar D, Ghosh S, Kulshreshtha R. HIF-inducible miR-191 promotes migration in breast cancer through complex regulation of TGFbeta-signaling in hypoxic microenvironment. Scientific reports. 2015; 5:9650.
39. Nagpal N, Ahmad HM, Molparia B, Kulshreshtha R. MicroRNA-191, an estrogen-responsive microRNA, functions as an oncogenic regulator in human breast cancer. Carcinogenesis. 2013; 34:1889–1899.
40. Li R, Gao K, Luo H, Wang X, Shi Y, Dong Q, Luan W, You Y. Identification of intrinsic subtype-specific prognostic microRNAs in primary glioblastoma. Journal of experimental & clinical cancer research. 2014; 33:9.
41. Dave N, Chow LM, Gudelsky GA, LaSance K, Qi X, Desai PB. Preclinical pharmacological evaluation of letrozole as a novel treatment for gliomas. Molecular cancer therapeutics. 2015; 14:857–864.
42. Tivnan A, Zakaria Z, O’Leary C, Kögel D, Pokorny JL, Sarkaria JN, Prehn JH. Inhibition of Multidrug resistance protein 1 (MRP1) improves chemotherapy drug response in primary and recurrent glioblastoma multiforme. Frontiers in Neuroscience. 2015; 9.
43. Zakaria Z, Tivnan A, Flanagan L, Murray DW, Salvucci M, Stringer BW, Day BW, Boyd AW, Kogel D, Rehm M, O’Brien DF, Byrne AT, Prehn JH. Patient-derived glioblastoma cells show significant heterogeneity in treatment responses to the inhibitor-of-apoptosis-protein antagonist birinapant. British journal of cancer. 2016; 114:188–198.
44. Boon WC, Chow JD, Simpson ER. The multiple roles of estrogens and the enzyme aromatase. Prog Brain Res. 2010; 181:209–232.
45. Stocco C. Tissue physiology and pathology of aromatase. Steroids. 2012; 77:27–35.
46. Lephart ED. A review of brain aromatase cytochrome P450. Brain Res Brain Res Rev. 1996; 22:1–26.
47. Buzdar A, Douma J, Davidson N, Elledge R, Morgan M, Smith R, Porter L, Nabholtz J, Xiang X, Brady C. Phase III, multicenter, double-blind, randomized study of letrozole, an aromatase inhibitor, for advanced breast cancer versus megestrol acetate. Journal of clinical oncology. 2001; 19:3357–3366.
48. Demers LM. Effects of Fadrozole (CGS 16949A) and Letrozole (CGS 20267) on the inhibition of aromatase activity in breast cancer patients. Breast Cancer Res Treat. 1994; 30:95–102.
49. Miller WR, Larionov A, Anderson TJ, Evans DB, Dixon JM. Sequential changes in gene expression profiles in breast cancers during treatment with the aromatase inhibitor, letrozole. Pharmacogenomics J. 2012; 12:10–21.
50. Neville-Webbe HL, Coleman RE, Holen I. Combined effects of the bisphosphonate, zoledronic acid and the aromatase inhibitor letrozole on breast cancer cells in vitro: evidence of synergistic interaction. British journal of cancer. 2010; 102:1010–1017.
51. Tominaga T, Adachi I, Sasaki Y, Tabei T, Ikeda T, Takatsuka Y, Toi M, Suwa T, Ohashi Y. Double-blind randomised trial comparing the non-steroidal aromatase inhibitors letrozole and fadrozole in postmenopausal women with advanced breast cancer. Annals of oncology. 2003; 14:62–70.
52. Parry PV, Engh JA. CD90 is identified as a marker for cancer stem cells in high-grade gliomas using tissue microarrays. Neurosurgery. 2012; 70:N23–24.
53. Tivnan A, Orr WS, Gubala V, Nooney R, Williams DE, McDonagh C, Prenter S, Harvey H, Domingo-Fernandez R, Bray IM, Piskareva O, Ng CY, Lode HN, et al. Inhibition of neuroblastoma tumor growth by targeted delivery of microRNA-34a using anti-disialoganglioside GD2 coated nanoparticles. PloS one. 2012; 7:e38129.
54. Di Leva G, Piovan C, Gasparini P, Ngankeu A, Taccioli C, Briskin D, Cheung DG, Bolon B, Anderlucci L, Alder H, Nuovo G, Li M, Iorio MV, et al. Estrogen mediated-activation of miR-191/425 cluster modulates tumorigenicity of breast cancer cells depending on estrogen receptor status. PLoS genetics. 2013; 9:e1003311.
55. Dave N, Gudelsky GA, Desai PB. The pharmacokinetics of letrozole in brain and brain tumor in rats with orthotopically implanted C6 glioma, assessed using intracerebral microdialysis. Cancer Chemother Pharmacol. 2013; 72:349–357.
56. Burke M, R L, Brim H. Central Nervous System: Drug Delivery to Treat. (New York: Wiley & Sons).
57. Grabrucker AM, Ruozi B, Belletti D, Pederzoli F, Forni F, Vandelli MA, Tosi G. Nanoparticle transport across the blood brain barrier. Tissue barriers. 2016; 4:e1153568.
58. Smith JE, Sapsford KE, Tan W, Ligler FS. Optimization of antibody-conjugated magnetic nanoparticles for target preconcentration and immunoassays. Anal Biochem. 2011; 410:124–132.
59. Balzeau J, Pinier M, Berges R, Saulnier P, Benoit JP, Eyer J. The effect of functionalizing lipid nanocapsules with NFL-TBS.40–63 peptide on their uptake by glioblastoma cells. Biomaterials. 2013; 34:3381–3389.
60. Bago JR, Pegna GJ, Okolie O, Mohiti-Asli M, Loboa EG, Hingtgen SD. Electrospun nanofibrous scaffolds increase the efficacy of stem cell-mediated therapy of surgically resected glioblastoma. Biomaterials. 2016; 90:116–125.
61. Martinez-Quintanilla J, He D, Wakimoto H, Alemany R, Shah K. Encapsulated stem cells loaded with hyaluronidase-expressing oncolytic virus for brain tumor therapy. Molecular therapy. 2015; 23:108–118.
62. Pellegatta S, Poliani PL, Stucchi E, Corno D, Colombo CA, Orzan F, Ravanini M, Finocchiaro G. Intra-tumoral dendritic cells increase efficacy of peripheral vaccination by modulation of glioma microenvironment. Neuro Oncol. 2010; 12:377–388.
63. Rainov NG, Kramm CM, Banning U, Riemann D, Holzhausen HJ, Heidecke V, Burger KJ, Burkert W, Korholz D. Immune response induced by retrovirus-mediated HSV-tk/GCV pharmacogene therapy in patients with glioblastoma multiforme. Gene therapy. 2000; 7:1853–1858.
64. Kim AJ, Woodworth GF, Boylan NJ, Suk JS, Hanes J. Highly compacted pH-responsive DNA nanoparticles mediate transgene silencing in experimental glioma. J Mater Chem B Mater Biol Med. 2014; 2:8165–8173.
65. Clark PA, Bhattacharya S, Elmayan A, Darjatmoko SR, Thuro BA, Yan MB, van Ginkel PR, Polans AS, Kuo JS. Resveratrol targeting of AKT and p53 in glioblastoma and glioblastoma stem-like cells to suppress growth and infiltration. J Neurosurg. 2016:1–13.
66. Elyakim E, Sitbon E, Faerman A, Tabak S, Montia E, Belanis L, Dov A, Marcusson EG, Bennett CF, Chajut A, Cohen D, Yerushalmi N. hsa-miR-191 is a candidate oncogene target for hepatocellular carcinoma therapy. Cancer research. 2010; 70:8077–8087.
67. Liu Z, Huang S. Inhibition of miR-191 contributes to radiation-resistance of two lung cancer cell lines by altering autophagy activity. Cancer Cell Int. 2015; 15:16.
68. Zhang XF, Li KK, Gao L, Li SZ, Chen K, Zhang JB, Wang D, Tu RF, Zhang JX, Tao KX, Wang G, Zhang XD. miR-191 promotes tumorigenesis of human colorectal cancer through targeting C/EBPbeta. Oncotarget. 2015; 6:4144–4158. doi: 10.18632/oncotarget.2864.
69. Murphy AC, Weyhenmeyer B, Noonan J, Kilbride SM, Schimansky S, Loh KP, Kogel D, Letai AG, Prehn JH, Murphy BM. Modulation of Mcl-1 sensitizes glioblastoma to TRAIL-induced apoptosis. Apoptosis. 2014; 19:629–642.
70. Hetschko H, Voss V, Horn S, Seifert V, Prehn JH, Kogel D. Pharmacological inhibition of Bcl-2 family members reactivates TRAIL-induced apoptosis in malignant glioma. Neuro Oncol. 2008; 86:265–272.
71. Barrantes-Freer A, Kim E, Bielanska J, Giese A, Mortensen LS, Schulz-Schaeffer WJ, Stadelmann C, Bruck W, Pardo LA. Human glioma-initiating cells show a distinct immature phenotype resembling but not identical to NG2 glia. J Neuropathol Exp Neurol. 2013; 72:307–324.
72. Barrantes-Freer A, Renovanz M, Eich M, Braukmann A, Sprang B, Spirin P, Pardo LA, Giese A, Kim EL. CD133 Expression Is Not Synonymous to Immunoreactivity for AC133 and Fluctuates throughout the Cell Cycle in Glioma Stem-Like Cells. PloS one. 2015; 10:e0130519.
73. Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, Bayani J, Head R, Lee M, Bernstein M, Squire JA, Smith A, Dirks P. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell stem cell. 2009; 4:568–580.
74. Mora-Huertas CE, Fessi H, Elaissari A. Polymer-based nanocapsules for drug delivery. International journal of pharmaceutics. 2010; 385:113–142.
75. Mondal N, Pal TK, Ghosal SK. Development and validation of a spectrophotometric method for estimation of letrozole in bulk and pharmaceutical formulation. Pharmazie. 2007; 62:597–598.