
INTRODUCTION
Colorectal cancer (CRC) remains the third leading cause of cancer death in both men and women in the United States with an estimated 134,490 new cases and 49,190 deaths in 2016 [1]. Recent advances in the treatment of metastatic CRC (mCRC) have identified improved outcomes with the addition of epidermal growth factor receptor (EGFR)-targeting agents to conventional combination cytotoxic therapy in patients with extended RAS wild-type tumors. In contrast, activating mutations in the RAS gene (KRAS or NRAS, present in approximately 50% of cases of mCRC) and BRAF gene (present in about 5% of mCRC patients) have been associated with lack of clinically meaningful benefit or harm when anti-EGFR therapy is employed [2]. The identification of candidates for anti-EGFR therapy through the exclusion of RAS and BRAF mutations in mCRC serves as a model of selecting optimal therapy based on patient genomic profiles and molecular phenotypes.
Several decades of genomic studies, including the use of more recent next-generation sequencing (NGS), have expedited the search of genetic alterations for potential therapeutic targeting in CRC [3, 4]. Recently, comprehensive molecular characterization of 224 colorectal tumors was performed by The Cancer Genome Atlas (TCGA) Network [5]. Sixteen percent of colorectal tumors were found to be hypermutated and more commonly found in the right colon with 75% of these cases demonstrating expectedly high microsatellite instability (MSI-H). Twenty-four genes were identified to have significant mutations of interest including APC, SMAD4, TP53, PIK3CA, and KRAS mutations, as expected. Interestingly, mutations, deletions, or amplifications of the ERRB gene family were found in 19% of tumors. In sum, this genomic analysis identified several molecular alterations that are considered targetable, including mediators of dysregulated WNT, RAS, and PI3K pathways such as ERRB2, ERRB3, MEK, AKT, MTOR, IGF2, and IGFR.
The recent identification of gene mutations and amplifications of potential significance for therapeutic purposes has led us to investigate the genomic profiles of mCRC patients using NGS (FoundationOne). Here, we describe a single-institution experience in reporting results from comprehensive genomic analysis of tumors from 138 mCRC patients. We aim to characterize genetic alterations present in our study population that have known correlates to prognosis, therapeutic resistance, and potential therapeutic targets in mCRC. In this study, we also report the existence of concurrent gene mutations rarely described in the literature and novel mutations and amplifications that can lead to targeting outside of National Comprehensive Cancer Network (NCCN) standard treatments.
RESULTS
Study population
The molecular results from FoundationOne testing of tumors from 138 mCRC patients are summarized in Table 1. The median age of our study group was 56 years (range 27-88) with 59.4% (82) males and 40.6% (56) females. The most common ethnicity was White (85, 61.6%) followed by Asian (29, 21.0%). The most common sites of primary were sigmoid colon (33.3%), rectum (19.6%), and cecum (15.2%). Sixty-eight patients (49.3%) had KRAS mutations, 9 patients (6.5%) had BRAF mutations, 3 (2.2%) had NRAS mutations, 1 (0.7%) had an ARAF mutation, 1 (0.7%) had a RAF-1 mutation, 7 (5.1%) had ERRB2 amplifications, 25 (18.1%) had PIK3CA mutations, 15 (10.9%) had PTEN mutations, 4 (2.9%) had AKT mutations, and 3 (2.2%) had MET amplifications.
Table 1: Demographics/patient characteristics
Characteristic (n = 138) |
Frequency (%) |
|---|---|
Age (at initial diagnosis) |
Median (range) |
56 (27-88) |
|
Sex |
|
Male |
82 (59.4%) |
Female |
56 (40.6%) |
Ethnicity |
|
White |
85 (61.6%) |
Asian |
29 (21.0%) |
Black |
7 (5.1%) |
Other |
3 (2.2%) |
Unknown |
14 (10.1%) |
Primary disease site |
|
Sigmoid colon |
46 (33.3%) |
Rectal |
27 (19.6%) |
Cecum |
21 (15.2%) |
Rectosigmoid |
13 (9.4%) |
Colon NOS |
31 (22.5%) |
Stage (at diagnosis) |
|
II |
5 (3.6%) |
III |
16 (11.6%) |
IV |
117 (84.8%) |
Relapsed disease |
|
Yes |
52 (37.7%) |
No |
86 (62.3%) |
KRAS alterations |
68 (49.3%) |
G12D |
23 (16.7%) |
G13D |
10 (7.2%) |
G12V |
8 (5.8%) |
G12S |
7 (5.1%) |
G12C |
6 (4.3%) |
A146T |
3 (2.2%) |
G12A |
2 (1.4%) |
Amplification |
2 (1.4%) |
Q61H |
1 (0.7%) |
Q61K |
1 (0.7%) |
Q61L |
1 (0.7%) |
K117N |
1 (0.7%) |
R68S |
1 (0.7%) |
A146V |
1 (0.7%) |
A146V^sub |
1 (0.7%) |
BRAF alterations |
9 (6.5%) |
V600E |
5 (3.6%) |
D594G |
1 (0.7%) |
G466V |
1 (0.7%) |
G469E |
1 (0.7%) |
L597R |
1 (0.7%) |
NRAS alterations |
|
Q61K |
2 (1.4%) |
Amplification |
1 (0.7%) |
ARAF mutation |
|
S214F |
1 (0.7%) |
RAF1 mutation |
|
S257L |
1 (0.7%) |
ERBB2 alterations |
|
Amplification |
7 (5.1%) |
MET alterations |
|
Amplification |
3 (2.2%) |
AKT1/AKT2 mutations |
|
AKT1 |
3 (2.2%) |
AKT2 |
1 (0.7%) |
Total number of clinically significant alterations |
Median (range) 5 (1-25) |
MSI |
|
MSS |
121 (87.7%) |
MSI-L |
0 (0.0%) |
MSI-H |
2 (1.4%) |
Unknown/not reported |
15 (10.9%) |
NOS: Not otherwise specified; MSI: Microsatellite instability; MSS: Microsatellite stable; MSI-L: MSI low; MSI-H: MSI high.
RAS mutations
Overall, RAS mutations were present in 51.4% of our mCRC patients, RAF mutations were seen in 7.2%, of which RAS+RAF concurrent mutations were seen in 1.4%. The remainder (42.8%) were RAS/RAF wild type (Figure 1). The most common RAS mutations were KRAS mutations of exon 2 (codons 12 and 13) including G12D (32.4%), G13D (14.1%), G12V (11.3%), G12S (9.9%), G12C (8.5%), and G12A (2.8%, Figure 2). Beyond the well-established point mutations in codons 12 and 13 of exon 2 of KRAS, we identified mutations in codon 61 of exon 3 (Q61H, 1.4%; Q61K, 1.4%; Q61L, 1.4%), codon 117 of exon 4 (K117N, 1.4%), and codon 146 of exon 4 (A146V, 1.4%; A146V^sub, 1.4%; A146T, 4.2%). Two mutations (2.8%) in codon 61 (exon 3) of NRAS were also detected. Altogether, these non-KRAS exon 2 mutations constitute 15.5% of RAS mutations.
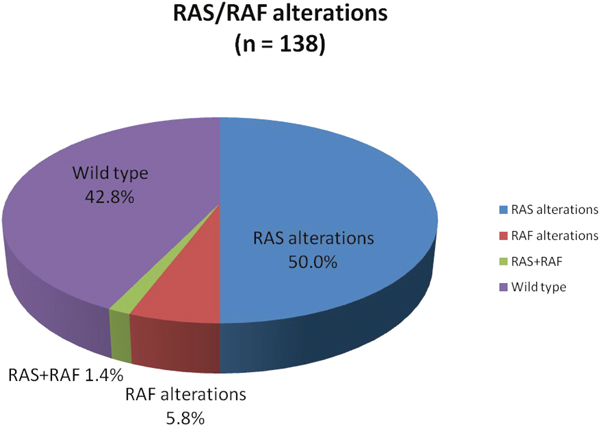
Figure 1: Proportion of RAS, RAF, RAS+RAF mutations, and RAS/RAF wild type status identified by comprehensive genomic profiling. RAS+RAF mutations are not included in RAS or RAF percentages.
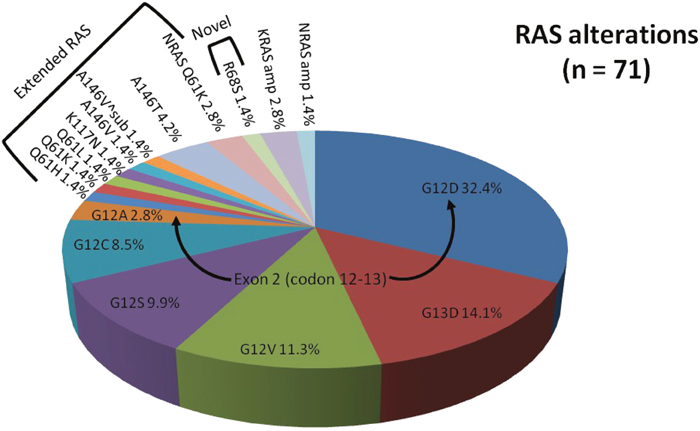
Figure 2: Proportion of RAS alterations identified by comprehensive genomic profiling. Arrows denote common mutations of exon 2 (codon 12-13). Brackets denote panel of extended RAS mutations or novel RAS mutation.
In our patient population, 2 KRAS amplifications (2.8%) and 1 NRAS amplification (1.4%) were identified. One patient was a 51-year-old female with KRAS amplified rectal cancer with synchronous diffuse metastases (lung and liver). Her best overall response to standard first-line combination chemotherapy (5-fluorouracil (5-FU) and irinotecan or FOLFIRI) plus anti-EGFR therapy (panitumumab) was stable disease (SD) for 6 months. The other patient with KRAS amplification was a 51-year-old male diagnosed with right-sided colon cancer and synchronous metastases to the liver and peritoneum who had rapid progression on first- and second-line non-anti-EGFR based therapies. Our 74-year-old male patient with NRAS amplification presented with poorly differentiated rectosigmoid adenocarcinoma and synchronous diffuse metastases (liver, mesentery, and bones) and experienced progressive disease (PD) at 2 months on second-line FOLFIRI + cetuximab. Notably, a novel KRASR68S1 alteration (Figure 2) was identified in a 41-year-old female (1.4%) with rectal cancer and synchronous metastases to the liver and retroperitoneal and supraclavicular lymph nodes who experienced PD at 2 months on anti-EGFR therapy with second-line irinotecan + cetuximab.
RAF mutations
A total of 11 RAF mutations (1 concurrent BRAF+RAF1 mutation) were found in 7.2% of our patients (Figure 3). Of these, BRAFV600E activating mutations (exon 15) were the most common single mutations present (40.0%). One activating BRAFL597Ralteration (exon 15) was identified (10.0%) in a 56-year-old male with bulky rectal adenocarcinoma with synchronous metastases that progressed through 9 months of first-line anti-EGFR therapy. One activating ARAFS214F alteration was also identified (10.0%) in our series of RAF mutations. This 60-year-old male patient developed multiple recurrences of rectal adenocarcinoma including, most recently, metastatic disease to the lung treated with neoadjuvant 5-FU, oxaliplatin, and irinotecan (FOLFOXIRI) followed by metastatectomy; he remains in clinical remission. A dual BRAFV600E+KRASA164V^subalterationwas present (10.0%) in an elderly male (age 72) with poorly differentiated right-sided colon cancer with synchronous metastases on first-line systemic combination therapy without anti-EGFR agents. Here an oncogenic RAS alteration was paired with a known activating BRAF mutation.
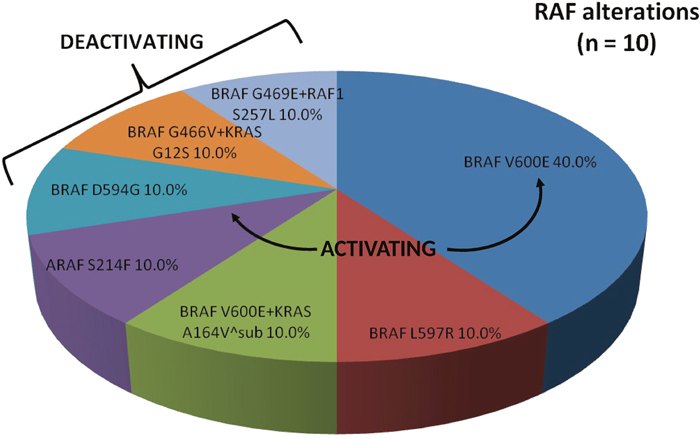
Figure 3: Proportion of RAF alterations identified by comprehensive genomic profiling. Arrows denote known activating mutations. Brackets denote known deactivating mutations.
Deactivating mutations in BRAFD594G(10.0%), BRAFG466V concurrent with KRASG12S (10.0%), and BRAFG469E concurrent with RAF1S257L (10.0%) were also identified. Our patient with a deactivating BRAFD594G mutation was a 59-year-old male with moderately-poorly differentiated right-sided colon cancer with diffuse metastases that was refractory to all standard of care chemotherapy, including FOLFIRI + cetuximab, and ultimately died from progressive disease. Interestingly, he was noted to have a concurrent MET amplification. The patient with a deactivating BRAFG466V mutation concurrent with an activating KRASG12S mutation was a 51-year-old male with right-sided colon cancer with diffuse metastases that progressed with carcinomatosis while on FOLFOX and immediately following salvage debulking surgery with hyperthermic chemotherapy. Notably, he currently has achieved ongoing partial response (PR) on third-line FOLFIRI and bevacizumab (41+ cycles). Our 66-year-old female with dual deactivating BRAFG469E and activating RAF1S257L mutation presented with a right colon cancer with synchronous metastases to bone, liver, lung, and peritoneum. Her disease was refractory to FOLFOX + bevacizumab and is currently on second-line FOLFIRI + bevacizumab with a clinical benefit.
ERBB2 amplifications
Seven patients (5.1%) were found to have ERRB2 amplified tumors with one having a concurrent KRASG12D mutation (Figure 4). The majority of these tumors were MSS (87.5%) with HER2 copy numbers that ranged from 9-190 (Table 2). Notably, all ERRB2 amplified tumors were located in the rectosigmoid colon as its primary disease site. Four patients with RAS wild-type ERBB2 amplification received anti-EGFR therapy, 3 experienced SD ≥ 4 months (2 first-line and 1 second-line) and 1 (second-line) experienced a PR lasting for 5 months as their best overall response to anti-EGFR therapy. The concurrent ERRB2 amplified and KRASG12D mutated tumor was found in a 58-year-old male with moderately differentiated rectal adenocarcinoma with synchronous solitary liver metastasis treated with neoadjuvant 5-FU, oxaliplatin (FOLFOX) followed by hepatic resection and resection of primary – he is currently under surveillance and without evidence of disease.
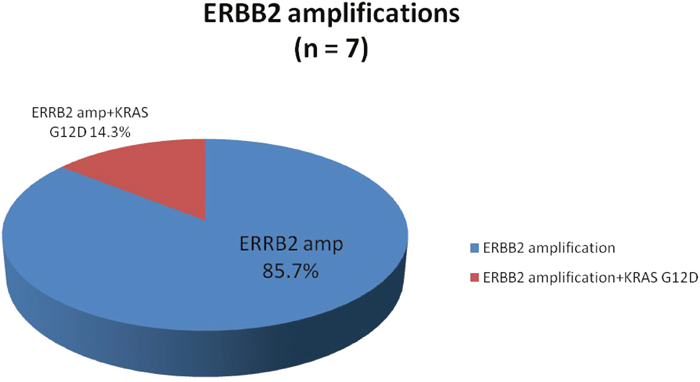
Figure 4: Proportion of ERBB2 amplifications identified by comprehensive genomic profiling.
Table 2: Subset of ERBB2 amplifications
Copy number |
Dual mutations |
Age |
Race/sex |
Primary disease site |
Lines of systemic therapy |
Best overall response to anti-EGFR |
MSI |
|---|---|---|---|---|---|---|---|
9 |
55 |
Latino/M |
Rectum |
2 |
SD (4 mo) |
NR |
|
12 |
88 |
Middle Eastern/M |
Sigmoid |
1 |
MSS |
||
37 |
50 |
White/F |
Rectum |
2 |
MSS |
||
77 |
KRAS G12D |
58 |
White/M |
Rectum |
2 |
MSS |
|
93 |
60 |
White/M |
Rectosigmoid |
2 |
PR (5 mo) |
MSS |
|
189 |
48 |
White/M |
Sigmoid |
4 |
SD (4 mo) |
MSS |
|
190 |
71 |
Asian/M |
Sigmoid |
2 |
SD (5 mo) |
MSS |
EGFR: Epidermal growth factor receptor; MSI: Microsatellite instability; SD: Stable disease; NR: Not reported; MSS: Microsatellite stable; PR: Partial response.
AKT1/2 mutations
Three patients (2.2%) had AKT1E17K mutations while 1 patient (0.7%) had an AKT2E17K mutation (Table 3). Of these, a majority had concurrent mutations (75%) and tumors located in the right colon (75%). One AKT1E17K mutated tumor was found to have concurrent BRAFV600E +KRASA164V^subalterations with phenotype described above. One AKT1E17K mutated tumor had concurrent alterations in KRASA146T+PIK3CAG106V and was found in a 61-year-old male with initial right-sided colon cancer that recurred with metastases to the liver showing moderately differentiated colon adenocarcinoma. His tumor was characterized by aggressive features, including metastatic disease recurrence following a diagnosis of stage I disease, and development of bony metastases within the first year of recurrence. A concurrent AKT2E17K+KRASG12C altered tumor was found in a 57-year-old female with originally moderately differentiated sigmoid adenocarcinoma that was resected but recurred with metastases to the retroperitoneal lymph nodes currently on first-line FOLFIRI + bevacizumab.
Table 3: Subset of AKT1/2 mutations
Variant |
Dual mutations* |
Age |
Race/Sex |
Primary disease site |
Lines of systemic therapy |
MSI |
|---|---|---|---|---|---|---|
E17K (AKT1) |
KRAS A146T |
61 |
Black/M |
Right colon, NOS |
2 |
NR |
E17K (AKT1) |
BRAF V600E, KRAS A146^sub |
72 |
White/M |
Ascending colon |
1 |
MSS |
E17K (AKT1) |
69 |
White/F |
Ascending colon |
1 |
MSS |
|
E17K (AKT2) |
KRAS G12C |
57 |
Asian/F |
Sigmoid |
1 |
MSS |
*For concurrent RAS and BRAF mutations only, NOS: Not otherwise specified; MSI: Microsatellite instability; NR: Not reported; MSS: Microsatellite stable.
PIK3CA and PTEN mutations
In total, we identified 25 patients (18.1%) with PIK3CA alterations in our cohort (Table 4). The most common primary disease sites included cecum (36.0%), sigmoid colon (12.0%), and rectum (12.0%). Notably, right-sided colon cancers comprised nearly half (48.0%) of tumors with PIK3CA alterations. The most commonly identified variants were E545K (24.0%, exon 9), E542K (12.0% exon 9), E110del (8.0%), and Q546K (8.0%). Tumors with PIK3CA alterations frequently had concurrent mutations in the RAS-RAF-MAPK signaling pathway. A majority (19 or 76.0%) had concurrent mutations in KRAS (G12D 36.0%, G12S 12.0%, G13D 8.0%, and A146T 8.0%). Two patients (8.0%) with PIK3CA tumors were found to have concurrent deactivating BRAF mutations (G466V and G469E). Notably, these 2 patients had additional alterations in KRASG12S and RAF1S257L, respectively, with phenotypes described above. We also identified additional alterations in the PTEN-PIK3CA-AKT signaling pathway in our group of PIK3CA altered tumors (Figure 5). Five patients (20.0%) with PIK3CA altered tumors also had PTEN mutations, while 1 patient (4.0%) had a dual PIK3CA and AKT1 mutated tumor. Of note, 1 female patient (age 55) with a dual PIK3CA and PTEN mutation had a rectal tumor demonstrating MSI-H and developed a solitary liver metastasis that has since been resected and treated with adjuvant FOLFOX – she is currently in remission.
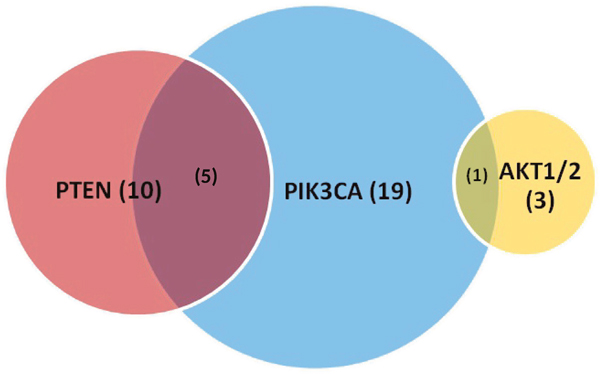
Figure 5: Proportion of PIK3CA, PTEN, AKT1/2 mutations identified by comprehensive genomic profiling with overlap. Values in parentheses represent numbers and not percentages.
Table 4: Subset of PIK3CA alterations
Characteristic (n = 25) |
Frequency (%) |
|---|---|
Age (at initial diagnosis) |
Median (range) 55 (30-81) |
Sex |
|
Male |
14 (56.0%) |
Female |
11 (44.0%) |
Ethnicity |
|
White |
17 (68.0%) |
Asian |
2 (8.0%) |
Black |
2 (8.0%) |
Other/unknown |
4 (16.0%) |
Primary disease site |
|
Right colon |
12 (48.0%) |
Transverse colon |
2 (8.0%) |
Left colon (includes sigmoid) |
8 (32.0%) |
Rectum |
3 (12.0%) |
Variant |
|
E545K (exon 9) |
6 (24.0%) |
E542K (exon 9) |
3 (12.0%) |
E110del |
2 (8.0%) |
Q546K |
2 (8.0%) |
C901F |
1 (4.0%) |
C420R |
1 (4.0%) |
C420R+E726K |
1 (4.0%) |
E545G^sub+R108H^sub |
1 (4.0%) |
G106V |
1 (4.0%) |
H1047L (exon 20) |
1 (4.0%) |
H1047Y (exon 20) |
1 (4.0%) |
N107del |
1 (4.0%) |
N345K |
1 (4.0%) |
P104L |
1 (4.0%) |
P104_V105del |
1 (4.0%) |
R88Q |
1 (4.0%) |
Concurrent KRAS mutations |
19 (76.0%) |
G12D |
9 (36.0%) |
G12S |
3 (12.0%) |
G13D |
2 (8.0%) |
A146T |
2 (8.0%) |
G12V |
1 (4.0%) |
G12C |
1 (4.0%) |
A146V |
1 (4.0%) |
Concurrent PTEN mutations |
5 (20.0%) |
H93Y^sub |
1 (4.0%) |
L316fs*7 |
1 (4.0%) |
L57fs*6+N323fs*2 |
1 (4.0%) |
R130Q+R142W |
1 (4.0%) |
Splice site 1008_1026+5del24 |
1 (4.0%) |
Concurrent BRAF mutations |
|
G466V |
1 (4.0%) |
G469E |
1 (4.0%) |
Concurrent AKT1 mutation |
|
E17K |
1 (4.0%) |
MSI |
|
MSS |
21 (84.0%) |
MSI-L |
0 (0.0%) |
MSI-H |
1 (4.0%) |
Unknown/Not reported |
3 (12.0%) |
MSI: Microsatellite instability; MSS: Microsatellite stable; MSI-L: MSI low; MSI-H: MSI high.
MET amplifications
Three patients (2.2%) in our series had MET amplifications (Table 5). Two-thirds of these tumors were MSS, located in the right colon, and associated with concurrent mutations in RAS or RAF genes. One 67-year-old male was initially diagnosed with right-sided colon cancer (KRASG13D+MET alterations present) and synchronous liver metastases. His course has been punctuated by recurrent metastases to the liver and lungs despite several systemic and regional therapies. Another right-sided colon cancer was identified with both a deactivating BRAFD594G mutation and MET amplification with aggressive phenotype described above. A third patient was a 27-year-old male with primary rectal adenocarcinoma that recurred with metastases to the liver and retroperitoneal lymph nodes and refractory to capecitabine + irinotecan + cetuximab. In particular, 2 of 3 patients with MET amplications and RAS/BRAFV600E wild-type tumors were refractory to anti-EGFR-based therapies.
Table 5: Subset of MET amplifications
Variant |
Dual Mutations* |
Age |
Race/Sex |
Smoker |
Primary disease site |
Lines of systemic therapy |
MSI |
|---|---|---|---|---|---|---|---|
MET |
BRAF D594G |
59 |
Asian/M |
Yes |
Right colon, NOS |
4 |
MSS |
MET |
27 |
White/M |
Yes |
Rectum |
2 |
NR |
|
MET |
KRAS G13D |
67 |
Asian/M |
No |
Ascending colon |
1 |
MSS |
*For concurrent RAS and BRAF mutations only, MSI: Microsatellite instability; NOS: Not otherwise specified; NR: Not reported; MSS: Microsatellite stable.
Hypermutant status
The majority of our 138 patients with mCRC had tumors with <9 clinically significant alterations (121 or 87.7%) as described by FoundationOne reports (Table 6). The majority of these tumors were located in the left colon and all were MSS. Fourteen patients (10.1%) had 9-16 total alterations while only 3 patients (2.2%) were allocated to the highest number of clinically significant alterations category (17-25). Notably, 2 patients with MSI-H tumors were identified in the highest number of alterations group. One 55-year-old female patient was found to have a dual PIK3CA and PTEN mutated rectal tumor with phenotype described previously. Tumor mutational burden (TMB) from FoundationOne report showed a high TMB of 33 mutations per megabase (Mb). The other was a 47-year-old female with KRASG12V mutated metastatic rectal cancer that has progressed through 3 lines of systemic therapy and currently on anti-PD-1 therapy with pembrolizumab with a clinical response. Again, TMB corroborated her findings of a relatively hypermutated tumor with a TMB of 31 mutations/Mb. The third patient with a hypermutant FoundationOne profile had a MSS tumor with an associated POLEV411L mutation. Interestingly, this patient was elderly (age 80), had a right colon tumor, and had a recurrence pattern consistent with locoregional recurrence. This patient demonstrated a TMB of 122 mutations/Mb, which was the highest among the cohort.
Table 6: Total number of clinically significant alterations
No. of alterations |
Frequency (%) |
Age |
Sex |
Race |
Primary disease site |
MSI |
|---|---|---|---|---|---|---|
<9 |
121 (87.7%) |
Median (range) |
F 48 (39.7%) |
White 74 (61.2%) |
Right colon 28 (23.1%) |
MSS 121 (100%) |
9-16 |
14 (10.1%) |
Median (range) |
F 6 (42.9%) |
White 8 (57.1%) |
Right colon 6 (42.9%) |
MSS 13 (92.9%) |
17-25 |
3 (2.2%) |
Mean 59.7 |
F 2 (66.6%) |
White 3 (100%) |
Rectum 2 (66.6%) |
MSI-H 2 (66.6%) |
MSI: Microsatellite instability; NOS: Not otherwise specified; MSS: Microsatellite stable; NR: Not reported; MSI-H: MSI high.
DISCUSSION
Comprehensive molecular characterization of 138 tumors from patients with mCRC was performed via NGS (FoundationOne) in this single-institution retrospective study. Overall, 51.4% and 7.2% of our patients with mCRC were shown to carry RAS and RAF mutations, respectively, which is concordant with frequencies historically reported in mCRC [2]. The majority of our RAS mutations were KRAS mutations of exon 2 (codons 12 and 13), which represent those identified in initial phase III trials that predicted lack of benefit from anti-EGFR therapy in mCRC [6, 7]. We also found that 15.5% of all RAS mutations in our population comprised a panel of extended RAS mutations. This is also consistent with recent data from the PRIME and CRYSTAL clinical trials, where exon 3 and 4 KRAS and exons 2, 3, and 4 NRAS mutations reflected 14-17% of RAS mutations [8, 9]. Identifying these rare RAS mutations has major clinical significance, given their association with anti-EGFR resistance [10].
Notably, we identified 2 KRAS amplifications and 1 NRAS amplification that are extremely rare and poorly characterized. These were found in 3 patients with diffusely metastatic CRC progressive through several lines of systemic therapy including anti-EGFR therapy.
Putative high-level amplifications of NRAS were observed in <1% of cases in TCGA dataset though its significance in CRC remains poorly described [5]. KRAS amplifications have been associated with acquired resistance to EGFR inhibitors cetuximab or panitumumab in CRC preclinical models [11]. To our knowledge, we are the first to report a novel KRASR68S1 alteration that was associated with a particularly aggressive phenotype and PD at 2 months on anti-EGFR therapy with cetuximab.
The majority of RAF mutations found in our population were BRAFV600E activating mutations (exon 15), which have been historically associated with poorer survival, resistance to chemotherapy, and lack of clinical benefit with anti-EGFR therapy in mCRC [12–15]. We also identified a lone BRAFL597Ralteration (exon 15), which is poorly described in CRC but has been shown to similarly activate RAF-MEK-ERK signaling in melanoma in vitro [16]. Of note, this patient received 9 months of first-line anti-EGFR therapy though our sample size of 1 precludes any meaningful generalizations. One ARAFS214F alteration was also identified in a patient whose course has been characterized by multiple recurrences of rectal cancer. Mutations in ARAF have been linked as oncogenic drivers in lung adenocarcinoma, and are exceedingly rare in CRC and comprise approximately 2% of cases in the CRC dataset from TCGA [5, 17]. Treatment with the oral RAF inhibitor, sorafenib, has demonstrated prolonged response in a case of refractory non-small-cell lung cancer and rapid responses in patients with refractory histiocytic neoplasms bearing somatic ARAF mutations [17, 18].
Despite a previous conception that KRAS and BRAF mutations are mutually exclusive, we found 1 dual BRAFV600E+KRASA164V^submutated tumor that, in our case, was associated with poor prognostic features [19]. One case of concurrent BRAFG466V+KRASG12S mutation and one patient with a concurrent BRAFG469E+RAF1S257L mutation were present in our cohort. BRAF mutants G466V and G469E have been shown to represent variants with impaired or complete loss of kinase activity in vitro [20, 21]. Nevertheless, it has been shown that tumorigenesis is promoted in the presence of deactivating BRAF mutations through oncogenic RAS mutation and/or CRAF (or RAF-1) signaling [21, 22]. In our study, one deactivating BRAFG466V mutation was paired with an oncogenic KRASG12S mutation, and one deactivating BRAFG469E mutation was paired with an oncogenic RAF1S257L alteration, supporting the notion of an evolutionary adaptation in the cancer genome to overcome BRAF mutations with impaired function. In both cases, there were associated features of poor prognosis though the dual BRAFG466V+KRASG12S mutated tumor has seen disease control recently on 41 cycles of FOLFIRI and bevacizumab, which may argue for varying degrees of relative contribution from each mutation on tumor phenotype. Interestingly, one patient with deactivating BRAFD594Gmutation was refractory to all lines of treatment, including anti-EGFR therapy, and ultimately died of aggressive disease. This is at odds with recent reports suggesting that BRAFD594G mutation may be an indicator of good prognosis [23]. It is unclear whether this patient’s concurrent MET amplification may have contributed to his overall poor prognosis and therapeutic resistance.
ERBB2 (HER2/neu) amplifications were found in 5.1% of our mCRC patients with the majority in KRAS wild-type tumors (except for 1 with a concurrent ERRB2+KRASG12D alteration). Another FoundationOne analysis of >10,000 cases of gastrointestinal malignancies identified HER2 amplifications and mutations in 3.0% and 4.8%, respectively, of cases from the CRC cohort [24]. Our patients with HER2 amplified tumors appeared to have shortened clinical benefit with anti-EGFR therapy, which is consistent with the recent phase II HERACLES trial where none of the patients with HER2 amplified, RAS/BRAF wild-type metastatic colorectal tumors had a response to anti-EGFR therapy [25]. Similar to the preponderance of left colon primary tumors in the HERACLES trial, all of our HER2 amplified tumors were located in the rectosigmoid colon. In short, the identification of HER2 amplifications in patients with RAS/BRAF wild-type metastatic colorectal tumors is of major significance given the clinical benefit derived from dual HER2-directed therapy including trastuzumab + lapatinib (HERACLES) or trastuzumab + pertuzumab (MyPathway) [25, 26].
PIK3CA, PTEN, and AKT mutations were identified in 18.1% (25), 10.9% (15), and 2.9% (4) of our mCRC patients, respectively. Many of these patients had metastatic tumors associated with aggressive features. In addition, 75% of AKT mutated tumors were located in the right colon, almost half (48.0%) of PIK3CA mutated tumors were right-sided colon cancers, and concurrent mutations in RAS-RAF-MAPK or PTEN-PIK3CA-AKT signaling were common. For example, 19 patients (76.0%) with PIK3CA mutations also had concurrent KRAS mutations while 5 (20.0%) and 1 (4.0%) with PIK3CA altered tumors also had concurrent PTEN and AKT1 mutations, respectively. Mutations in mediators of the PTEN-PIK3CA-AKT signaling pathway in CRC have been associated with poorer prognosis and lack of clinical response to anti-EGFR therapy [27, 28]. For PIK3CA mutations, in particular, prior studies have demonstrated that exon 9 mutations had no effect while exon 20 mutations were associated with resistance to anti-EGFR therapy [29]. However, this differential effect by exon has not been supported by recent meta-analysis [30]. Given the high rate of concurrent RAS mutations seen with PIK3CA and related pathway mutations, a definitive association between resistance to EGFR inhibition and PTEN-PIK3CA-AKT pathway mutations is difficult to make. Further studies are needed to resolve this issue.
Three patients (2.2%) demonstrated MET amplifications associated with poor prognostic features. MET amplification and increased c-MET expression have also been associated with an aggressive phenotype and therapeutic resistance, particularly to MEK inhibition, in mCRC [31, 32]. Interestingly, we have observed anti-EGFR refractoriness in 2 of our patients with MET amplifications despite the presence of a RAS- wild-type phenotype and lack of activating BRAF mutations. This is consistent with preclinical data suggesting MET activation as a mechanism of resistance to anti-EGFR therapy [33].
We lastly identified 3 patients (2.2%) with tumors categorized in the highest number of clinically significant alterations group (17-25) that also demonstrated high TMB as per FoundationOne. TMB categories per FoundationOne testing have been validated in melanoma patients treated with PD-1 blockade [34]. Response to PD-1 inhibitors was significantly superior in patients with high TMB (>23.1 mutations/MB) compared to intermediate or low TMB (3.2-23.1 mutations/MB and <3.2 mutations/MB, respectively). Furthermore, a recent phase II study showed that patients with advanced urothelial cancer who responded to the programmed death ligand 1 (PD-L1) inhibitor atezolizumab had a significantly higher TMB (median 12.4 mutations/Mb) than non-responders (median 6.4 mutations/Mb, p < 0.0001) [35]. Two patients had MSI-H tumors while 1 hypermutant tumor was MSS and harbored a POLE mutation. Interestingly, 42.9% of tumors with 9-16 clinically significant alterations were located in the right colon while one-third of tumors with 17-25 total alterations were located in the right colon; tumors with <9 number of alterations were predominantly located in the left colon. In the CRC dataset from TCGA, 75% of hypermutated tumors arose from the right colon yet not all of them were MSI-H [5]. Mutations in polymerase ε or POLE were found among 25% of hypermutated tumors in this cohort. Mutations in POLE have been shown to contribute to an ultramutated yet MSS phenotype in colorectal tumors [36]. A recent NGS study confirmed that increasing mutational load correlated with MSI yet colorectal tumors with the highest mutational burden that were distinct from MSI tumors all harbored POLE mutations [37]. Furthermore, mismatch repair-deficiency or MSI has recently been shown to predict clinical benefit to immune checkpoint blockade with anti-PD-1 therapy in mCRC [38]. The characterization of mutational load in CRC may serve as a better indicator than MSI status in determining a hypermutant profile that could predict benefit from immunotherapy. Our findings are hypothesis generating and offer support to consider molecular analysis of tumors to determine the total number of alterations as a potential correlate to MSI and candidacy for anti-PD-1 therapy in mCRC.
Future studies of larger size and, ideally, prospective design will be helpful in corroborating associations between molecular alterations of interest described in our study and prognosis, resistance to EGFR inhibition, and/or ability to be targeted for therapy in mCRC. Comparative genomic analyses have identified a high level of concordance particularly for RAS, BRAF, and PIK3CA mutations between colorectal primary and metastatic tumors [39, 40]. However, other molecular alterations may differ based on the site of tumor and/or exposure to chemotherapy [41–44]. Although such mixed results are likely dependent on the specific mutation that is profiled, other factors including specimen integrity and sampling method may also contribute to heterogeneity. Indeed, further analyses are needed to describe the concordance or discordance of other mutations across tumor sites and treatment effects in mCRC, and careful consideration in design will be needed in order to account for confounding factors as described above.
In conclusion, comprehensive genomic profiling can uncover gene alterations beyond conventional RAS or RAF mutant subtypes that predict resistance to anti-EGFR therapy and in identifying potential therapeutic targets outside of NCCN standard treatments in mCRC. ERBB2 amplified tumors commonly originate from the rectosigmoid colon, are predominantly RAS/BRAF wild-type, and may predict benefit to HER2-directed therapy. Hypermutant tumors or tumors with POLE mutations may predict benefit to anti-PD-1 therapy. Our findings are hypothesis generating and warrant further investigation in larger datasets and in prospective settings.
MATERIALS AND METHODS
Study patients and tumor samples
Patients with advanced or metastatic (stage IV) colorectal cancer treated at the Gastrointestinal Medical Oncology Clinic at City of Hope National Medical Center (Duarte, CA) between April 2013 and February 2016 were screened for this study. Eligibility criteria was limited to those who underwent expanded genomic tumor analysis by FoundationOne. There were no exclusions to tumor histology, medical comorbidities, previous treatment or lines of prior therapy, or performance status. Comprehensive genomic profiling was conducted through NGS via FoundationOne (Foundation Medicine, Inc., Cambridge, MA) with reports generated from April 2013 to February 2016. The study was approved by the Institutional Review Board (IRB).
Next-generation sequencing
Comprehensive genomic analysis was conducted on tumor samples (formalin-fixed paraffin-embedded) retrieved from surgical resection, core needle biopsies, or excisional biopsies and delivered to Foundation Medicine, Inc. The NGS assay performed by FoundationOne has been previously described and validated [45]. The initial whole-genome shotgun library construction and hybridization-based capture of 4,557 exons from 287 cancer-related genes and 47 introns from 19 genes with frequent DNA rearrangements has since been expanded to identify genetic alterations across the coding regions of 315 cancer-related genes and introns from 28 genes commonly rearranged in solid cancers.
Study design
Retrospective analysis of genetic mutations, amplifications, or alterations present in our cohort of 138 patients with mCRC was performed through test results provided in an integrative report available via FoundationICE (Interactive Cancer Explorer). Patient demographics including age, sex, ethnicity, site of primary, stage at diagnosis, and number of previous treatments were obtained from chart abstraction of each patient’s electronic medical record (EMR). Microsatellite instability classified as stable (MSS), low (MSI-L), or high (MSI-H) were abstracted from pathology reports and response to anti-EGFR therapy, when available, was described according to Response Evaluation Criteria in Solid Tumors (RECIST) criteria and obtained from medical records [46]. The total number of clinically significant alterations for each patient was determined by tallying the sum of alterations included in the panel of clinically significant variants provided by FoundationICE reports and arbitrarily categorized into 3 groups (<9, 9-16, and > 16 total number of alterations). We defined hypermutant tumors as those in the highest number of mutations group that were also found to have high TMB as validated by FoundationOne (high >23.1 mutations/MB, intermediate 3.2-23.1 mutations/MB, and low <3.2 mutations/MB) [34].
Statistical analyses
All statistical analyses performed were descriptive and no formal statistical hypotheses were assessed. The sample size was determined by the total number of mCRC patients with FoundationOne results available. All descriptive statistics were conducted in Excel with associated formulas and functions.
CONFLICTS OF INTEREST
All authors declare no potential conflicts of interest.
GRANT SUPPORT
The research described was supported by the City of Hope Cancer Center Support Grant (CCSG) and CCSG-supported Core Facilities.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7-30.
2. Fakih MG. Metastatic colorectal cancer: current state and future directions. J Clin Oncol. 2015; 33:1809-1824.
3. Clark CR, Starr TK. Mouse models for the discovery of colorectal cancer driven genes. World J Gastroenterol. 2016; 22:815-822.
4. Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011; 6:479-507.
5. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330-337.
6. Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008; 26:1626-1634.
7. Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008; 359:1757-1765.
8. Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013; 369:1023-1034.
9. Van Cutsem E, Lenz HJ, Köhne CH, Heinemann V, Tejpar S, Melezínek I, Beier F, Stroh C, Rougier P, van Krieken JH, Ciardiello F. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol. 2015; 33:692-700.
10. Sorich MJ, Wiese MD, Rowland A, Kichenadasse G, McKinnon RA, Karapetis CS. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol. 2015; 26:13-21.
11. Valtorta E, Misale S, Sartore-Bianchi A, Nagtegaal ID, Paraf F, Lauricella C, Dimartino V, Hobor S, Jacobs B, Ercolani C, Lamba S, Scala E, Veronese S, et al. KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int J Cancer. 2013; 133:1259-1265.
12. Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008; 26:5705-5712.
13. Van Cutsem E, Köhne CH, Láng I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, Schlichting M, Zubel A, Celik I, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011; 29:2011-2019.
14. Tveit KM, Guren T, Glimelius B, Pfeiffer P, Sorbye H, Pyrhonen S, Sigurdsson F, Kure E, Ikdahl T, Skovlund E, Fokstuen T, Hansen F, Hofsli E, et al. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: the NORDIC-VII study. J Clin Oncol. 2012; 30:1755-1762.
15. Seymour MT, Brown SR, Middleton G, Maughan T, Richman S, Gwyther S, Lowe C, Seligmann JF, Wadsley J, Maisey N, Chau I, Hill M, Dawson L, et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncol. 2013; 14:749-759.
16. Dahlman KB, Xia J, Hutchinson K, Ng C, Hucks D, Jia P, Atefi M, Su Z, Branch S, Lyle PL, Hicks DJ, Bozon V, Glaspy JA, et al. BRAF (L597) mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer Discov. 2012; 2:791-797.
17. Imielinski M, Greulich H, Kaplan B, Araujo L, Amann J, Horn L, Schiller J, Villalona-Calero MA, Meyerson M, Carbone DP. Oncogenic and sorafenib-sensitive ARAF mutations in lung adenocarcinoma. J Clin Invest. 2014; 124:1582-1586.
18. Diamond EL, Durham BH, Haroche J, Yao Z, Ma J, Parikh SA, Wang Z, Choi J, Kim E, Cohen-Aubart F, Lee SC, Gao Y, Micol JB, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov. 2016; 6:154-165.
19. Kaczirek K, Ciuleanu TE, Vrbanec D, Marton E, Messinger D, Liegl-Atzwanger B, Wrba F, Knittelfelder R, Lindner E, Zielinski CC, Streubel B, Brodowicz T. FOLFOX4 plus cetuximab for patients with previously untreated metastatic colorectal cancer according to yumor RAS and BRAF mutation status: updated analysis of the CECOG/CORE 1.2.002 study. Clin Colorectal Cancer. 2015; 14:91-98.
20. Houben R, Becker JC, Kappel A, Terheyden P, Bröcker EB, Goetz R, Rapp UR. Constitutive activation of the Ras-Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J Carcinog. 2004; 3:6.
21. Smalley KS, Xiao M, Villanueva J, Nguyen TK, Flaherty KT, Letrero R, Van Belle P, Elder DE, Wang Y, Nathanson KL, Herlyn M. CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations. Oncogene. 2009; 28:85-94.
22. Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, Marais R. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010; 140:209-221.
23. Cremolini C, Di Bartolomeo M, Amatu A, Antoniotti C, Moretto R, Berenato R, Perrone F, Tamborini E, Aprile G, Lonardi S, Sartore-Bianchi A, Fontanini G, Milione M, et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann Oncol. 2015; 26:2092-2097.
24. Bose R, Ali S, Jain N, Gowen K, Bertotti A, Murray E, Ross JS, Beattie MS, Hurwitz H, Hainsworth JD. Frequency of HER2 mutations and amplification in GI malignancies and ability of pertuzumab to overcome neuregulin1 mediated drug resistance to a HER2 tyrosine kinase inhibitor in colon cancer. J Clin Oncol. 2016; 34:Abstr 630.
25. Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, Bergamo F, Zagonel V, Leone F, Depetris I, Martinelli E, Troiani T, Ciardiello F, Racca P, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016; 17:738-46. doi: 10.1016/S1470-2045(1016)00150-00159.
26. Hurwitz H, Hainsworth JD, Swanton C, Perez EA, Sweeney C, Burris HA, Spigel D, Meric-Bernstam F, Strickland DK, Leon L, Brammer M, Beattie MS, Bose R. Targeted therapy for gastrointestinaI (GI) tumors based on molecular profiles: early results from MyPathway, an open-label phase IIa basket study in patients with advanced solid tumors. J Clin Oncol. 2016; 34:Abstr 653.
27. De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011; 12:594-603.
28. Ogino S, Nosho K, Kirkner GJ, Shima K, Irahara N, Kure S, Chan AT, Engelman JA, Kraft P, Cantley LC, Giovannucci EL, Fuchs CS. PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. J Clin Oncol. 2009; 27:1477-1484.
29. De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010; 11:753-762.
30. Therkildsen C, Bergmann TK, Henrichsen-Schnack T, Ladelund S, Nilbert M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: a systematic review and meta-analysis. Acta Oncol. 2014; 53:852-864.
31. Bradley CA, Dunne P, McArt D, Arthur K, McQuaid S, Salto-Tellez M, Johnston P, Van Schaeybroeck S. The role of c-MET/HGF signaling as a critical mediator of an invasive and resistant phenotype in colorectal cancer. Cancer Res. 2015; 75:4018.
32. Zeng ZS, Weiser MR, Kuntz E, Chen CT, Khan SA, Forslund A, Nash GM, Gimbel M, Yamaguchi Y, Culliford AT, D'Alessio M, Barany F, Paty PB. c-Met gene amplification is associated with advanced stage colorectal cancer and liver metastases. Cancer Lett. 2008; 265:258-269.
33. Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A, Zecchin D, Apicella M, Migliardi G, Galimi F, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013; 3:658-673.
34. Johnson DB, Frampton GM, Rioth MJ, Yusko E, Ennis R, Fabrizio D, Greenbowe JR, Ali SM, Frederick DT, Puzanov I, Balko JM, Cates J, Ross JS, et al. Hybrid capture-based next-generation sequencing (HC NGS) in melanoma to identify markers of response to anti-PD-1/PD-L1. J Clin Oncol. 2016; 34:Abstr 105.
35. Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O'Donnell PH, Balmanoukian A, Loriot Y, Srinivas S, Retz MM, Grivas P, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016; 387:1909-1920.
36. Briggs S, Tomlinson I. Germline and somatic polymerase ε and δ mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol. 2013; 230:148-153.
37. Stadler ZK, Battaglin F, Middha S, Hechtman JF, Tran C, Cercek A, Yaeger R, Segal NH, Varghese AM, Reidy-Lagunes DL, Kemeny NE, Salo-Mullen EE, Ashraf A, et al. Reliable detection of mismatch repair deficiency in colorectal cancers using mutational load in next-generation sequencing panels. 2016; 34:2141-2147. doi: 10.1200/JCO.2015.1265.1067.
38. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015; 372:2509-2520.
39. Brannon AR, Vakiani E, Sylvester BE, Scott SN, McDermott G, Shah RH, Kania K, Viale A, Oschwald DM, Vacic V, Emde AK, Cercek A, Yaeger R, et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014; 15:454.
40. Vakiani E, Janakiraman M, Shen R, Sinha R, Zeng Z, Shia J, Cercek A, Kemeny N, D'Angelica M, Viale A, Heguy A, Paty P, Chan TA, et al. Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J Clin Oncol. 2012; 30:2956-2962.
41. Alagaratnam S, Brunsell TH, Danielsen SA, Sveen A, Eknæs M, Hektoen M, Røsok B, Bjørnbeth BA, Nesbakken A, Lothe R. Heterogeneity of colorectal cancers and multiple liver metastases by mutation and copy number profiling. Cancer Res. 2016; 76:Abstr 2396.
42. Kogita A, Yoshioka Y, Sakai K, Togashi Y, Sogabe S, Nakai T, Okuno K, Nishio K. Inter- and intra-tumor profiling of multi-regional colon cancer and metastasis. Biochem Biophys Res Commun. 2015; 458:52-56.
43. Lee SY, Haq F, Kim D, Jun C, Jo HJ, Ahn SM, Lee WS. Comparative genomic analysis of primary and synchronous metastatic colorectal cancers. PLoS One. 2014; 9:e90459.
44. Sveen A, Løes IM, Alagaratnam S, Nilsen G, Høland M, Lingjærde OC, Sorbye H, Berg KC, Horn A, Angelsen JH, Knappskog S, Lønning PE, Lothe RA. Intra-patient inter-metastatic genetic heterogeneity in colorectal cancer as a key determinant of survival after curative liver resection. PLoS Genet. 2016; 12:e1006225.
45. Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, Sun J, Juhn F, Brennan K, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013; 31:1023-1031.
46. Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000; 92:205-216.