
INTRODUCTION
Hepatocellular carcinoma (HCC) is a malignant tumor of the liver. It is the third most common cause of cancer-related death worldwide [1, 2]. Asian countries (particularly China) account for 55% of all newly diagnosed HCC cases and more than 50% of HCC deaths worldwide [3]. The 5-year survival rate for HCC patients is 15% (localized, 28%; regional, 7%; and distant, 3%), and 80-90% of patients present with advanced and unresectable tumors at the time of diagnosis due to widespread intra- and extra-hepatic metastasis [4]. Thus, it is critical to elucidate the molecular mechanisms underlying HCC progression and develop novel treatment strategies. Several drugs have shown efficacy in HCC including the multi-kinase inhibitor Sorafenib (targets Raf, VEGFR, PDGFRβ, c-Kit, RET, and other kinases), which is approved for clinical use. However, the therapeutic limitation has also been demonstrated in several clinical trials [5, 6].
Actinidia chinensis Planch root extract (acRoots) is a traditional Chinese medicine that has been used for the treatment of various types of cancer including HCC [7]. The principal compounds are triterpenes, which exhibit remarkable anticancer effects in HCC [7-9]. Other compounds have also shown cytotoxic activity including the phenolic constituents and isomeric flavonoids [10, 11]. Although the anticancer effects of acRoots have been observed clinically, the mechanisms underlying the effects are not fully understood.
Many recent studies have described metabolic alterations in proliferating cancer cells [12]. Using a non-targeted metabolic profiling strategy based on liquid chromatography-mass spectrometry, Huang et al. demonstrated that the predominant metabolic alternations in HCC included increased glycolysis, gluconeogenesis, and β-oxidation with reduced tricarboxylic acid cycle and Δ-12 desaturase [13]. These metabolic alterations provide the nutrients and energy to support the uncontrolled proliferation of malignant cells. Therefore, therapeutics that target these metabolic pathways in cancer cells may be effective in HCC.
Triterpenes from Alismatis rhizoma were shown to have a lipid-lowering effect on mice with high-fat diet-induced hyperlipidemia [14]. Therefore, we hypothesized that acRoots could alter metabolic processes in HCC cells. Here, we show that acRoots inhibits cholesterol metabolism in HCC cell lines through upregulation of PCSK9.
RESULTS
acRoots inhibits proliferation and regulates metabolic pathways in HCC
To assess the effects of acRoots on HCC cells, we treated human HCC cell lines (LM3 and HepG2), and normal liver cells (HL-7702) with various doses of acRoots and analyzed cell viability in response to treatment. CCK-8 assays revealed that acRoots treatment inhibited cell viability in a dose-dependent manner (Figure 1A, 1B, and Supplementary Figure 1A). These effects were observed at a dose of 5 mg/mL in LM3 and HepG2 cells, and at a dose of 30 mg/mL in HL-7702 cells. LM3 cells were selected for gene expression profiling. The results of mRNA profiling suggested that acRoots induced variations in the expression of genes involved in the immune response, inflammation, proliferation, cell cycle control, and metabolism in LM3 cells (Supplementary Figure 1B). We focused on genetic variations in metabolic genes annotated to the term “metabolic process” (GO:0008152) and related children in Gene Ontology (http://amigo1.geneontology.org/) (Supplementary Data sets 1 and 2). We quantified the expression of 711 metabolic genes in LM3 cells that showed at least a two-fold change in expression compared to the untreated control group after treatment with any concentration of acRoots (Figure 1C, Supplementary Data set 3). Distribution maps were plotted to show differences in mRNA levels induced by various doses of acRoots. These data indicated acRoots inhibited the expression of metabolic genes in a dose-dependent manner (Figure 1D and 1E). Hierarchical clustering analysis yielded eight clusters based on the similarity of the drug concentration profiles (Figure 1F). The Database for Annotation, Visualization, and Integrated Discovery (DAVID) software was then used to determine which Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were enriched [15, 16]. We found that several members of the steroid and terpenoid backbone biosynthesis pathways were significantly up-regulated in LM3 cells (Figure 1G, Supplementary Figure 1C and 1D), suggesting that acRoots altered steroid metabolism in HCC cells.
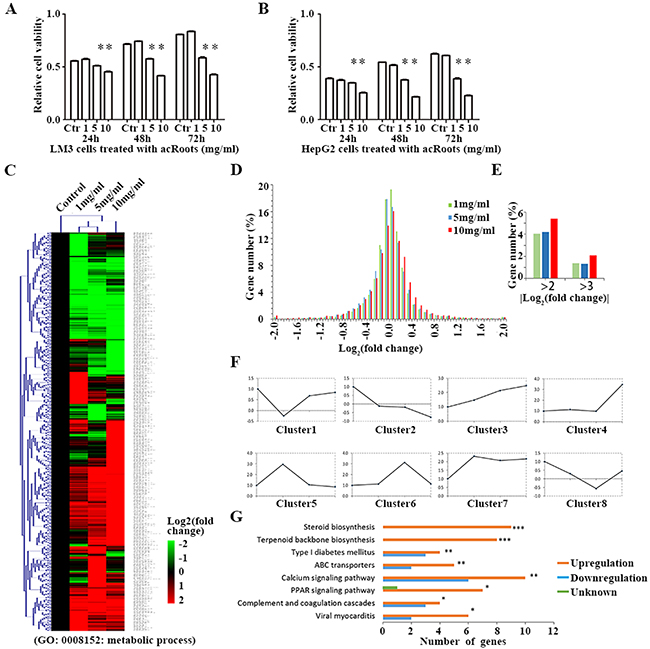
Figure 1: Metabolic gene profiling of LM3 cells treated with acRoots. A, B. Proliferation ability of LM3 and HepG2cells after treatment with acRoots; C. Hierarchical clustering of all metabolic genes regulated by acRoots in LM3 cells. The genes are annotated to the term “metabolic process” (GO: 0008152) or its children in Gene Ontology; D, E. Distribution map of mRNA changes induced by a concentration gradient of acRoots; F. Eight classes based on the similarity of the drug concentration profiles: clusters 2 and 3, corresponding to acRoots-induced up- or down-regulation of genes in a dose-dependent manner; G. Enrichment of KEGG pathways in the metabolic gene profiling analysis was determined using the DAVID software. Benjamini-Hochberg adjusted p values are shown for each indicated bar (***p < 0.0001, **p < 0.05, *p < 0.1).
Expression analysis of metabolic genes in LM3 cells
Of the eight clusters, clusters 2 and 3 (Supplementary Data sets 4 and 5) contained genes that were either up- or down-regulated by acRoots in a dose-dependent manner. We selected five genes from each of the two clusters that had a signal > 6.0 in the mRNA and the flag was the p value (representing a higher expression) to validate by quantitative reverse transcription PCR (qRT-PCR) (Figure 2 and Supplementary Figure 2). The results were consistent with those of the mRNA profiling, which validated the accuracy of the mRNA chip. Interestingly, PCSK9 was the most significantly up-regulated metabolic gene by acRoots in LM3 cells (Figure 2B and 2C).
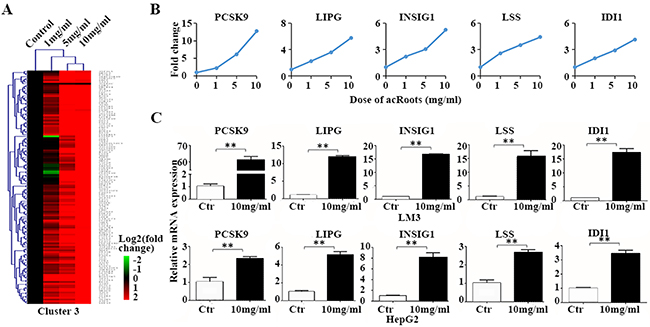
Figure 2: Metabolic genes up-regulated in LM3 cells by acRoots in a dose-dependent manner. A. acRoots dose profiles for cluster 3; B. Example genes from cluster 3: five genes for which the mRNA array signal was > 6.0 and the flag was the p value (representing a higher expression); C. Validation of the metabolic gene profiles for the example genes by qRT-PCR in LM3 and HepG2 cells treated with acRoots. Data represent the mean ± SD of triplicate samples; **p < 0.05.
Cholesterol signaling in LM3 cells treated with acRoots
PCSK9 is a secreted serine protease that regulates the post-transcriptional degradation of the low density lipoprotein (LDL) receptor (LDLR) [17] to reduce cholesterol uptake. To determine whether acRoots altered cholesterol metabolism in HCC cells in a PCSK9-dependent process, we analyzed the molecular network (www.string-db.org) and signaling pathways associated with PCSK9 based on previous reports (Figure 3A and 3B) [18]. The corresponding changes in the mRNA level are shown next to the gene symbols (Figure 3B). Molecular network analysis indicated that sterol regulatory element binding transcription factor 2 (SREBF2) and LDLR were the closest links to PCSK9. Signaling pathway analysis revealed that LDLR and 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), which are involved in cholesterol uptake and synthesis, respectively, were down-regulated in a dose-dependent manner, suggesting the possibility that acRoots inhibited cholesterol uptake and synthesis by altering LDLR and HMGCR expression in HCC.
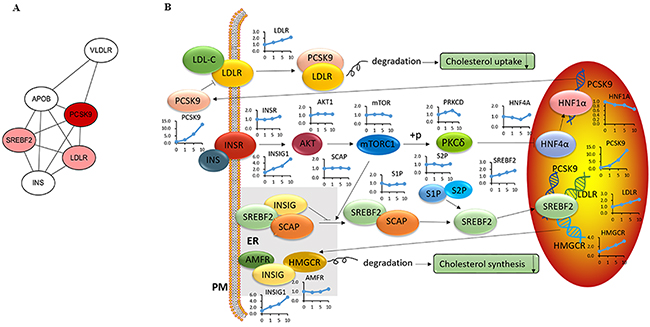
Figure 3: Modulation of the cholesterol signaling pathway in LM3 cells treated with acRoots. A. PCSK9 molecular network; Red represents a close relationship; B. PCSK9 signaling pathway and associated changes in mRNA levels assessed by microarray analysis.
acRoots promotes PCSK9 expression and inhibits cholesterol metabolism
To validate the results of the pathway analysis, we investigated the expression levels of the associated genes and cholesterol levels after treatment of HCC cells with acRoots. We determined that the level of PCSK9 mRNA increased in acRoots-treated compared to untreated cells in a dose- and time-dependent manner (Figure 4A). Additionally, acRoots induced PCSK9 expression in other HCC cell lines (Figure 4B). LDLR is a target of PCSK9, and previous studies have generally reported an inverse relationship with PCSK9. However, we did not observe a decrease in the LDLR mRNA level concomitant with up-regulation of PCSK9 expression in acRoots-treated LM3 cells (Figure 4C). Therefore, we examined whether acRoots could alter the protein levels of total LDLR and/or cell surface LDLR expression. As predicted, acRoots treatment attenuated total and cell surface LDLR expression in a dose-dependent manner, indicating that acRoots-regulated LDLR reduction at the post-transcriptional level (Figures 4D, 4E, and 4F). We analyzed whether acRoots altered cholesterol metabolism as expected based on the pathway analysis. We evaluated 3,3’-dioctadecylindocarbocyanine-labeled low-density lipoprotein (Dil-LDL) uptake and the total cellular cholesterol level in LM3 cells. These data indicated that acRoots attenuated cholesterol uptake and intra-cellular cholesterol levels in a dose-dependent manner (Figures 4G, 4H, 4I, and Supplementary Figure 3).
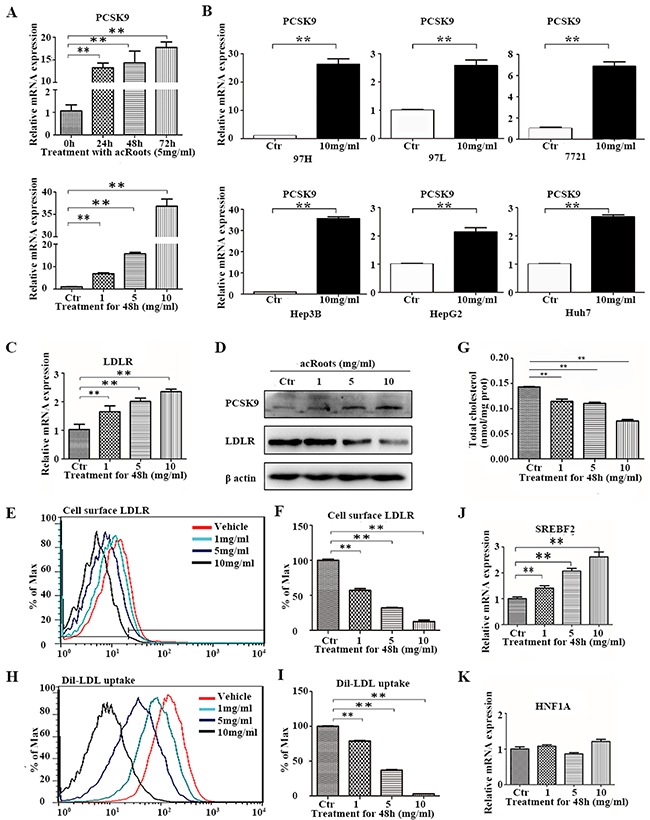
Figure 4: Alterations in PCSK9-related signaling pathways and cholesterol metabolism in LM3 cells in response to acRoots. A. Changes in PCSK9 mRNA in LM3 cells treated with different doses of acRoots at the indicated time points; B. Increased PCSK9 expression in other HCC cell lines treated with acRoots; C, D. Detection of LDLR mRNA and protein expression. Inverse relationship between PCSK9 and total LDLR protein in acRoots-treated LM3 cells quantified by western blotting; E, F. Levels of cell surface LDLR expression quantified by flow cytometry analysis; G, H. Dil-LDL uptake analyzed by flow cytometry; I. Attenuation of the intracellular cholesterol level in complete medium by acRoots; J, K. Changes in the mRNA levels of transcription factors that regulate PCSK9 expression. Data represent the mean ± SD of triplicate samples; **p < 0.05.
Signaling pathway analysis revealed a potential upstream regulator SREBF2 of acRoots-induced PCSK9 expression. Previous studies have also demonstrated that the SREBF2 and hepatocyte nuclear factor 1α (HNF1A) transcription factors bind to the PCSK9 promoter and promote PCSK9 transcription. Here, we showed that SREBF2 was up-regulated in a acRoots dose-dependent manner, suggesting that acRoots promoted PCSK9 expression by up-regulating SREBF2 rather than HNF1A (Figure 4J and 4K).
PCSK9-regulated signaling pathway promotes the effect of acRoots on HCC cells
To determine whether PCSK9 had a role in cholesterol metabolism and cell proliferation, we overexpressed PCSK9 in LM3 cells. LM3-PCSK9 cells exhibited > 100-fold higher levels of PCSK9 mRNA and protein (Figure 5A). Forty-eight hours after transfection, LM3 cells were treated with acRoots (5mg/ml) for another forty-eight hours, and then LDLR expression, Dil-LDL uptake, the total cholesterol concentration, and cell viability were assessed. We selected this concentration of acRoots because it was sufficient to induce PCSK9 expression without obvious toxicity (Figure 1A, 4A, and Supplementary Figure 1A). Interestingly, PCSK9 overexpression further decreased acRoots-mediated LDLR expression, Dil-LDL uptake, the total cholesterol concentration, and cell viability (Figure 5B, 5C, 5D and 5E).
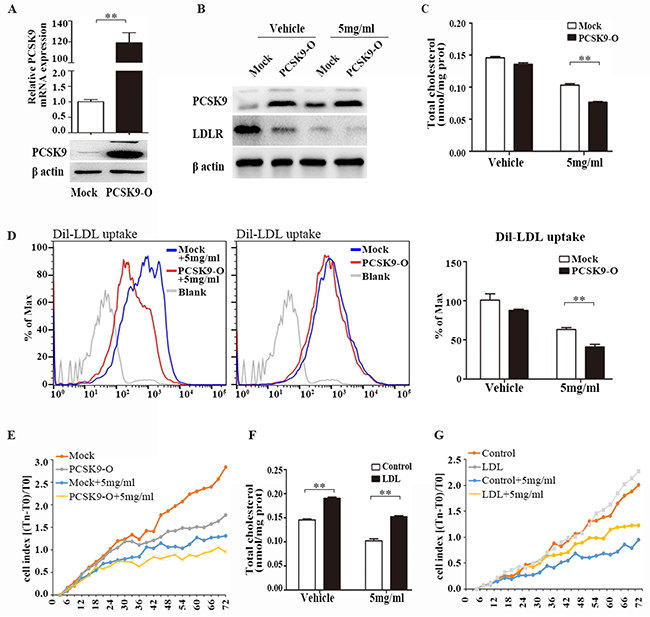
Figure 5: PCSK9 overexpression promotes the effect of acRoots. Overexpression of PCSK9 in LM3 cells confirmed by qRT-PCR and western blot A. Forty-eight hours after transfection, LM3 cells were treated with acRoots (5mg/ml) for another forty-eight hours, then LDLR proteins expression B. total cholesterol concentration C. Dil-LDL uptake D. and cell viability E. were analyzed; LDL at 200ug/ml increases intracellular cholesterol contents F. LDL significantly rescued acRoots-reduced cell viability G. Data represent the mean ± SD of triplicate samples; **p < 0.05.
Moreover, we detected the influence of cholesterol accumulation in acRoots-reduced cell viability by using human LDL, which containing 49% cholesterol, could increases intracellular cholesterol contents in HCC cells (Figure 5F). As shown in Figure 5G, LDL at 200ug/ml partially rescued acRoots-reduced cell viability (Figure 5G). These data suggested that PCSK9-regulated signaling pathway promotes the effect of acRoots on HCC cells.
PCSK9 knockdown inhibits acRoots regulation of cholesterol metabolism
We next investigated whether PCSK9 directly regulated cholesterol metabolism and cell proliferation in HCC cells in response to acRoots treatment, we knocked-down PCSK9 in LM3 cells. The efficacy of PCSK9 silencing by the selected siRNA was confirmed by real-time qRT-PCR (Figure 6A, 6B, and 6C). Forty-eight hours after transfection, LM3 cells were treated with acRoots (5 mg/mL) and LDLR expression, Dil-LDL uptake, total cellular cholesterol, and cell proliferation measured after 48 hours. As expected, the inhibition of total LDLR and surface LDLR protein expression by acRoots was significantly rescued by PCSK9-siRNA, while the LDLR mRNA level was still slightly up-regulated by acRoots (Figure 6D, 6E, 6F, and 6G). Additionally, knockdown of PCSK9 ameliorated the effects of acRoots on Dil-LDL uptake, cellular cholesterol levels, and cell proliferation (Figures 6H, 6I, 6J, and 6K).
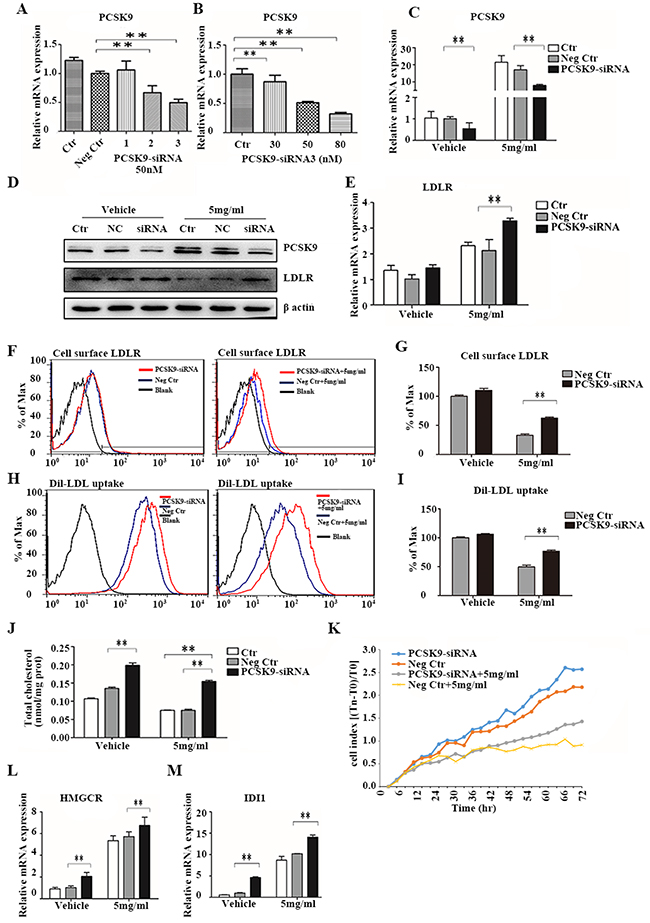
Figure 6: PCSK9 knockdown inhibits the effect of acRoots. A, B. Identification of efficient PCSK9-siRNA sequences and measurement of the relative PCSK9 concentration 48 hours after transfection using qRT-PCR; C-L. LM3 cells transfected with PCSK9-siRNA for 48 hours were treated with acRoots (5 mg/mL) for an additional 48 hours and PCSK9 mRNA and protein, total LDLR (C, D, E), and cell surface LDLR expression quantified (F, G), Dil-LDL uptake (H, I), total cellular cholesterol levels (J), cell proliferation (K), mRNA levels of HMGCR (L), and mRNA levels of IDI1 M. Data represent the mean ± SD of triplicate samples; **p < 0.05.
The intracellular cholesterol concentration depends on the rate of cholesterol uptake and synthesis. PCSK9 knockdown improved cellular cholesterol uptake resulting in an increase in the total cellular cholesterol level. We investigated whether PCSK9 knockdown also induced cholesterol synthesis. The mRNA levels of IDI1 and HMGCR, which are involved in cholesterol synthesis, were analyzed by qRT-PCR. These results indicated that PCSK9 knockdown resulted in an increase in IDI1 and HMGCR levels in both acRoots-treated and untreated LM3 cells (Figure 6L and 6M). Thus, silencing of PCSK9 increases cholesterol synthesis in HCC cells.
The cholesterol-lowering effect of acRoots is independent of INSIG1
INSIG1 expression was altered after treatment of LM3 cells with acRoots (Figure 2B and 3B). INSIG1 expression was up-regulated by acRoots in a dose- but not time-dependent manner (Figure 7A and 7B). INSIG1 expression was also increased in response to acRoots treatment in other HCC cell lines (Figure 7C, 7D, 7E, 7F, 7G, and 7H). The expression of INSIG1 is induced by insulin. INSIG1 encodes an endoplasmic reticulum membrane protein that regulates cholesterol metabolism. This protein may act as a scaffold to recruit the reductase that stimulates ubiquitination and proteasomal degradation of HMGCR. This results in inhibition of cholesterol synthesis because HMGCR is the rate-limiting enzyme of this pathway. We hypothesized that the cholesterol-lowering effect of acRoots could result from up-regulation of INSIG1. To test this hypothesis, we silenced INSIG1 in LM3 cells. The efficacy of siRNA-mediated silencing was confirmed by real-time qRT-PCR (Figure 7I and 7J). Forty-eight hours after transfection with the selected INSIG1-siRNA, LM3 cells were treated with acRoots (5 mg/mL) and the total intracellular cholesterol level analyzed after 48 hours (Figure 7K). These data indicated that INSIG1 knockdown did not rescue the cholesterol-lowering effect of acRoots.
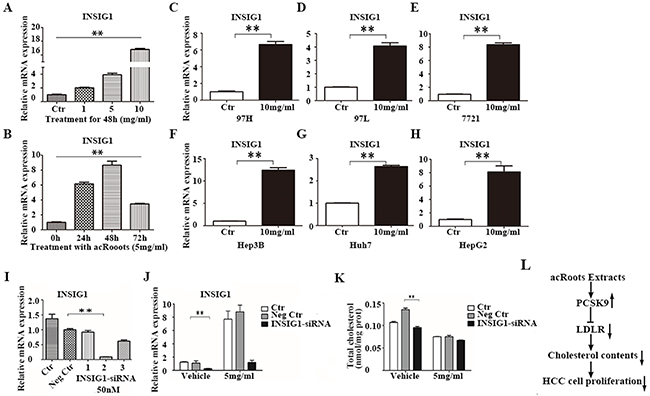
Figure 7: The cholesterol-lowering effect of acRoots is independent of INSIG1. A, B. Changes in PCSK9 mRNA levels in LM3 cells after treatment with the different doses of acRoots at the indicated time points; C-H. Increased INSIG1 expression in other HCC cell lines treated with acRoots; I. Identification of efficient INSIG1-siRNA sequences by qRT-PCR 48 hours after transfection; J. LM3 cells transfected with PCSK9-siRNA for 48 hours were treated with acRoots (5 mg/mL) for an additional 48 hours and the relative mRNA levels of INSIG1 quantified; K. LM3 cells transfected with PCSK9-siRNA for 48 hours were treated with acRoots (5 mg/mL) for an additional 48 hours to quantify the relative levels of total cellular cholesterol; L. Schematic representation of the working model. Data represent the mean ± SD of triplicate samples; **p < 0.05.
DISCUSSION
Metabolic alterations are a hallmark of cancer cells. The most prominent metabolic alterations are a switch to glycolysis in the presence of oxygen [19] and de novo lipid synthesis [20, 21]. These pathways produce the nutrients to support uncontrolled cancer cell proliferation. Although our understanding of the metabolic alterations that drive cancer progression has improved, there is still a lack of effective therapeutic strategies. The traditional Chinese medicine acRoots is widely used as crude drug to treat various cancers [22] including liver, lung, and breast cancer. However, the mechanisms underlying the anti-tumor effects of acRoots have not been elucidated. In this study, we demonstrated that acRoots reduces HCC cell viability, and inhibits cholesterol uptake and synthesis at a dose that is not toxic to normal liver cells. These data suggest that acRoots has anti-tumor efficacy.
Cholesterol has a critical role in multiple cellular functions including membrane synthesis, steroid synthesis, lipid raft formation, maintenance of membrane fluidity, and transmembrane signaling [23]. Intracellular cholesterol is generated by dietary uptake through the LDLR and from endogenous cholesterol biosynthesis [24]. In normal mammalian cells, cholesterol metabolism is maintained at a steady level through complex regulatory mechanisms. However, the strong requirement for cholesterol to support the growth and proliferation of malignant cells results in increased cholesterol synthesis and uptake [25, 26]. We demonstrated that acRoots inhibited cholesterol metabolism and proliferation in LM3 cells in a dose-dependent manner, suggesting that acRoots may suppress tumor growth by lowering cholesterol levels. KEGG pathway analysis also revealed that the mRNA levels of several metabolic genes involved in steroid synthesis and terpenoid backbone biosynthesis were up-regulated in response to acRoots treatment in a dose-dependent manner (Supplementary Figure 1C and 1D). These results suggest acRoots inhibits intracellular cholesterol levels through upregulation of PCSK9, which in turn induces a metabolic feedback loop in LM3 cells.
PCSK9 is a secreted serine protease that is involved in the post-transcriptional regulation of LDL (promotes intracellular degradation in acidic subcellular compartments) [27]. LDLR is the receptor for serum LDL. Therefore, PCSK9-induced degradation of LDLR results in reduced intracellular cholesterol uptake and increased serum LDL-cholesterol levels [28, 29]. Decreased PCSK9 expression and increased LDLR expression have been observed in HCC tissues, which may provide a constant supply of cholesterol in the HCC microenvironment [30]. Here, we demonstrated that acRoots promotes PCSK9 expression and inhibits LDLR expression at the post-translational level. PCSK9 knockdown rescued the cholesterol-lowering and anti-proliferative effects of acRoots, suggesting that PCSK9 has a key role in cholesterol metabolism and that PCSK9 and LDLR expression are inversely correlated.
SREBF2, a membrane-bound transcription factor, regulates cholesterol homeostasis [31]. SREBF2 is activated in response to reduced cholesterol levels and up-regulates the expression of genes responsible for cholesterol synthesis and uptake. SREBF2 activates PCSK9 and LDLR transcription through binding to functional sterol regulatory elements in the promoters of these genes [32]. HNF1A, a liver-enriched basal transcription factor, cooperates with SREBF2 to promote PCSK9 transcription [33]. Insulin stimulation of mammalian target of rapamycin complex 1 (mTORC1) was previously shown to initiate a signaling cascade that results in nuclear exclusion of HNF1A and decreased PCSK9 transcription. Conversely, inhibition of hepatic mTORC1 in the mouse liver by rapamycin or insulin receptor knockdown stimulates PCSK9 expression and reduces LDLR levels [18]. We observed no changes in the expression of proteins in the insulin/mTORC1/HNF1A pathway in LM3 cells after treatment with acRoots. However, we did observe dose-dependent upregulation of SREBF2 expression in response to acRoots treatment, suggesting that SREBF2 is the upstream transcription factor that regulates acRoots-induced expression of PCSK9.
INSIG1 is also up-regulated by acRoots. The INSIG1 protein recruits the reductase that promotes degradation of HMGCR and inhibits cholesterol synthesis [34]. In the present study, acRoots treatment resulted in decreased total cholesterol levels. Therefore, we hypothesized that the cholesterol-lowering effect of acRoots resulted from the inhibition of cholesterol uptake by PCSK9 and cholesterol synthesis by INSIG1. However, our data indicated that INSIG1 knockdown failed to rescue the cholesterol-lowering effect of acRoots.
acRoots extract consists of many compounds. According to previous reports, the inhibition of cell viability might be a result of multiple components, including triterpenes, flavonoids and phenolics [10, 11]. In the present study, we first demonstrated the role of acRoots in cholesterol metabolism. However, the concrete components involved in regulating cholesterol metabolism are still unknown. Further isolation and identification of the bioactive components would be conducted in our subsequent work.
In conclusion, our data indicate that acRoots significantly enhances PCSK9 expression, which decreases LDLR expression at the post-transcriptional level, inhibits LDL uptake, lowers the total intracellular cholesterol concentration, and suppresses cell viablity in LM3 cells (Figure 7L). Additionally, silencing of PCSK9, but not INSIG1, attenuates the cholesterol-lowering effect of acRoots, suggesting that PCSK9 has a key role in acRoots-regulated cholesterol metabolism in HCC cells. The effects of acRoots on cholesterol metabolism must be further analyzed in vivo to validate the anti-tumor efficacy.
MATERIALS AND METHODS
Cell lines and cell culture
The HCCLM3, MHCC97H, and MHCC97L cell lines were established at the Liver Cancer Institute of Fudan University. HepG2, SMMC-7721, Huh7, Hep3B, and HL-7702 cells were purchased from the Cell Bank of the Institute of Biochemistry and Cell Biology, China Academy of Sciences, Shanghai, China. Cells were cultured in DMEM with high glucose (Gibco BRL, NY, USA) supplemented with 10% fetal bovine serum (Gibco BRL, NY, USA). All cells were incubated at 37˚C in a humidified atmosphere containing 5% CO2.
Preparation of acRoots
Actinidia chinensis Planch roots were chopped and immersed in ten volumes of double distilled water. The mixture was then heated to 100˚C and incubated for 1 hour. Finally, the decoction was filtered. The drug was obtained after two cycles of this procedure and was concentrated to generate a 1 g/mL working solution.
Cell viability and proliferation assays
HCC cell viability was assessed using CCK-8 assays (Cell Counting Kit-8, Dojindo, Japan). Briefly, 4,000 cells were seeded in 96-well culture plates. The cells were allowed to attach for 6 hours. We then added 100 mL CCK-8 assay buffer to each well after 0, 24, 48, and 72 hours. The optical density (450 nm) was measured in each well using a microplate reader (BIO-TEK, Winooski, VT, USA) according to the manufacturer’s instructions. Three independent experiments were performed.
Changes in cell proliferation were recorded using Cell-IQ, an integrated, fully automated system (Chip-Man Technologies Ltd., Finland) as described previously [35]. LM3 cells were plated onto 24-well plates at an appropriate density. The system automatically selected three or four visual fields in each well and recorded images every 30 minutes for 4 days. The proliferation rate was analyzed with the Cell-IQ Analyser Pro-Write V.AN 2.3.0 (Chip-Man Technologies Ltd.).
Gene expression profiling
LM3 cells were treated with acRoots (0, 1, 5, and 10 mg/mL concentrations) for 72 hours and total RNA extracted using the TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). Total RNA was amplified and labeled using the One-color Low Input Quick Amp Labeling Kit (Agilent Technologies, Santa Clara, CA, USA), according to the manufacturer’s instructions. Labeled cRNA was purified using the RNeasy Mini Kit (Qiagen, Hilden, Germany). We then hybridized 1.65 μg Cy3-labeled cRNA to the arrays using the Agilent Gene Expression Hybridization Kit (Agilent) in a hybridization oven (Agilent) for 17 hours. Following the incubation, the slides were washed in staining dishes (Thermo Scientific, Waltham, MA, USA) with the Gene Expression Wash Buffer (Agilent) and scanned on an Agilent microarray scanner using the default settings. Data were extracted using the Feature Extraction software version 10.7 (Agilent). Raw data were normalized using a quantile algorithm and the Gene Spring software version 11.0 (Agilent).
Gene set annotation by metabolic process
In order to identify all metabolic genes regulated by acRoots in LM3 cells, we developed a filtering strategy based on the following criteria: a) All metabolism-related genes must have been annotated to the term “metabolic process” (GO: 0008152) or its children in Gene Ontology (http://amigo1.geneontology.org/) (Supplementary Data set 1 and 2); and b) The metabolic genes regulated by acRoots in LM3 cells were required to exhibit at least a two-fold change in response to any concentration of acRoots relative to the untreated control group (Supplementary Data set 3). A correlational formula (VLOOKUP) was used to correlate the data from Supplementary Data set 1 with that of Supplementary Data set 4 and identify all metabolic genes regulated by acRoots in LM3 cells (Supplementary Data set 3).
Pathway analysis
The DAVID software was used to determine KEGG pathway enrichment. All metabolic genes regulated by acRoots in LM3 cells were queried in the DAVID database. Pathway information was downloaded from the KEGG database (Supplementary Data set 6).
RNA isolation and real-time qRT-PCR analysis
Total RNA was isolated from LM3 cells with the TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. Complementary DNA was synthesized from 500 ng RNA using the Reverse Transcription Reagent Kit (TaKaRa, Shiga, Japan). All primers are shown in Supplementary Table 1. Real-time qRT-PCR analysis was performed using an Applied Biosystems 7500 Real-Time PCR System. Values were derived from at least three independent experiments performed in duplicate and normalized to β-actin expression. The2-△△Ct method was used for the data analysis [36].
Western blot analysis
Western blotting was performed as described [37]. The primary antibodies (anti-PCSK9, and anti-LDLR) were purchased from Abcam (Cambridge, MA, USA). The anti-β-actin antibody was obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). The secondary antibodies were either anti-mouse or anti-rabbit HRP-linked. Blots were developed using the Pierce ECL Western Blotting Substrate (Pierce Biotechnology, IL, USA).
Cell transfection
Gene silencing was performed by transfection LM3 cells with siRNA oligonucleotides (GenePharma, Shanghai, China). All siRNA sequences are shown in Supplementary Table 2. For transfections in 12-well plates, 1.0 × 105 cells were seeded per well and the cells transfected using the siPORTTMNeoFXTM Transfection Agent (Invitrogen) according to the manufacturer’s protocol. Efficient siRNA oligonucleotides were detected by qRT-PCR. PCSK9 overexpression in LM3 cells was performed by transfecting the cells with the indicated PCSK9 constructs (GenePharma) using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Triplicate samples were analyzed in all experiments and all experiments were repeated at least three times.
Flow cytometry
Cell surface LDLR was detected by flow cytometry. Following treatment, LM3 cells were detached from the plates by scraping and washed with phosphate-buffered saline (PBS) containing 5% bovine serum albumin for 30 minutes at room temperature. Cells were then washed with PBS, incubated with an anti-LDLR antibody (Abcam) at room temperature for 1 hour, and then incubated with an Alexa Fluor 488-conjugated goat anti-rabbit IgG secondary antibody (Abcam) for 30 minutes at room temperature. Background fluorescence (control) was quantified using cells incubated with an isotype rabbit IgG antibody (Abcam) for 1 hour and Alexa Fluor 488-conjugated goat anti-rabbit IgG for 30 minutes. The cells were washed and resuspended in PBS, and then analyzed by flow cytometry using an FL1 emission filter (FACScan, BD Biosciences, San Jose, CA, USA). The data were analyzed using the FlowJo software.
Dil-LDL (Biomedical Technologies Inc., USA) uptake was representative of total cellular cholesterol uptake. Following treatment of LM3 cells, the media was exchanged to serum-free and incubated with 10 ug/mL Dil-LDL at 37˚C for 12 hours. The cells were detached with trypsin, washed, and the resuspended in PBS for flow cytometry analysis using an FL2 emission filter (FACScan, BD Biosciences, San Jose, CA, USA). Data were analyzed using the FlowJo software (CA, USA).
LDL preparation
LDL was purchased from AngYu Biotechnologies (Shanghai, China). It is isolated from blood bank produced human plasma. It is purified via ultracentrifugation to homogeneity determined by agarose gel electrophoresis. Each lot is analyzed on agarose gel electrophoresis for migration versus LDL.
Measurement of total cellular cholesterol
Total cellular cholesterol was measured with the Cholesterol Quantitation Kit from Applygen (Peking, China) according to the manufacturer’s protocol. The activities of cholesterol esterase and cholesterol oxidase, and the amount of H2O2 produced were quantified using this colorimetric assay. The values were normalized against the total protein concentration in each sample, which was estimated using Bicinchoninic Acid assays (Beyotime Biotechnology, China).
Statistical analysis
Statistical analysis was performed using the SPSS16.0 statistical software (SPSS Inc, Chicago, IL, USA). Student’s t-tests were used to assess significant differences between the study groups. The level of significance was p < 0.05. All experiments were performed at least three times.
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.
FUNDING
This study was sponsored by grants from the National Natural Science Foundation of China (Nos. 81272732 and 81572395), the Shanghai Leading Talent Projects (No. 048, 2013), the Shanghai Leading Academic Discipline Project (Project Number: B115), and the Shanghai Outstanding Academic Leaders (Project Number: 14XD1401100).
REFERENCES
1. Venook AP, Papandreou C, Furuse J, de Guevara LL. The incidence and epidemiology of hepatocellular carcinoma: a global and regional perspective. Oncologist. 2010; 15:5-13.
2. Singh S, Singh PP, Roberts LR, Sanchez W. Chemopreventive strategies in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2014; 11:45-54.
3. Wang FS, Fan JG, Zhang Z, Gao B, Wang HY. The global burden of liver disease: the major impact of China. Hepatology. 2014; 60:2099-2108.
4. Howlader N, Noone A, Krapcho M, Garshell J, Miller D, Altekruse S, Kosary C, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, et al. SEER Cancer Statistics Review, 1975-2011, National Cancer Institute https://seer.cancer.gov/publications/csr.html.
5. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, Luo R, Feng J, Ye S, Yang TS, Xu J, Sun Y, Liang H, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009; 10:25-34.
6. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008; 359:378-390.
7. Zhu WJ, Yu DH, Zhao M, Lin MG, Lu Q, Wang QW, Guan YY, Li GX, Luan X, Yang YF, Qin XM, Fang C, Yang GH, Chen HZ. Antiangiogenic triterpenes isolated from Chinese herbal medicine Actinidia chinensis Planch. Anticancer Agents Med Chem. 2013; 13:195-198.
8. Cheng QL, Li HL, Huang ZQ, Chen YJ, Liu TS. 2beta, 3beta, 23-trihydroxy-urs-12-ene-28-olic acid (TUA) isolated from Actinidia chinensis Radix inhibits NCI-H460 cell proliferation by decreasing NF-kappaB expression. Chem Biol Interact. 2015; 240:1-11.
9. Ku C, Wang Y, Lin H, Lu S, Lin J. Corosolic acid inhibits hepatocellular carcinoma cell migration by targeting the VEGFR2/Src/FAK pathway. PLoS One. 2015; 10:e126725. 2015; 10:e126725.
10. Chang J, Case R. Cytotoxic phenolic constituents from the root of Actinidia chinensis. Planta Med. 2005; 71:955-959.
11. Zhao T, He J, Wang X, Ma B, Wang X, Zhang L, Li P, Liu N, Lu J, Zhang X. Rapid detection and characterization of major phenolic compounds in Radix Actinidia chinensis Planch by ultra-performance liquid chromatography tandem mass spectrometry. J Pharm Biomed Anal. 2014; 98:311-320.
12. Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. 2014; 15:243-256.
13. Huang Q, Tan Y, Yin P, Ye G, Gao P, Lu X, Wang H, Xu G. Metabolic characterization of hepatocellular carcinoma using nontargeted tissue metabolomics. Cancer Res. 2013; 73:4992-5002.
14. Li S, Jin S, Song C, Chen C, Zhang Y, Xiang Y, Xu Y, Feng Y, Wan Q, Jiang H. The metabolic change of serum lysophosphatidylcholines involved in the lipid lowering effect of triterpenes from Alismatis rhizoma on high-fat diet induced hyperlipidemia mice. J Ethnopharmacol. 2016; 177:10-18.
15. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44-57.
16. Dennis GJ, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003; 4:P3.
17. Lopez D. PCSK9: an enigmatic protease. Biochim Biophys Acta. 2008; 1781:184-191.
18. Ai D, Chen C, Han S, Ganda A, Murphy AJ, Haeusler R, Thorp E, Accili D, Horton JD, Tall AR. Regulation of hepatic LDL receptors by mTORC1 and PCSK9 in mice. J Clin Invest. 2012; 122:1262-1270.
19. WARBURG O. On the origin of cancer cells. Science. 1956; 123:309-314.
20. Nelson ER, Chang CY, McDonnell DP. Cholesterol and breast cancer pathophysiology. Trends Endocrinol Metab. 2014; 25:649-655.
21. Lu M1, Hu XH, Li Q, Xiong Y, Hu GJ, Xu JJ, Zhao XN, Wei XX, Chang CC, Liu YK, Nan FJ, Li J, Chang TY, et al. A specific cholesterol metabolic pathway is established in a subset of HCCs for tumor growth. J Mol Cell Biol. 2013.
22. Zuo LL, Wang ZY, Fan ZL, Tian SQ, Liu JR. Evaluation of Antioxidant and Antiproliferative Properties of Three Actinidia (Actinidia kolomikta, Actinidia arguta, Actinidia chinensis) Extracts in Vitro. Int J Mol Sci. 2012; 13:5506-5518.
23. Nelson ER, Chang C, McDonnell DP. Cholesterol and breast cancer pathophysiology. Trends Endocrinol Metab. 2014; 25:649-655.
24. Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006; 22:129-157.
25. de Boussac H, Pommier AJ, Dufour J, Trousson A, Caira F, Volle DH, Baron S, Lobaccaro JM. LXR, prostate cancer and cholesterol: the good, the bad and the ugly. Am J Cancer Res. 2013; 3:58-69.
26. Danilo C, Frank PG. Cholesterol and breast cancer development. Curr Opin Pharmacol. 2012; 12:677-682.
27. Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, Cohen JC, Hobbs HH. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat a of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. 2007; 282:18602-18612.
28. Park SW, Moon YA, Horton JD. Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J Biol Chem. 2004; 279:50630-50638.
29. Lagace TA, Curtis DE, Garuti R, McNutt MC, Park SW, Prather HB, Anderson NN, Ho YK, Hammer RE, Horton JD. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. 2006; 116:2995-3005.
30. Bhat M, Skill N, Marcus V, Deschenes M, Tan X, Bouteaud J, Negi S, Awan Z, Aikin R, Kwan J, Amre R, Tabaries S, Hassanain M, et al. Decreased PCSK9 expression in human hepatocellular carcinoma. Bmc Gastroenterol. 2015; 15:176.
31. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002; 109:1125-1131.
32. Dong B, Wu M, Li H, Kraemer FB, Adeli K, Seidah NG, Park SW, Liu J. Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J Lipid Res. 2010; 51:1486-1495.
33. Li H, Dong B, Park SW, Lee HS, Chen W, Liu J. Hepatocyte nuclear factor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J Biol Chem. 2009; 284:28885-28895.
34. Jo Y, Debose-Boyd RA. Control of cholesterol synthesis through regulated ER-associated degradation of HMG CoA reductase. Crit Rev Biochem Mol Biol. 2010; 45:185-198.
35. Fang X, Li K, Tao X, Chen C, Wang X, Wang L, Wang DC, Zhang Y, Bai C, Wang X. Effects of phosphoinositide 3-kinase on protease-induced acute and chronic lung inflammation, remodeling, and emphysema in rats. Chest. 2013; 143:1025-1035.
36. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402-408.
37. Dong Y, Xie X, Wang Z, Hu C, Zheng Q, Wang Y, Chen R, Xue T, Chen J, Gao D, Wu W, Ren Z, Cui J. Increasing matrix stiffness upregulates vascular endothelial growth factor expression in hepatocellular carcinoma cells mediated by integrin beta1. Biochem Biophys Res Commun. 2014; 444:427-432.