
INTRODUCTION
Breast cancer (BC) is the most common cancer among women and one of the leading causes of cancer-related deaths worldwide [1–3]. It is a clinically heterogeneous disease, with at least five subtypes, according to the St Gallen International Expert Consensus in 2013 [4]: luminal A-like, luminal B-like/HER2-negative, luminal B-like/HER2-positive, HER2-positive (non-luminal) and triple-negative. Although BC incidence remains high, an increase in overall survival (OS) has been attributed to advances in early detection programmes and therapeutic approaches directed against molecular biomarkers, such as hormone receptors and HER2, which are overexpressed and amplified in luminal and HER2 subtypes, respectively. From the therapeutic point of view, their cell-signalling transduction abilities have been successfully abolished by administration of tamoxifen and trastuzumab, respectively [5–7]. Nevertheless, BC prognosis is quite variable and approximately 20-30% of early-stage cases will eventually experience recurrence and develop distant metastasis. Currently, however, there is no acceptable method for monitoring patients who are likely to progress [8]. BC is thought to result from the presence of certain abnormal genetic and epigenetic changes in tumour suppressor genes, such as TP53 or BRCA1, and proto-oncogenes, like members of the PI3K signalling pathway, among others [4, 9]. A thorough understanding of the mechanisms responsible for BC development and progression is still needed to identify prognostic biomarkers.
Gene expression-based approaches have added significant prognostic and predictive value to pathological staging, histological grade and standard molecular marker identification [10]. However, the high cost of expression profiling and the molecular instability of mRNA have limited its clinical use, so expression-based BC classification has not become a routine method [11]. In fact, the most recent consensus [4] agreed a BC classification based on the expression of various immunohistochemical markers (presence or absence of oestrogen, progesterone and HER2 receptors and Ki67). Thus, although distinguishing BC subtypes by immunohistochemical markers is considered the gold standard, there is an urgent need to identify new and well-defined prognostic biomarkers to stratify BC patients with good and poor prognosis [12].
Epigenetic alterations are common molecular abnormalities in cancer, including DNA methylation, alterations in microRNA profiling and post-translational modifications of histones [13]. Over the past decade, aberrant DNA methylation has been recognised as one of the most common molecular abnormalities in BC [14, 15]. Methylation of certain genes has been related to clinical and pathological characteristics of breast tumours, and is considered a biomarker of diagnosis [16], hormone receptor [17] and HER2 [18] status, response to tamoxifen [17] and chemotherapy [19], metastases during follow-up [14], and has demonstrated its value as a predictor of survival [17, 20].
The CHL1 gene (Close Homolog of L1, also known as L1CAM2, Entrez Gene accession number 10752) encodes a member of the L1 family of neural cell adhesion molecules essential for the brain development and involved in signal transduction pathways. Some of these proteins, such as L1CAM, are expressed in a wide range of tissues in addition to the brain, and are known to play an important role in carcinogenesis and progression in a variety of human cancers by overexpression and association with poor prognosis [2, 21]. Interestingly, L1CAM upregulation promotes cell adhesion and migration and is associated with shorter progression-free survival (PFS) and overall survival (OS) in BC [22–24].
However, very few studies have focused on the role of CHL1 in cancer [2, 21]. There is weak evidence that CHL1 expression is downregulated at the mRNA level in BC tissues relative to non-cancerous breast tissues [21], but nothing is known about the causes of this silencing. The biological role of CHL1 in BC has been reported in only a single study, in which, in addition to confirming CHL1 downregulation at the mRNA and protein levels in BC tissues and cell lines, the authors found that overexpression of CHL1 impaired cell proliferation and invasion, while CHL1 depletion caused the opposite effect in vitro, and promoted tumour formation in vivo [2]. Nevertheless, the clinical value of CHL1 silencing in human tissues as a potential biomarker of prognosis remains to be elucidated. The aim of this study was to determine the mechanisms and clinical implications of CHL1 downregulation in BC.
RESULTS
CHL1 hypermethylation is present in BC
To determine the methylation status of the CHL1 gene, three CpG sites in its promoter were pyrosequenced in a series of 142 breast tumours, 45 paired tumour and adjacent-to-tumour tissues, and 19 non-neoplastic breast tissues from reduction mammoplasties (Supplementary Figure 1). Since pyrosequencing provides a quantitative measure of methylation, the optimal cut-off value distinguishing statistically between the unmethylated and methylated status of each of the CpG sites was estimated by ROC curve analysis: 17.5% methylation for CpG1, 4.5% methylation for CpG2, and 9.5% for CpG3 (Table 1). We also considered that a case had hypermethylated CHL1 when the three tested CpG sites simultaneously showed methylation percentages above their cut-off values. In contrast, non-neoplastic breast samples displayed very low percentages of methylation (< 11%) (Figure 1). Importantly, non-neoplastic adjacent-to-tumour tissues harboured significantly lower methylation levels in all CpG sites than tumour tissues, but slightly higher levels than those of non-neoplastic tissues (Figure 1). Interestingly, this epigenetic alteration was maintained across all BC subtypes (Supplementary Figure 2).
Table 1: Methylation status of CHL1 in breast samples
|
CpG1 |
CpG2 |
CpG3 |
All CpGs |
|---|---|---|---|---|
Median % methylation in breast tumours (range) |
18 |
5 |
5 |
|
Median % methylation in adjacent-to-tumour tissues (range) |
6 |
1 |
1 |
|
Median % methylation in non-neoplastic breast samples (range) |
5 |
0 |
0 |
|
Cut-off value (related to PFS) |
17.5 |
4.5 |
9.5 |
Above cut-off in all CpGs |
PFS: progression-free survival.
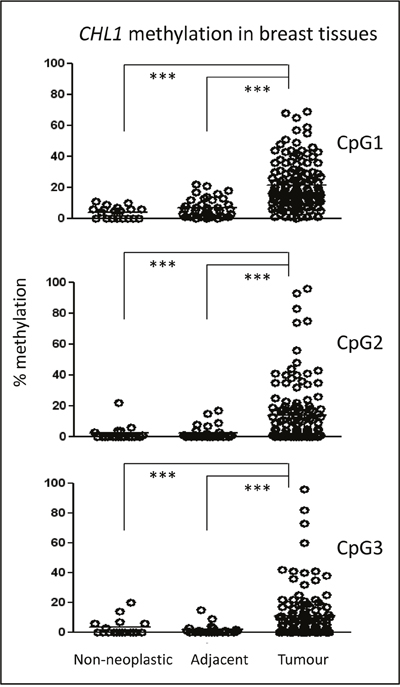
Figure 1: Epigenetic status of CHL1 in BC patients. The methylation of three CpG sites in the CHL1 gene promoter was interrogated by pyrosequencing in a series of 142 BC cases, 45 adjacent-to-tumour tissues, and 19 non-neoplastic mammary tissues from reduction mammoplasties. The horizontal lines in each group represent the median of the series. (***, p < 0.001).
These results indicate, for the first time, that a subset of breast tumours has higher levels of CHL1 gene methylation than do adjacent-to-tumour tissues and non-neoplastic samples.
CHL1 protein expression pattern in mammary tissues
Since DNA methylation is a well-known mechanism of gene expression regulation, the expression pattern of the CHL1 protein was measured by immunohistochemistry in 57 BC tissues, their adjacent-to-tumour counterparts and 20 non-neoplastic tissues from reduction mammoplasties. We found a significantly higher level of expression in both types of non-neoplastic cells relative to tumour cells, being slightly lower in adjacent-to-tumour than in non-neoplastic tissue (Figure 2 and Supplementary Figure 3A). Although the predicted location of CHL1 protein is the cell membrane, the pattern of expression was cytoplasmic without nuclear or membrane expression (Supplementary Figure 3B), even when using two different antibodies (data not shown). Furthermore, the same cytoplasmic pattern with a lack of membrane staining was observed by immunofluorescence in CHL1-expressing immortalized but non-neoplastic mammary cells (Supplementary Figure 3C).
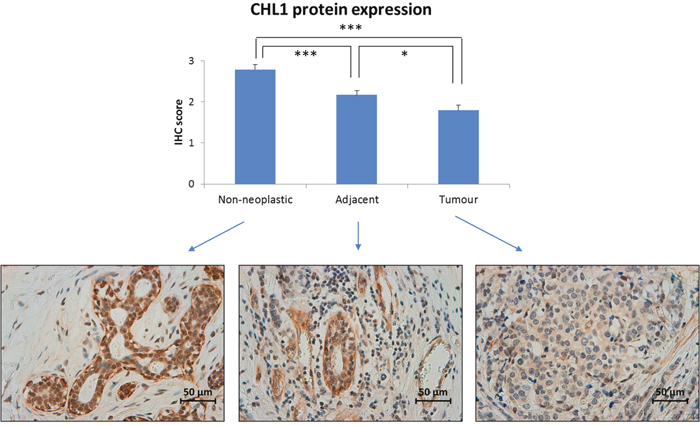
Figure 2: CHL1 protein expression in BC. Immunohistochemistry was employed to measure CHL1 expression in 57 paired breast tumour and adjacent-to-tumour samples, along with 20 non-neoplastic tissues from reduction mammoplasties. It was scored as: 0, no expression; 1: weak expression; 2: intermediate expression; and 3: strong expression (***, p < 0.001; *, p < 0.05). Images were acquired at 400x magnification using a Leica DMD 108 digital microscope (Leica, Wetzlar, Germany).
These results are consistent with the epigenetic pattern we observed: breast tumours, with higher methylation levels, displayed a lower level of protein expression than did non-neoplastic tissues.
CHL1 expression can be modulated by epigenetic drugs in BC cell lines
The methylation status of the CpG1 site in the CHL1 gene promoter that had been examined by pyrosequencing in BC samples was analysed in a panel of four BC cell lines and one immortalized but non-neoplastic mammary cell line. We found that the majority of BC cell lines had higher levels of methylation overall than non-neoplastic HBL-100 cells. Accordingly, mRNA levels of CHL1 were lower in BC cell lines than in non-neoplastic HBL-100 cells, as assessed by qRT-PCR. In fact, we observed a strong and significant correlation between CHL1 methylation and expression (Spearman’s correlation coefficient = - 0.9; p = 0.037) (Figure 3A), suggesting that CHL1 expression may also be regulated by methylation in vitro in BC.
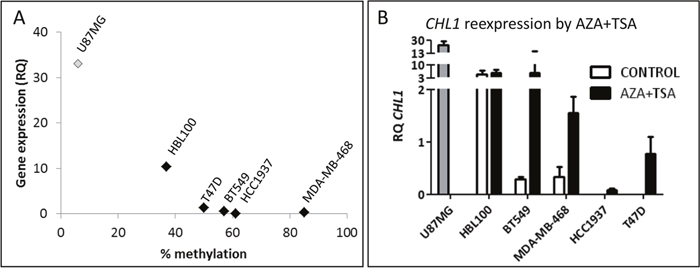
Figure 3: Molecular status of CHL1 in BC cell lines. A. Correlation between CHL1 hypermethylation in the CpG1 site and expression in BC cell lines (Spearman’s correlation coefficient = -0.9; p = 0.037), measured by pyrosequencing and qRT-PCR, respectively. U-87 MG cells were used as a positive control for CHL1 expression, but were not included in the correlation analysis (RQ, relative quantification). B. Restoration of CHL1 expression in BC cell lines by treatment with 4 μM 5-aza-dC for 72 h and 300 nM TSA for 24 h (AZA+TSA), measured by qRT-PCR.
In order to test whether CHL1 expression can be modulated by epigenetic mechanisms, all cell lines were treated with two epigenetic drugs. We found by qRT-PCR that AZA+TSA treatment restored CHL1 expression in all BC cell lines (Figure 3B), while single treatment was not as effective as the drug combination (data not shown).
These results indicate that the hypermethylation contributes to the regulation of CHL1 expression in BC, and that it can be dynamically modulated by in vitro epigenetic treatments.
CHL1 silencing promotes cell proliferation and migration of non-neoplastic mammary cells
To determine the effect of CHL1 silencing in BC, we inhibited CHL1 expression in the only mammary tissue-derived cell line expressing high levels of CHL1: the immortalized but non-neoplastic HBL-100 cell line. To this end, we inserted two shRNAs against CHL1 and one scramble shRNA into the pHIV1-SIREN-PuroR plasmid, and lentiviruses were produced upon transfection in 293T cells. HBL-100 cells were then transduced and selected with puromycin. Western blot showed that the shCHL1_1 was more efficient at depleting CHL1 protein than shCHL1_2 (Figure 4A). Concomitantly, shCHL1_1, but not shCHL1_2, significantly enhanced HBL-100 cell proliferation (Figure 4B) and migration (Figure 4C), but not invasion (Figure 4D).
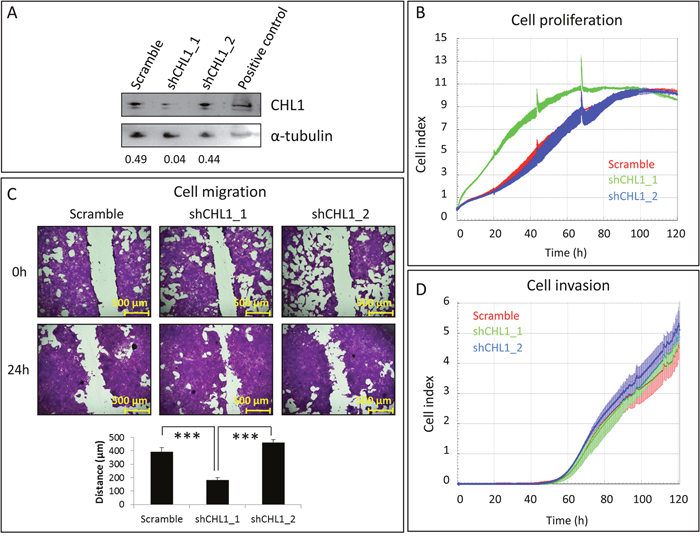
Figure 4: Effects of CHL1 silencing on immortalized but non-neoplastic mammary cells. A. HBL-100 cells were transduced with pHIV1-SIREN+scramble, pHIV1-SIREN+shCHL1_1, or pHIV1-SIREN+shCHL1_2 and selected with puromycin for 11 days. CHL1 silencing efficiency was checked by western blot, using α-tubulin as a loading control. Numbers indicate the amount of CHL1 relative to that of α-tubulin, as measured by densitometry. B. Cell proliferation was measured by RTCA for 5 additional days upon CHL1 silencing. C. Effects of CHL1 knockdown on cell migration were measured for 24 h. Images were acquired at 50x magnification with NIS-Elements, using an Olympus BX51 microscope (Olympus, Tokyo, Japan). D. Cell invasion was measured by RTCA for 3 additional days after CHL1 silencing.
These observations indicate that CHL1 silencing could be important for in vitro breast tumour cell growth.
CHL1 hypermethylation predicts BC progression
Finally, we aimed to examine the clinical value of CHL1 hypermethylation in our series of 142 BC patients (Supplementary Table 1). Using the cut-off values of CHL1 methylation mentioned above, we found that the methylation status of the CHL1 gene was very significantly associated with shorter PFS (p < 0.001) (Figure 5), but not with OS (data not shown). We confirmed in our series that other well-known prognostic factors, such as lymph node involvement, histological grade and stage, substantially contributed to a shorter PFS (Supplementary Figure 4). Therefore, the independent impact of CHL1 hypermethylation on progression, regardless of those important clinical variables, was tested in a Cox regression model. Importantly, we found that methylation in all the studied CpG sites in the CHL1 promoter was still very significantly associated with poor PFS (p = 0.001), irrespective of age and stage (Table 2). The other clinical parameters significantly correlated with PFS (histological grade and lymph node involvement) were not included in the Cox regression model due to their association with the stage (p < 0.001). We also observed that the methylated status of all tested CpG sites of CHL1 promoter displayed a hazard ratio of 5 (Table 2).
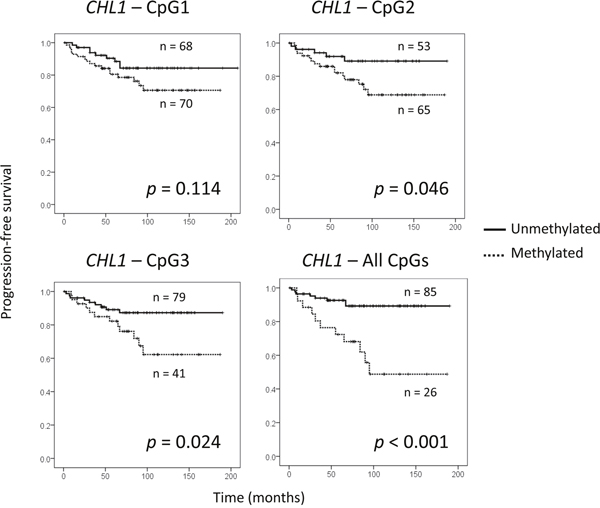
Figure 5: Prognostic value of CHL1 hypermethylation in a series of 142 BC patients. (A) Association between shorter periods of progression-free survival and CHL1 hypermethylation in each of the three CpG sites analysed, and in all of them simultaneously. Cut-off values for hypermethylation were calculated by ROC analysis: 17.5% for CpG1; 4.5% for CpG2; 9.5% for CpG3.
Table 2: CHL1 hypermethylation as an independent prognostic factor
Variable |
Hazard ratio |
p-value |
|---|---|---|
Age |
1.012 |
0.586 |
Stage |
2.406 |
0.118 |
CHL1 hypermethylation – all CpGs |
5.061 |
0.001 |
Cox regression model shows the independent effect of each prognostic factor on progression-free survival (CI, confidence interval).
These results suggest that CHL1 hypermethylation is of independent value as a predictor of shorter PFS in BC.
DISCUSSION
The CHL1 gene has been described as being downregulated in BC tissues with biological effects on cell proliferation in both in vitro and in vivo BC models [2, 21]. However, the mechanisms underlying CHL1 silencing and its potential clinical role have not previously been explored. This gene is located on the short arm of chromosome 3 (band 3p26), a commonly deleted region in malignant peripheral nerve sheath tumours [25], nasopharyngeal carcinomas [26] and oral squamous cell carcinomas, in which the loss of this region is of prognostic value [27]. In BC, besides being deleted, this region has also been reported to harbour candidate tumour suppressor genes [21]. Deletions in one allele are usually accompanied by hypermethylation of the other [28]. In this study, we show for the first time that the CHL1 gene, located in this region, is silenced in a subset of invasive BC cases due to promoter hypermethylation, but not in adjacent-to-tumour tissue and non-neoplastic tissue from mammoplasties. This epigenetic alteration has been found by pyrosequencing, a technique that yields quantitative measurements of methylation in contrast to methylation-specific PCR [29–33]. Consistent with the observed hypermethylation, we have found that tumour cells in BC tissues have a lower level of CHL1 expression compared with non-neoplastic adjacent-to-tumour cells. Importantly, this is the first report of the immunohistochemical pattern of CHL1 expression in invasive BC.
The biological role of CHL1 silencing in BC was analysed in the only mammary tissue-derived cell line expressing high levels of CHL1 mRNA available to us: an immortalized but non-neoplastic mammary cell line, HBL-100. All BC cell lines displayed very low CHL1 expression, as previously described [2]. We also demonstrated that CHL1 hypermethylation can be reversed by epigenetic treatment, since demethylating agents and histone deacetylase inhibitors modulated dynamics of CHL1 expression in vitro and restored its silenced status in BC-derived cell lines. By reducing CHL1 protein levels with shRNA, a dramatic increase in non-neoplastic HBL-100 cell proliferation and migration was found (around 2-fold), but not in cell invasion, suggesting that CHL1 might act as a tumour suppressor gene in the early stages of BC development. Interestingly, the expression of L1CAM, another neural cell adhesion molecule from the same family as CHL1, has been described to promote BC cell adhesion and migration in vitro, while cell invasion was also unaffected [24]. Our results are consistent with the only study to date to demonstrate the biological role of CHL1 in BC [2], in which tumour cell proliferation and invasion were suppressed and stimulated by overexpression and depletion of CHL1, respectively, due to its interaction with the cytoskeleton [22]. The new in vitro finding observed in our study is that CHL1 knockdown can also affect non-neoplastic cell proliferation, other than tumour cell spreading, as reported by He et al [2]. It has been proposed that during initial growth, CHL1 is silenced in tumour cells to facilitate in situ tumour growth, acting as a tumour suppressor gene; CHL1 is then re-expressed on the edge of the tumour mass and around tumour vessels to promote migration and local invasive growth, and acts as an oncogene to initiate the metastatic process [21].
The clinical role of CHL1 hypermethylation in invasive BC has also been studied here for the first time. In this context, a cut-off value of CHL1 hypermethylation has been established to stratify unmethylated and methylated cases, as seen with MGMT hypermethylation, which has been useful for predicting both PFS and OS in glioblastoma [34]. Importantly, we observed that CHL1 hypermethylation was very significantly associated with shorter PFS in our large series of BC patients. Accordingly, an association between low CHL1 mRNA levels and unfavourable histological grade was previously reported in a small series of breast tumours [2]. Most importantly, CHL1 hypermethylation was an independent prognostic factor in our series that predicted shorter PFS, regardless of other crucial factors in BC prognosis, such as age or stage. Thus, testing CHL1 hypermethylation by pyrosequencing, an easy-to-implement technique that returns an achievable and quantitative measurement [35], could have a significant clinical impact in BC patients.
In conclusion, our results show for the first time that CHL1 promoter is hypermethylated in BC and that this epigenetic alteration, established with a quantitative cut-off value by pyrosequencing, is an independent prognostic biomarker in invasive BC.
MATERIALS AND METHODS
Patient samples
We analysed a series of 142 formalin-fixed, paraffin-embedded samples from BC patients alongside 20 non-neoplastic mammary samples from reduction mammoplasties. Paired adjacent-to-tumour tissue was available in 57 cases. All patients were diagnosed with primary invasive breast cancer between 1996 and 2006 in the Pathology Department of the Complejo Hospitalario de Navarra (Navarra Public Health System, Pamplona, Spain). Pathological and clinical characteristics are summarised in Supplementary Table 1. All tumours were surgically removed and staged according to their size, histological grade and lymph node involvement, and diagnosis was reclassified using the recommended criteria of the St Gallen International Expert Consensus in 2013 [4], considering a Ki-67 threshold of 14% [36], and upon microscopic evaluation by two independent observers with expertise in breast pathology. It was ensured that all cases harboured at least 70% tumour cells. None of the patients had received radiotherapy or chemotherapy before surgery. The study was approved by the Regional Clinical Research Ethics Committee and samples were obtained in accordance with the current Spanish legislation regarding written informed consent.
Cell lines and treatments
A panel of four human BC cell lines was used in this study: T-47D (luminal-like) and BT-549 (triple-negative) were purchased from the American Type Cell Collection (ATCC, Rockville, MD, USA); HCC-1937 and MDA-MB-468 (all from the triple-negative subtype) were obtained from the Leibniz Institute DSMZ - German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). Additionally, one immortalized but non-tumorigenic human mammary epithelial cell line (HBL-100) was obtained from the ATCC (Rockville, MD, USA). Two cell lines derived from other tissues were used (all from ATCC, Rockville, MD, USA): human embryonic kidney 293T cells, which were used for transfection experiments; and U-87 MG, derived from human malignant glioma, which was used as a positive control for CHL1 expression. All these cell lines were grown in RPMI-1640 or DMEM, supplemented with 10% foetal bovine serum and 1% penicillin/streptomycin (all from Life Technologies, Carlsbad, CA, USA), at 37°C in a humidified atmosphere with 5% CO2.
All cell lines at low passage were treated with the demethylating agent 5-aza-2′-deoxycytidine (AZA) and the histone deacetylase inhibitor trichostatin A (TSA) (both from Sigma-Aldrich, St Louis, MO, USA). Briefly, cells were seeded at a density of 1x105 cells/ml, allowed to attach overnight, and treated with 4 μM AZA for 72 h added freshly every 24 h, 300 nM TSA for 24 h, or the combination of the two drugs for the last 24 h, using PBS as a vehicle control.
DNA extraction, bisulphite conversion and pyrosequencing
To determine the methylation status of the CHL1 gene, DNA was extracted from formalin-fixed, paraffin-embedded breast tumours, non-neoplastic mammary tissues and BC cell lines using a QIAamp DNA FFPE Tissue kit (Qiagen, Hilden, Germany). Bisulphite conversion of DNA was performed to transform non-methyl cytosines into thymidines, while methyl cytosines remained intact. 500 ng of DNA were treated with freshly prepared bisulphite using an EZ DNA Methylation-Gold kit (Zymo Research, Irvine, CA, USA), following the manufacturer’s instructions. Pyrosequencing was carried out to analyse the methylation of three CpG sites in the promoter of the CHL1 gene (Supplementary Figure 1). For this purpose, first, PCR amplification was performed using Immolase DNA polymerase (BioLine, London, UK) in a final volume of 30 μl containing 2 μl of bisulphite modified DNA and the primers indicated in Supplementary Table 2. Amplification conditions were: initial DNA polymerase activation at 95°C for 10 min, followed by 50 cycles at 95°C for 30 s, 58–60°C for 30 s and 72°C for 30 s, and a final extension at 72°C for 7 min. The amplicons were resolved by electrophoresis using 2% (w/v) agarose gel in 1x Tris-borate-EDTA buffer, stained using SYBR Red Safe (Life Technologies, Carlsbad, CA, USA) and visualized in a standard transilluminator (ChemiDoc XRS, Bio-Rad Laboratories, Hercules, CA, USA). DNA methylation analysis was quantified as follows: 20 μl of PCR products were immobilized with Streptavidin Sepharose HP Beads (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA) using a Vacuum Prep Workstation. This was followed by annealing (80°C for 2 min) the sequencing primers (Supplementary Table 2) and pyrosequencing in a PyroMark Q24 using PyroMark Gold Q24 reagents and PyroQ-CpGTM Software (v.1.0.11) (all from Qiagen, Hilden, Germany). Results were analysed using PyroMark Q24 software in CpG analysis mode.
Immunohistochemistry
3-μm sections of 57 BC tumours and their matched adjacent-to-tumour counterparts, along with 20 non-neoplastic tissues from reduction mammoplasties, were placed on slides and then deparaffinized, hydrated and treated to block endogenous peroxidase activity. After incubating with the primary rabbit polyclonal CHL1 antibody (ab106269, Abcam, Cambridge, UK) at 1:800 dilution for 20 min (antigen retrieval at 90°C for 20 min, pH = 6.0), the antibody was developed using a Bond Polymer Refine Detection kit (Leica, Wetzlar, Germany) and visualized with diaminobenzidine. The pattern of expression was evaluated blind by two independent observers. The intensity of expression was ascribed to one of four categories: 0, no expression; 1, weak expression; 2, intermediate expression; 3, strong expression. Images were acquired with a Leica DMD 108 digital microscope (Leica, Wetzlar, Germany).
Immunofluorescence
In order to explore the CHL1 expression pattern in cultured cells, the immortalized but non-neoplastic mammary HBL-100 cells were seeded on coverslips and allowed to attach overnight. Then, cells were fixed with 4% paraformaldehyde, blocked with 5% foetal bovine serum in PBS at room temperature for 1 h, and incubated with anti-CHL1 primary antibody (ab106269, Abcam, Cambridge, UK) at 4°C overnight at 1:800 dilution, and with AlexaFluor 488 secondary anti-rabbit (1:200) and AlexaFluor 594 Phalloidin (1:500) (both from Life Technologies, Carlsbad, CA, USA) at room temperature for 1 h. Finally, samples were mounted on slides with DAPI to counterstain nuclei. Confocal microscopy was performed with a Leica TCS SP5 laser scanning microscope (AOBS) (Leica, Wetzlar, Germany) using excitation wavelengths of 488 nm (for FITC) and 561 nm (for Texas Red).
RNA extraction and quantitative reverse transcription PCR (qRT-PCR)
qRT-PCR was performed to measure the levels of CHL1 expression in BC-derived cell lines and to check the restoration of gene expression by AZA+TSA treatment. To this end, first, total RNA was extracted and purified using an RNeasy Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. 500 ng of total RNA were retrotranscribed using a PrimeScript™ RT Reagent Kit (TaKaRa, Otsu, Japan) at 37°C for 15 min and 85°C for 5 s. 1 μl of the resulting cDNA was placed in a 96-well plate with 0.5 μl TaqMan probes (CHL1: Hs00544069_m1 from Life Technologies, Carlsbad, CA, USA; and GAPDH: Hs.PT.39a.22214836, from IDT, Coralville, Iowa, USA) and 19 μl of mix were included in the Premix Ex Taq™ kit (TaKaRa, Otsu, Japan). PCR amplification was performed in triplicate using the Quant Studio 12K Flex (Life Technologies, Carlsbad, CA, USA) under thermal cycler conditions of 95°C for 30 s and 40 cycles at 95°C for 5 sec and 60°C for 34 s. The cycle threshold (Ct) values were calculated using Quant Studio software (Life Technologies, Carlsbad, CA, USA), and the relative quantification (RQ) was calculated by the ΔCt method (RQ = 2−ΔCt), using GAPDH as the endogenous control gene.
CHL1 silencing in immortalized but non-neoplastic mammary cells
To study the functional role of CHL1 in BC, HBL-100 cells were transduced using lentivirus containing short-hairpin RNAs (shRNAs) against CHL1. For their construction, two sequences targeting CHL1 (shCHL1_1: 5’-GCAGCAATATTAGCGAGTATAT-3’ and shCHL1_2: 5’-GCGTCCATTGATACAAACCAAA-3’) and one scramble sequence (5’-GCAACAAGATGAAGAGCACCAA-3’) were inserted into the pHIV1-SIREN-PuroR plasmid [37] through digestion with BamHI and EcoRI restriction enzymes (Life Technologies, Carlsbad, CA, USA) and ligation with the T4 DNA ligase enzyme (New England Biolabs, Ipswich, MA, USA). Plasmids were purified using the Qiagen Plasmid Midi kit (Qiagen, Hilden, Germany) and sequenced to check the ligation. Lentiviruses containing the scramble, shCHL1_1 or shCHL1_2 were produced by the three-plasmid cotransfection method in 293T cells: p8.91, encoding HIV-1 structural proteins; pVSVg, encoding the vesicular stomatitis virus surface glycoprotein; and the constructed plasmids (scramble, shCHL1_1 or shCHL1_2). Media containing viruses were recovered and filtered every day for a week, ultracentrifuged at 25,000 rpm at 4°C for 2 h and stored at -80°C until used. Since the plasmid contains the puromycin resistance gene for mammalian cell selection, sensitivity to this antibiotic was first tested in HBL-100 cells, and an optimal concentration of 1 μg/ml was chosen from a wide range of possibilities. HBL-100 cells were then transduced with 5 μl of each lentivirus for 24 h (multiplicity of infection: 2.5 lentiviral particles/cell), and once the lentiviral particles had been removed, puromycin was added to the culture medium and cells were maintained for 2 weeks for selection.
Western blot
CHL1 silencing efficiency was checked by western blot. Upon puromycin selection, cells transduced with the scramble, shCHL1_1 or shCHL1_2 were harvested, lysed with 30 μl of RIPA buffer (Sigma-Aldrich, St Louis, MO, USA) containing a cocktail of protease inhibitors (Roche, Basel, Switzerland). After centrifugation at 8000 x g for 10 min at 4°C, proteins contained in the supernatants were quantified using the Protein DC kit (Bio-Rad, Hercules, CA, USA) in an Epoch plate reader (BioTek, Winooski, VT, USA) and following manufacturer’s recommendations. For western blot, 80 μg of proteins were resolved by SDS-PAGE in a 10% gel and transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA, USA). The membrane was blocked with 5% non-fat milk and incubated with the anti-CHL1 antibody (ab106269, Abcam, Cambridge, UK) at a 1:500 dilution, overnight and at 4°C. It was then incubated with the secondary anti-rabbit antibody (Bio-Rad, Hercules, CA, USA) at 1:3000 for 1 h at room temperature. The signal was detected with the SuperSignal West Pico Chemiluminiscent Substrate kit (Thermo Scientific, Rockford, IL, USA) in a ChemiDoc (Bio-Rad, Hercules, CA, USA) using the ImageLab software. The α-tubulin antibody (T-6074 from Sigma-Aldrich, St Louis, MO, USA) and the secondary anti-mouse antibody were used at 1:10000 and 1:2000, respectively, for 30 min, as a loading control. Finally, the intensity of bands was quantitated by densitometric analysis using ImageJ software.
Real-time cell analysis (RTCA) of cell proliferation and invasion
To evaluate the functional role of CHL1 in cell proliferation, HBL-100 cells transduced with scramble and two shCHL1 were seeded (1x104 cells/well) into 400 μl of medium in an E-plate L8 device (iCELLigence system, ACEA Biosciences, San Diego, CA, USA), after measuring the background in 100 μl of medium. The invasion assays were performed in CIM-16 plates with 8-μm-pore membranes (ACEA Biosciences, San Diego, CA, USA). Wells were coated with 30 μl of 5% Matrigel (BD Biosciences, San Jose, CA, USA) and allowed to gel at 37°C and 5% CO2 for 4 h. Then, the lower chamber wells were filled with 160 μl of medium containing 10% foetal bovine serum and the top chamber wells with 40 μl of serum-free medium. The two portions were assembled together and allowed to equilibrate for 1 h at 37°C and 5% CO2. Cells were incubated for 16 h in 0.05% foetal bovine serum media; for seeding, the cells were rinsed with PBS, trypsinized and resuspended in serum-free medium. A total of 4 × 104 cells/well were seeded onto the top chamber of CIM-16 plates and placed into the xCELLigence system (ACEA Biosciences, San Diego, CA, USA) for data collection after background measurement.
Two replicates for each condition were analysed. Cell attachment, spreading, proliferation and invasion were monitored by RTCA for 3-5 days, on the basis of changes in cell-sensor impedance, as previously described [33, 38].
Cell migration
To examine the role of CHL1 silencing on cell migration, HBL-100 cells transduced with the scramble, shCHL1_1 and shCHL1_2 were seeded into 6-well plates at a density of 2x105 cells/well. When they had nearly reached confluency, cells were serum-starved for 8 h, then three scratches were made in the cell monolayer with a 10-μl pipette tip, and cells were washed twice with PBS 1X. Some cells were harvested here (time, 0 h), while others were maintained for 24 h in a culture medium containing 5% foetal bovine serum, as previously described [22]. Finally, harvested cells were fixed with paraformaldehyde and stained with crystal violet (Sigma-Aldrich, St Louis, MO, USA), and 10 pictures were taken with an Olympus BX51 microscope (Olympus, Tokyo, Japan). The length of the scratch in each picture was determined using NIS-Elements software from more than 10 measurements taken from each picture.
Statistical analysis
Demographic, clinical and pathological data were summarised as frequencies (and percentages) and means or medians (and ranges) ± standard error of the mean, as appropriate. All statistical analyses were carried out using IBM SPSS Statistics v20. The optimal cut-off values identifying the methylated or unmethylated status of each CpG were estimated using ROC curve analysis. Across several cut-off points, Youden’s index was chosen as the best cut-off value by considering maximum sensitivity and specificity. The optimal cut-off values predicting OS and PFS were estimated as previously described [34]. Methylation levels in tumour, adjacent-to-tumour and non-neoplastic tissues were compared using the Kruskal-Wallis test. Methylation in tumours and their adjacent counterparts was compared by a paired t-test. Differences in immunohistochemical expression were analysed with the Mann-Whitney and Wilcoxon tests. Correlation between methylation and expression in BC cell lines was assessed by Spearman’s correlation coefficient: U-87 MG cells were excluded from this analysis. The effects of CHL1 silencing on cell proliferation, migration and invasion were compared using two-tailed unpaired t-tests at different times (0, 24, 48, 72, 96, 120 h). Finally, Kaplan-Meier plots and log-rank tests were used to examine the association of CHL1 hypermethylation with PFS and OS. A multivariate Cox regression model was fitted to test the independent contribution of each variable to patient outcome. Hazard ratios and 95% confidence intervals were used to estimate the effect of each variable on the outcome. Association between clinical variables was tested with the χ2 test.
ACKNOWLEDGMENTS
The authors wish to thank Ana Aramendía, Valle Coca and Leticia Sanjosé (Biobank in Navarrabiomed. Departmento de Salud-UPNA. IdiSNA, Pamplona, Spain) for their excellent technical assistance with the immunohistochemical stainings. We thank Dr Berta Ibáñez (Methodology Unit, Navarrabiomed. Departmento de Salud-UPNA. IdiSNA, Pamplona, Spain) for her exceptional help with the statistical analyses. We are also indebted to Dr Agustín Fernández and Dr Gustavo Fernández Bayón (Cancer Epigenetics Group, University Institute of Oncology in Asturias, IUOPA, Oviedo, Spain) for their assistance and training in pyrosequencing and bioinformatics techniques. We also thank the Breast Cancer Patients’ Association in Navarra (SARAY) for their support.
CONFLICTS OF INTEREST
The authors disclose no potential conflicts of interest.
FUNDING
This work has been funded in competitive calls by the Spanish Institute of Health and FEDER (PI14/00579), the Basque Foundation for Healthcare Research and Innovation (BIO-11-CM-013), La Caixa Foundation (70789) and the Breast Cancer Patients’ Association in Navarra (SARAY). EMS is the recipient of a grant from the Spanish Ministry of Economy and Competitiveness (PTA2015-11895-I); NPJ was the recipient of a predoctoral grant from the Department of Health of the Government of Navarra; DE is funded by a Miguel Servet fellowship from the Spanish Institute of Health.
Author contributions
EMS designed and performed the experiments, analysed the data and wrote the manuscript; SM, AUG, IMS, IBL and NPJ performed the experiments; AC and DGS evaluated the immunohistochemistry cases; FVG and JJI provided clinical data; PLS and ME were responsible for the cell invasion experiments; DE provided vital reagents and revised the manuscript; DM carried out confocal microscopy; DGS conceived the research and revised the manuscript.
REFERENCES
1. Stefansson OA, Moran S, Gomez A, Sayols S, Arribas-Jorba C, Sandoval J, Hilmarsdottir H, Olafsdottir E, Tryggvadottir L, Jonasson JG, Eyfjord J, Esteller M. A DNA methylation-based definition of biologically distinct breast cancer subtypes. Mol Oncol. 2015; 9:555-568.
2. He LH, Ma Q, Shi YH, Ge J, Zhao HM, Li SF, Tong ZS. CHL1 is involved in human breast tumorigenesis and progression. Biochem Biophys Res Commun. 2013; 438:433-438.
3. Smith RA, Andrews K, Brooks D, DeSantis CE, Fedewa SA, Lortet-Tieulent J, Manassaram-Baptiste D, Brawley OW, Wender RC. Cancer screening in the United States, 2016: A review of current American Cancer Society guidelines and current issues in cancer screening. CA Cancer J Clin. 2016; 66:96-114.
4. Goldhirsch A, Winer EP, Coates AS, Gelber RD, Piccart-Gebhart M, Thürlimann B, Senn HJ and members P. Personalizing the treatment of women with early breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann Oncol. 2013; 24:2206-2223.
5. Forbes JF, Cuzick J, Buzdar A, Howell A, Tobias JS, Baum M, Arimidex T, A.one or in Combination (ATAC) Trialists' Group. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 100-month analysis of the ATAC trial. Lancet Oncol. 2008; 9:45-53.
6. Slamon D, Eiermann W, Robert N, Pienkowski T, Martin M, Press M, Mackey J, Glaspy J, Chan A, Pawlicki M, Pinter T, Valero V, Liu MC, Sauter G, von Minckwitz G, Visco F, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med. 2011; 365:1273-1283.
7. Senkus E, Kyriakides S, Ohno S, Penault-Llorca F, Poortmans P, Rutgers E, Zackrisson S, Cardoso F, Committee EG. Primary breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015; 26:v8-30.
8. Huo D, Clayton WM, Yoshimatsu TF, Chen J, Olopade OI. Identification of a circulating MicroRNA signature to distinguish recurrence in breast cancer patients. Oncotarget. 2016; 7:55231-55248. doi: 10.18632/oncotarget.10485.
9. Guo T, Ren Y, Wang B, Huang Y, Jia S, Tang W, Luo Y. Promoter methylation of BRCA1 is associated with estrogen, progesterone and human epidermal growth factor receptor-negative tumors and the prognosis of breast cancer: A meta-analysis. Mol Clin Oncol. 2015; 3:1353-1360.
10. Sørlie T, Wang Y, Xiao C, Johnsen H, Naume B, Samaha RR, Børresen-Dale AL. Distinct molecular mechanisms underlying clinically relevant subtypes of breast cancer: gene expression analyses across three different platforms. BMC Genomics. 2006; 7:127.
11. Guiu S, Michiels S, André F, Cortes J, Denkert C, Di Leo A, Hennessy BT, Sorlie T, Sotiriou C, Turner N, Van de Vijver M, Viale G, Loi S, Reis-Filho JS. Molecular subclasses of breast cancer: how do we define them? The IMPAKT 2012 Working Group Statement. Ann Oncol. 2012; 23:2997-3006.
12. Bediaga NG, Acha-Sagredo A, Guerra I, Viguri A, Albaina C, Ruiz Diaz I, Rezola R, Alberdi MJ, Dopazo J, Montaner D, Renobales M, Fernández AF, Field JK, Fraga MF, Liloglou T, de Pancorbo MM. DNA methylation epigenotypes in breast cancer molecular subtypes. Breast Cancer Res. 2010; 12:R77.
13. Du J, Johnson LM, Jacobsen SE, Patel DJ. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol. 2015; 16:519-532.
14. Feng W, Shen L, Wen S, Rosen DG, Jelinek J, Hu X, Huan S, Huang M, Liu J, Sahin AA, Hunt KK, Bast RC, Shen Y, Issa JP, Yu Y. Correlation between CpG methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res. 2007; 9:R57.
15. Karsli-Ceppioglu S, Dagdemir A, Judes G, Ngollo M, Penault-Llorca F, Pajon A, Bignon YJ, Bernard-Gallon D. Epigenetic mechanisms of breast cancer: an update of the current knowledge. Epigenomics. 2014; 6:651-664.
16. Fang C, Wei XM, Zeng XT, Wang FB, Weng H, Long X. Aberrant GSTP1 promoter methylation is associated with increased risk and advanced stage of breast cancer: a meta-analysis of 19 case-control studies. BMC Cancer. 2015; 15:920.
17. Widschwendter M, Siegmund KD, Müller HM, Fiegl H, Marth C, Müller-Holzner E, Jones PA, Laird PW. Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res. 2004; 64:3807-3813.
18. Fiegl H, Millinger S, Goebel G, Müller-Holzner E, Marth C, Laird PW, Widschwendter M. Breast cancer DNA methylation profiles in cancer cells and tumor stroma: association with HER-2/neu status in primary breast cancer. Cancer Res. 2006; 66:29-33.
19. Wang L, Zeng H, Wang Q, Zhao Z, Boyer TG, Bian X, Xu W. MED12 methylation by CARM1 sensitizes human breast cancer cells to chemotherapy drugs. Sci Adv. 2015; 1:e1500463.
20. Shen Y, Wang Z, Loo LW, Ni Y, Jia W, Fei P, Risch HA, Katsaros D, Yu H. LINC00472 expression is regulated by promoter methylation and associated with disease-free survival in patients with grade 2 breast cancer. Breast Cancer Res Treat. 2015; 154:473-482.
21. Senchenko VN, Krasnov GS, Dmitriev AA, Kudryavtseva AV, Anedchenko EA, Braga EA, Pronina IV, Kondratieva TT, Ivanov SV, Zabarovsky ER, Lerman MI. Differential expression of CHL1 gene during development of major human cancers. PLoS One. 2011; 6:e15612.
22. Zhang J, Yang F, Ding Y, Zhen L, Han X, Jiao F, Tang J. Overexpression of L1 cell adhesion molecule correlates with aggressive tumor progression of patients with breast cancer and promotes motility of breast cancer cells. Int J Clin Exp Pathol. 2015; 8:9240-9247.
23. Schröder C, Schumacher U, Fogel M, Feuerhake F, Müller V, Wirtz RM, Altevogt P, Krenkel S, Jänicke F, Milde-Langosch K. Expression and prognostic value of L1-CAM in breast cancer. Oncol Rep. 2009; 22:1109-1117.
24. Li Y, Galileo DS. Soluble L1CAM promotes breast cancer cell adhesion and migration in vitro, but not invasion. Cancer Cell Int. 2010; 10:34.
25. Mantripragada KK, Spurlock G, Kluwe L, Chuzhanova N, Ferner RE, Frayling IM, Dumanski JP, Guha A, Mautner V, Upadhyaya M. High-resolution DNA copy number profiling of malignant peripheral nerve sheath tumors using targeted microarray-based comparative genomic hybridization. Clin Cancer Res. 2008; 14:1015-1024.
26. Chen J, Fu L, Zhang LY, Kwong DL, Yan L, Guan XY. Tumor suppressor genes on frequently deleted chromosome 3p in nasopharyngeal carcinoma. Chin J Cancer. 2012; 31:215-222.
27. Uchida K, Oga A, Nakao M, Mano T, Mihara M, Kawauchi S, Furuya T, Ueyama Y, Sasaki K. Loss of 3p26.3 is an independent prognostic factor in patients with oral squamous cell carcinoma. Oncol Rep. 2011; 26:463-469.
28. Kolla V, Zhuang T, Higashi M, Naraparaju K, Brodeur GM. Role of CHD5 in human cancers: 10 years later. Cancer Res. 2014; 74:652-658.
29. Buchegger K, Ili C, Riquelme I, Letelier P, Corvalán AH, Brebi P, Huang TH, Roa JC. Reprimo as a modulator of cell migration and invasion in the MDA-MB-231 breast cancer cell line. Biol Res. 2016; 49:5.
30. Labbozzetta M, Poma P, Vivona N, Gulino A, D'Alessandro N, Notarbartolo M. Epigenetic changes and nuclear factor-κB activation, but not microRNA-224, downregulate Raf-1 kinase inhibitor protein in triple-negative breast cancer SUM 159 cells. Oncol Lett. 2015; 10:3807-3815.
31. Minatani N, Waraya M, Yamashita K, Kikuchi M, Ushiku H, Kojo K, Ema A, Nishimiya H, Kosaka Y, Katoh H, Sengoku N, Tanino H, Sidransky D, Watanabe M. Prognostic significance of promoter DNA hypermethylation of cysteine dioxygenase 1 (CDO1) gene in primary breast cancer. PLoS One. 2016; 11:e0144862.
32. Yari K, Payandeh M, Rahimi Z. Association of the hypermethylation status of PTEN tumor suppressor gene with the risk of breast cancer among Kurdish population from Western Iran. Tumour Biol. 2015; 37:8145-8152.
33. Perez-Janices N, Blanco-Luquin I, Torrea N, Liechtenstein T, Escors D, Cordoba A, Vicente-Garcia F, Jauregui I, De La Cruz S, Illarramendi JJ, Coca V, Berdasco M, Kochan G, et al. Differential involvement of RASSF2 hypermethylation in breast cancer subtypes and their prognosis. Oncotarget. 2015; 6:23944-23958. doi: 10.18632/oncotarget.4062.
34. Villani V, Casini B, Pace A, Prosperini L, Carapella CM, Vidiri A, Fabi A, Carosi M. The prognostic value of pyrosequencing-detected MGMT promoter hypermethylation in newly diagnosed patients with glioblastoma. Dis Markers. 2015; 2015:604719.
35. Noehammer C, Pulverer W, Hassler MR, Hofner M, Wielscher M, Vierlinger K, Liloglou T, McCarthy D, Jensen TJ, Nygren A, Gohlke H, Trooskens G, Braspenning M, Van Criekinge W, Egger G, Weinhaeusel A. Strategies for validation and testing of DNA methylation biomarkers. Epigenomics. 2014; 6:603-622.
36. Cheang MC, Chia SK, Voduc D, Gao D, Leung S, Snider J, Watson M, Davies S, Bernard PS, Parker JS, Perou CM, Ellis MJ, Nielsen TO. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst. 2009; 101:736-750.
37. Lanna A, Henson SM, Escors D, Akbar AN. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat Immunol. 2014; 15:965-972.
38. Vizoso M, Ferreira HJ, Lopez-Serra P, Carmona FJ, Martínez-Cardús A, Girotti MR, Villanueva A, Guil S, Moutinho C, Liz J, Portela A, Heyn H, Moran S, Vidal A, Martinez-Iniesta M, Manzano JL, et al. Epigenetic activation of a cryptic TBC1D16 transcript enhances melanoma progression by targeting EGFR. Nat Med. 2015; 21:741-750.