
INTRODUCTION
Intramuscular myxoma is a benign soft tissue tumor characterized by bland spindle-shaped cells embedded in hypovascular, abundantly myxoid stroma. The pathologic entity was first delineated by Enzinger in 1965 [1] and his description has since been amply confirmed by others [2–5]. Intramuscular myxoma is a rare tumor that occurs mostly in adults and shows a predilection for women (70%). The reported annual incidence is 0.10–0.13/100.000 [6, 7]. The vast majority of patients are asymptomatic with the tumor appearing as a painless, slowly enlarging, palpable, well-defined, round mass 2 to 15 cm in diameter [8]. Almost half of the tumors are found in the thigh. Intramuscular myxomas are usually solitary lesions not associated with any other clinical abnormalities [6]. Multiple tumors are rare but may be associated with fibrous dysplasia, also in the Mazabraud syndrome [9–12].
The tumors have a gelatinous, lobulated cut surface [6]. The fibrous capsule is usually incomplete with most lesions showing infiltration into adjacent musculature [6]. Histologically, intramuscular myxomas are hypocellular, hypovascular, intensely mucoid, and basophilic in hematoxylin-eosin stained preparations. The four main histologic components are interstitial mucin, sparse spindle-shaped cells, fine fibrillary reticulin fibers, and varying numbers of strands or trabeculae of fibrous tissue [6]. Other benign myxoid tumors that can be confused with intramuscular myxomas include myxolipoma, myxoid neurofibroma, nerve sheath myxoma, chondroma with myxoid change, and nodular fasciitis. More importantly, intramuscular myxoma can also be diagnostically confused with low grade myxoid sarcomas such as myxofibrosarcoma, low grade fibromyxoid sarcoma, myxoid liposarcoma, and extraskeletal myxoid chondrosarcoma [13].
Genetic information on intramuscular myxomas is very limited. Only one tumor studied by banding cytogenetics and having an abnormal karyotype has been reported; the tumor had trisomy 18 as the sole anomaly [14]. Interphase cytogenetic analysis by fluorescent in situ hybridization and DNA flow cytometry showed a normal DNA content and no indication of numerical chromosome aberrations in the four intramuscular myxomas studied by Aoki et al. [15].
Molecular genetic analyses of the GNAS gene (20q13) have revealed activating missense mutations, R201H and R201C, in exon 8 at codon 201 of the GNAS gene in both solitary intramuscular myxoma and the multiple intramuscular myxomas of Mazabraud syndrome [16–18]. The mutation frequency has varied from study to study depending on detection method. Okamato et al. [13] used single strand conformation polymorphism (SSCP) methodology to find point mutations in five of six intramuscular myxomas (three with and two without fibrous dysplasia), mutations which were subsequently confirmed by sequence analysis (three R201H and two R201C). Delaney et al. [16] detected mutations in 8 of 28 (29%) intramuscular myxomas by conventional PCR followed by mutation-specific restriction enzyme digestion whereas 17 of 28 (61%) mutations were detected using COLD-PCR followed by mutation-specific restriction enzyme digestion.
Walther et al. [18] used conventional PCR followed by direct sequencing to detect GNAS mutation in 23 out of 63 (36 %) intramuscular myxomas corresponding to 52 % R201C and 48 % R201H missense mutations.
Here we present our karyotypic analysis of intramuscular myxomas as well as analysis of the GNAS gene in five of the tumors.
RESULTS
Karyotyping and fluorescence in situ hybridization (FISH) analyses
Abnormal karyotypes were found in 21 out of 68 tumors, 12 from female and 9 from male patients (Tables 1 and 2). Numerical aberrations only were seen in 12 tumors, whereas both numerical and structural rearrangements were found in 9. Almost all abnormal clones were pseudodiploid or near-diploid whereas one clone in case 11 was hyperhaploid. The vast majority (90 %) of cytogenetically abnormal tumors had simple karyotypes (1–3 chromosome changes) with only two tumors having complex karyotypes (6–7 aberrations) (cases 11 and 14). Two tumors (cases 3 and 20) had two cytogenetically unrelated clones; one with structural, the other with numerical chromosome aberrations.
Table 1: Information on the cytogenetically analyzed myxomas
Samples |
Analyzed Cases |
Abnormal |
Only numerical |
Both numerical and |
|---|---|---|---|---|
Female |
44 |
12 |
6 |
6 |
Male |
24 |
9 |
6 |
3 |
Total |
68 |
21 |
12 |
9 |
Table 2: Clinicopathological data on the intramuscular myxomas with abnormal karyotypes
Cases |
Sex/Age |
Site |
Largest diameter (cm) |
Karyotype |
|---|---|---|---|---|
1 |
F/36 |
Left shoulder |
2.5 |
47,XX,+3[2]/46,XX[23] |
2 |
M/54 |
Right thigh |
3 |
47,XY,+8[2]/46,XY[22] |
3 |
F/75 |
Left shoulder |
2 |
47,XX,+7[2]/46,XX,del(6)(q21)[2]/46,XX[11] |
4 |
F/61 |
Right thigh |
3 |
47,XX,+8[3]/46,XX[22] |
5 |
F/69 |
Right shoulder |
7.5 |
47,XX,+8[3]/46,XX[21] |
6 |
F/47 |
Left thigh |
Not available |
46,XX,del(8)(q13 or q13q22)[6]/46,XX[9] |
7 |
M/68 |
Right Shoulder |
6 |
45,X,-Y[3]/46,XY[23] |
8 |
F/55 |
Back |
5.5 |
47,XX,+8[2]/46,XX[14] |
9 |
F/68 |
Right thigh |
3 |
47,XX,+8[2]/46,XX[22] |
10 |
F/53 |
Right shoulder |
1.8 |
46,XX,der(1)inv(1)(p32q42)t(1;4)(q42;q21),der(4)t(1;4) [3]/46,XX[22] |
11 |
M/54 |
Left thigh |
3.5 |
29,X,-Y,+5,+7,+8,+12,+18,+19[20]/47,X,-Y,+4,+10[12]/46,XY[4] |
12 |
F/48 |
Right shoulder |
4 |
47,XX,+del(22)(q11)[16] |
13 |
M/34 |
Left chest wall |
4.7 |
46,XY,t(7;15)(q32;q15~22),t(11;17)(q23;q23)[15] |
14 |
F/67 |
Right upper arm |
3 |
45~46,XX,add(1)(p22),-5,der(9)t(5;9)(q11;p21),-17,+1~2mar[cp7]/46,XX[3] |
15 |
M/73 |
Left shoulder |
2 |
46,XY,t(2;8)(p21;q13~21),t(10;11)(p14~15;q12~13)[5]/46,XY[5] |
16 |
M/56 |
Right shoulder |
3 |
47~48,XY,+7[2][cp2]/46,XY[23] |
17 |
F/54 |
Left thigh |
4.2 |
47,XX,+X[2]/46,XX[23] |
18 |
M/46 |
Right thigh |
3.5 |
47,XY,+7[3]/46,XY[12] |
19 |
M/49 |
Abdominal wall |
2.6 |
45~47,XY,del(6)(q21q23),+8,tas(14;17)(pter;qter)[cp11]/46,XY[2] |
20 |
F/74 |
Left thigh |
3.6 |
46,XX,add(19)(p13),-21,+r[7]/47,XX,+7[2]/46,XX[9] |
21 |
M/70 |
Right thigh |
6.2 |
48,XY,+7,+9[5]/46,XY[20] |
The most clearly nonrandom involvement was of chromosome 8 seen in 9 tumors, 7 of which showed trisomy 8 whereas 2 had structural aberrations. Trisomy 8 was the sole anomaly in 5 myxomas (Figure 1). In both myxomas with structural aberrations of chromosome 8, bands 8q13-q22 were involved: one tumor (case 6) had a del(8)(q13 or q13q22) as the only cytogenetic change whereas the other (case 15) had the translocation t(2;8)(p21;q13~21) as the sole change (Table 2). Chromosome 7 was the second most frequently involved showing aberration in 6 tumors: 5 with numerical changes (trisomy 7; cases 3, 11, 16, 18, and 21) and one with a translocation t(7;15)(q32;q15~22) (case 13). A del(6)(q21) was found in two tumors (cases 3 and 19).

Figure 1: Karyotype of case 8 showing trisomy 8 as the sole anomaly.
In case 12, the karyotype was initially 47,XX,+mar. FISH with painting probes for chromosomes 19 and 22 showed that the marker contained material from chromosome 22 (data not shown). FISH with a breakapart probe for EWSR1 showed that the EWSR1 locus was not on the marker chromosome, nor was there any splitting of it (data not shown).
Analysis of GNAS expression and mutation
Expression analysis of the GNAS gene was for reasons of stored material shortage possible for cases 10–14 only (Figure 2). The GNAS locus has a highly complex imprinted expression pattern giving rise to transcripts (including non-coding ones) that are maternally, paternally, or biallelically expressed [19–21].
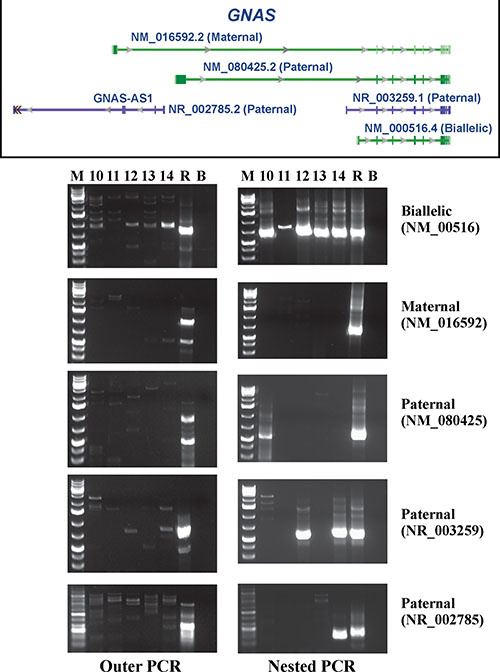
Figure 2: RT-PCR analysis for the expression of the biallelically, maternally, and three paternally expressed GNAS transcripts in cases 10–14. R is human universal reference total RNA. B is blank. M is 1kb Plus DNA ladder (GeneRuler, Fermentas).
Outer and nested RT-PCR amplified the biallelically expressed transcript (NM_000516) in the examined tumors (Figure 2). In none of the tumors was the maternally expressed transcript amplified (NM_016592). The paternally expressed transcript with accession number NM_080425 was detected in case 10 (Figure 2), whereas the paternally expressed transcript with accession number NR_003259 was found in cases 12 and 14. Only tumor 14 expressed the paternally expressed transcript with accession number NR_002785 (Figure 2).
The PCR products amplified in nested PCR using the primer set GNAS-379F1+GNAS-1040R1 corresponded to the biallelically expressed transcript with accession number NM_000516. Direct sequencing of these PCR products detected the R201C mutation in case 13 only (Figure 3). No mutations were found at codon 227.
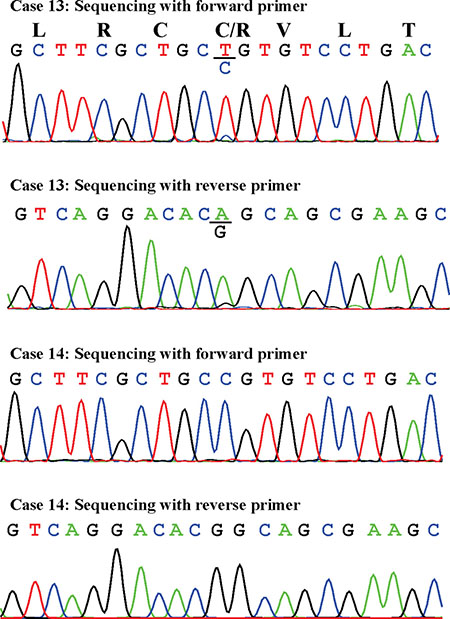
Figure 3: Partial sequence chromatogram of the cDNA fragment showing the mutation R201C in case 13 and the normal R201 in case 14. Sequences with both the forward and reverse primers are shown.
DISCUSSION
Information about the acquired genomic abnormalities of tumor cells, be it at the chromosomal or molecular level of resolution, is a powerful adjunct to microscopic tumor features in diagnostic pathology. The same information is also crucial to obtaining any deep understanding of tumorigenesis. The present study describes the largest series of cytogenetically analyzed intramuscular myxomas to date. It proves that acquisition of clonal chromosome aberrations is an integral part of the disease process inasmuch as 21 out of 68 intramuscular myxomas were shown to have clonal cytogenetic aberrations. Abnormalities of chromosome 8, mainly trisomy, followed by trisomy for chromosome 7 were the most common aberrations (Tables 1 and 2). Trisomy 8 is common also in cancer and may be found both alone and together with other aberrations [22]. Trisomy 8 as the sole abnormality occurs particularly in acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS), in 5–10% of cytogenetically abnormal cases [23]. The etiology, molecular mechanisms, and pathogenetic consequences behind this and other numerical chromosome aberrations remain unknown. However, a recent study indicated that AMLs with trisomy 8 as sole anomaly have distinct gene and microRNA-expression signatures [24]. In solid tumors, polysomy 8 was found in different diagnostic entities such as desmoid-type and superficial fibromatosis, clear cell sarcomas with t(12;22)(q13;q12), Ewing sarcomas, myxoid liposarcomas, synovial sarcomas, hepatoblastomas, Wilms’ tumor, and colorectal cancer [22]. Again, the etiology and pathogenetic consequences behind the change are unknown. The same goes for trisomy 7 which is also frequently seen in various both neoplastic and non-neoplastic disease lesions presenting as solid tumors [22, 25].
Intramuscular myxoma, and in particular its cellular variant, shows considerable overlapping histological features with grade I myxofibrosarcoma [26]. The “Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer” (https://cgap.nci.nih.gov/Chromosomes/Mitelman; database last updated on August 11, 2016) contains cytogenetic information on 84 myxofibrosarcomas of various grades. None of them had trisomy 8 or trisomy 7 as the sole cytogenetic abnormality.
Willems et al. [27] studied the genetic alterations and composition of extracellular matrix of intramuscular myxoma and grade I myxofibrosarcoma. Of the ten examined intramuscular myxomas, four did not have cytogenetic data whereas six had a normal karyotype. Of the ten myxofibrosarcomas, four did not have cytogenetic data, one had a normal karyotype, another had a balanced t(9;12) translocation, a third had monosomy of chromosome 21, and three had complex karyotypes. Moreover, five myxomas had mutation at codon 201 of GNAS. Neither myxomas nor myxofibrosarcomas had mutations of KRAS codon 12/13 or TP53, nor was there any significant difference in FOS expression between intramuscular myxoma and grade I myxofibrosarcoma. The only difference found between intramuscular myxoma and grade I myxofibrosarcoma was in the expression of decorin, a matrix proteoglycan, which was expressed in myxofibrosarcomas but not in myxomas [27]. Both decorin immunoreactivity and mRNA expression of DCN (the gene coding for decorin) were positive in the former but not in the latter [27]. In a recent study, on the other hand, Cates et al. [28] saw no difference in decorin immunoreactivity between myxoma and myxofibrosarcoma. In fact, none of the 19 potential diagnostic markers tested distinguished myxoma from myxofibrosarcoma [28]. The authors concluded that the distinction between these tumors must still be made based on morphologic criteria.
The GNAS locus has a highly complex imprinted expression pattern giving rise to maternally, paternally, and biallelically expressed transcripts (including non-coding ones) that are derived from four alternative promoters and 5’ exons [19–21]. An antisense transcript is produced from an overlapping locus on the opposite strand. One of the transcripts produced from this locus as well as its antisense transcript are paternally expressed noncoding RNAs that may regulate imprinting in the region (http://www.ncbi.nlm.nih.gov/gene/2778). Alternative splicing of downstream exons is also observed, resulting in different forms of the stimulatory G-protein alpha subunit, a key element in the classical signal transduction pathway linking receptor-ligand interactions with the activation of adenylyl cyclase and a variety of cellular responses (http://www.ncbi.nlm.nih.gov/gene/2778). In the present study, we found expression of the biallelic transcript with accession number NM_000516 which codes for guanine nucleotide binding protein alpha s long (GNASL), also known as alpha-S2, a form of the G-protein alpha subunit (http://www.ncbi.nlm.nih.gov/gene/2778). In contrast, none of the tumors expressed the maternally expressed transcript (accession number NM_016592). This transcript encodes secretogranin VI (SCG6, also known as NESP55) which localizes to large secretory vesicles of endocrine cells and neurons. Its coding region is within the most 5’ exon and does not overlap the coding regions used by other transcripts; thus, SCG6 has no similarity to isoforms of the G-protein alpha subunit. No consistent expression pattern was found for the three paternally expressed transcripts NM_080425, NR_003259, and NR_002785 (Figure 1).
Mutation analysis of the biallelically expressed transcript identified the mutation R201C in one out of five samples. This is in agreement with findings presented in previous reports that only some myxomas have mutations of codon R201 [16-18]. None of the examined myxomas had mutation of codon Q227 which was found in a small proportion of fibrous dysplasia tumors [29].
Apart from myxomas, R201C, R201H, and Q227R mutations of GNAS protein were found also in a variety of other neoplasms such as tumors of the kidney, thyroid, pituitary, leydig cells, adrenal cortex, and large bowel [30]. These mutations are thought to inhibit guanosine triphosphate hydrolysis resulting in constitutive activation of the stimulatory G-protein alpha subunit and its effector adenylate cyclase, leading to autonomous synthesis of cAMP [31, 32]. To study the role of GNAS in intestinal tumorigenesis, Wilson et al. [30] placed GNAS R201C under the control of the A33-antigen promoter (Gpa33) which is almost exclusively expressed in the intestines. GNAS R201C was found to cause augmentation of both the Wnt and ERK1/2 MAPK pathways in the intestinal epithelium of mice and the mutation co-operated with inactivation of Apc in intestinal tumor formation in vivo [30]. Collectively, the data suggest that mutations of GNAS can modify cell growth and may be oncogenic; however, how GNAS functions as an oncogene in the various tumors remains unclear.
In summary, our study showed that intramuscular myxomas are characterized by acquired numerical but occasionally also structural chromosome rearrangements, express the biallelic GNAS transcript but not the maternally expressed transcript, and that the mutated allele of GNAS is biallelically transcribed in the tumor cells.
MATERIALS AND METHODS
Patients
The material consisted of 68 samples from tumors diagnosed as intramuscular myxomas (Table 1), all surgically removed at The Norwegian Radium Hospital between 2000 and 2016. The patients, 46 females and 26 males, were from 34 to 78 years old with a median age of 55. The study was approved by the regional ethics committee (Regional komité for medisinsk forskningsetikk Sør-Øst, Norge, http://helseforskning.etikkom.no). Written informed consent was obtained from the patients to publication of the case details. The ethics committee’s approval included a review of the consent procedure. All patient information has been de-identified.
G-banding, karyotyping, and FISH
Fresh tissue from a representative area of the tumor was received and analyzed cytogenetically as part of our diagnostic routine. The samples were disaggregated mechanically and enzymatically with collagenase II (Worthington, Freehold, NJ, USA). The resulting cells were cultured and harvested using standard techniques. Chromosome preparations were G-banded with Wright’s stain (SigmaAldrich; St Louis, MO, USA) and examined. Metaphases were analyzed and karyograms prepared using the CytoVision computer assisted karyotyping system (Leica Biosystems, Newcastle, UK). The karyotypes were described according to the International System for Human Cytogenetics Nomenclature [33].
For case 12 (see below, Table 2), FISH was performed on metaphase spreads using painting probes for chromosomes 19 and 22 as well as an EWSR1 breakapart probe (Cytocell, Cambridge, UK).
RNA extraction and cDNA synthesis
Frozen (–80°C) tumor tissue adjacent to that used for cytogenetic analysis and histologic examination was available for cases 10-14 (Table 2). Total RNA was extracted using Trizol reagent according to the manufacturer’s instructions (Life Technologies, Oslo, Norway). cDNA was synthesized from this in a 20 μL reaction volume using iScript Advanced cDNA Synthesis Kit for RT-qPCR according to the manufacturer’s instructions (Bio-Rad Laboratories).
Analysis for GNAS expression
Nested RT-PCR was performed to assess the expression of GNAS. The primers used for PCR amplification and sequencing are listed in Table 3. The primer combinations, target fusion transcripts, and results of PCR amplifications are shown in Table 4. Human Universal Reference Total RNA was used as control (Clontech Laboratories, TaKaRa-Bio Group, Europe/SAS, Saint-Germain-en-Laye, France). According to the company’s information, it is a mixture of total RNAs from a collection of adult human tissues chosen to represent a broad range of expressed genes. Both male and female donors are represented. Four μL of cDNA (1 μL for the control sample) were used as template and amplified using the outer primer combination. One μL of the amplified products was used as template in nested PCR. For both outer and nested PCRs, the 25 μL reaction volume contained 12.5 μL of Premix Taq (TaKaRa-Bio Europe/SAS, Saint-Germain-en-Laye, France), template, and 0.4 μM of each of the forward and reverse primers (Tables 3 and 4). The PCRs were run on a C-1000 Thermal cycler (Bio-Rad Laboratories). PCR cycling consisted of an initial step of denaturation at 94°C for 30 sec followed by 35 cycles of 7 sec at 98°C, 30 sec at 60°C, 2 min at 72°C, and a final extension for 5 min at 72°C. Three μL of the PCR products were stained with GelRed (Biotium), analyzed by electrophoresis through 1.0% agarose gel, and photographed. DNA gel electrophoresis was performed using lithium borate buffer [34].
Table 3: Primers used for PCR amplification and sequencing
Oligo Name |
Sequence (5'->3') |
Position |
Accession number |
|---|---|---|---|
GNAS-356F1 |
CATGGGCTGCCTCGGGAACAG |
356–376 |
NM_000516.4 |
GNAS-379F1 |
AGACCGAGGACCAGCGCAACG |
379–399 |
NM_000516.4 |
GNAS-459F1 |
CAGGTCTACCGGGCCACGCAC |
459–479 |
NM_000516.4 |
GNAS-1088R1 |
GCTGCTGGCCACCACGAAGATG |
1109–1088 |
NM_000516.4 |
GNAS-1040R1 |
GATCCACTTGCGGCGTTCATCG |
1061–1040 |
NM_000516.4 |
GNAS-NR-193F1 |
AGGCGCTGCCTTGCGTGTGA |
193–212 |
NR_003259 |
GNAS-NR-213F1 |
GTGCACCTCACTCACATGTGCTGGA |
213–237 |
NR_003259 |
GNAS-1016F1-Mat |
CGTCGCTGCAAGCCAAAGAAGC |
1016–1037 |
NM_016592.2 |
GNAS-1089F1-Mat |
CCATCCGGCGTCACTAATGGAGG |
1089–1111 |
NM_016592.2 |
GNAS-2315F1-Pat |
AGATGGGCTACATGTGTACGCACCG |
2315–2339 |
NM_080425.2 |
GNAS-2223F1-Pat |
ACAGATGCGCAAAGAAGCCCTGG |
2223–2245 |
NM_080425.2 |
GNAS-841F1 |
GCTACGAACGCTCCAACGAGTA |
841–862 |
NM_000516.4 |
GNAS-921F1 |
GACTATGTGCCGAGCGATCA |
921–940 |
NM_000516.4 |
GNAS-982R1 |
TGTCCACCTGGAACTTGGTCT |
1002–982 |
NM_000516.4 |
GNAS-AS1-NR-249F |
GCAAGAAGATTTCCAGGGCTGGGA |
249–272 |
NR_002785.2 |
GNAS-AS1-NR-312F |
GGAGCAGCCCAGGATGGATAAGGA |
312–335 |
NR_002785.2 |
GNAS-AS1-NR-574R |
AACGGCAGCAATCTGGTAACGCAC |
597–574 |
NR_002785.2 |
GNAS-AS1-NR-652R |
CGGCCATTTTCAGCACGGGTAGA |
674–652 |
NR_002785.2 |
Table 4: Primer combinations for outer and nested PCR amplification of GNAS transcripts, accession number of the sequence and expression of the target amplification transcript
Outer PCR |
Nested PCR |
Amplification sequence |
Expression |
|---|---|---|---|
GNAS-356F1 + |
GNAS-379F1 + |
NM_000516.4 |
Biallelic |
GNAS-1016F1-Mat + |
GNAS-1089F1-Mat + |
NM_016592.2 |
Maternal |
GNAS-2223F1-Pat + |
GNAS-2315F1-Pat + |
NM_080425.2 |
Paternal |
GNAS-NR-193F1 + |
GNAS-NR-213F1 + |
NR_003259.1 |
Paternal |
GNAS-AS1-NR-249F + |
GNAS-AS1-NR-312F + |
NR_002785.2 |
Paternal |
Analysis for GNAS mutation
The PCR products which were amplified in nested PCR using the primer set GNAS-379F1+GNAS-1040R1 were purified using the Thermo Scientific GeneJET PCR purification Kit (Fisher Scientific, Oslo, Norway) and direct sequencing was performed using the light run sequencing service of GATC Biotech (https://www.gatc-biotech.com/en/index.html). The BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) program was used for computer analysis of sequence data against the reference sequence with accession number NM_000516.4.
ACKNOWLEDGMENTS
The authors thank Hege Kilen Andersen, Kristin Andersen, and Nina Øino for excellent technical assistance.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
FUNDING
This study was supported by grants from the Norwegian Radium Hospital Foundation.
Authors’ contributions
Ioannis Panagopoulos designed the research, evaluated the cytogenetic data, did the experiments, and wrote the manuscript. Ludmila Gorunova made the cytogenetic analyses. Ingvild Lobmeier and Bodil Bjerkehagen did the pathologic examinations. Sverre Heim supervised the project, evaluated the cytogenetics, and wrote the manuscript. All authors read and approved the final version of the manuscript.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Enzinger FM. Intramuscular Myxoma; a Review and Follow-up Study of 34 Cases. Am J Clin Pathol. 1965; 43:104–113.
2. Kindblom LG, Stener B, Angervall L. Intramuscular myxoma. Cancer. 1974; 34:1737–1744.
3. Miettinen M, Hockerstedt K, Reitamo J, Totterman S. Intramuscular myxoma—a clinicopathological study of twenty-three cases. Am J Clin Pathol. 1985; 84:265–272.
4. Nielsen GP, O’Connell JX, Rosenberg AE. Intramuscular myxoma: a clinicopathologic study of 51 cases with emphasis on hypercellular and hypervascular variants. Am J Surg Pathol. 1998; 22:1222–1227.
5. Miettinen M. Modern Soft Tissue Pathology. Tumors and Non-Neoplastic Conditions: Cambridge University Press. 2016.
6. Allen PW. Myxoma is not a single entity: a review of the concept of myxoma. Ann Diagn Pathol. 2000; 4:99–123.
7. Heymans O, Gebhart M, Alexiou J, de Saint Aubain N, Larsimont D. Intramuscular myxoma. Acta Chir Belg. 1998; 98:120–122.
8. Murphey MD, McRae GA, Fanburg-Smith JC, Temple HT, Levine AM, Aboulafia AJ. Imaging of soft-tissue myxoma with emphasis on CT, MR and comparison of radiologic and pathologic findings. Radiology. 2002; 225:215–224.
9. Aoki T, Kouho H, Hisaoka M, Hashimoto H, Nakata H, Sakai A. Intramuscular myxoma with fibrous dysplasia: a report of two cases with a review of the literature. Pathol Int. 1995; 45:165–171.
10. Gaumetou E, Tomeno B, Anract P. Mazabraud’s syndrome. A case with multiple myxomas. Orthop Traumatol Surg Res. 2012; 98:455–460.
11. Wirth WA, Leavitt D, Enzinger FM. Multiple intramuscular myxomas. Another extraskeletal manifestation of fibrous dysplasia. Cancer. 1971; 27:1167–1173.
12. Zoccali C, Teori G, Prencipe U, Erba F. Mazabraud’s syndrome: a new case and review of the literature. Int Orthop. 2009; 33:605–610.
13. Goldblum JR, Folpe AL, Weiss SW. Enzinger and Weiss´s soft tissue tumors Elsevier Saunders. 2014.
14. Meis-Kindblom JM, Sjogren H, Kindblom LG, Peydro-Mellquist A, Roijer E, Aman P, Stenman G. Cytogenetic and molecular genetic analyses of liposarcoma and its soft tissue simulators: recognition of new variants and differential diagnosis. Virchows Arch. 2001; 439:141–151.
15. Aoki T, Hisaoka M, Kouho H, Hashimoto H, Nakata H. Interphase cytogenetic analysis of myxoid soft tissue tumors by fluorescence in situ hybridization and DNA flow cytometry using paraffin-embedded tissue. Cancer. 1997; 79:284–293.
16. Delaney D, Diss TC, Presneau N, Hing S, Berisha F, Idowu BD, O’Donnell P, Skinner JA, Tirabosco R, Flanagan AM. GNAS1 mutations occur more commonly than previously thought in intramuscular myxoma. Mod Pathol. 2009; 22:718–724.
17. Okamoto S, Hisaoka M, Ushijima M, Nakahara S, Toyoshima S, Hashimoto H. Activating Gs(alpha) mutation in intramuscular myxomas with and without fibrous dysplasia of bone. Virchows Arch. 2000; 437:133–137.
18. Walther I, Walther BM, Chen Y, Petersen I. Analysis of GNAS1 mutations in myxoid soft tissue and bone tumors. Pathol Res Pract. 2014; 210:1–4.
19. Hayward BE, Bonthron DT. An imprinted antisense transcript at the human GNAS1 locus. Hum Mol Genet. 2000; 9:835–841.
20. Hayward BE, Kamiya M, Strain L, Moran V, Campbell R, Hayashizaki Y, Bonthron DT. The human GNAS1 gene is imprinted and encodes distinct paternally and biallelically expressed G proteins. Proc Natl Acad Sci USA. 1998; 95:10038–10043.
21. Hayward BE, Moran V, Strain L, Bonthron DT. Bidirectional imprinting of a single gene: GNAS1 encodes maternally, paternally, and biallelically derived proteins. Proc Natl Acad Sci USA. 1998; 95:15475–15480.
22. Heim S, Mitelman F. Cancer Cytogenetics: Chromosomal and Molecular Genetic Abberations of Tumor Cells: Wiley-Blackwell). 2015.
23. Paulsson K, Johansson B. Trisomy 8 as the sole chromosomal aberration in acute myeloid leukemia and myelodysplastic syndromes. Pathol Biol (Paris). 2007; 55:37–48.
24. Becker H, Maharry K, Mrozek K, Volinia S, Eisfeld AK, Radmacher MD, Kohlschmidt J, Metzeler KH, Schwind S, Whitman SP, Mendler JH, Wu YZ, Nicolet D, et al. Prognostic gene mutations and distinct gene- and microRNA-expression signatures in acute myeloid leukemia with a sole trisomy 8. Leukemia. 2014; 28:1754–1758.
25. Johansson B, Heim S, Mandahl N, Mertens F, Mitelman F. Trisomy 7 in nonneoplastic cells. Genes Chromosomes Cancer. 1993; 6:199–205.
26. van Roggen JF, McMenamin ME, Fletcher CD. Cellular myxoma of soft tissue: a clinicopathological study of 38 cases confirming indolent clinical behaviour. Histopathology. 2001; 39:287–297.
27. Willems SM, Mohseny AB, Balog C, Sewrajsing R, Briaire-de Bruijn IH, Knijnenburg J, Cleton-Jansen AM, Sciot R, Fletcher CD, Deelder AM, Szuhai K, Hensbergen PJ, Hogendoorn PC. Cellular/intramuscular myxoma and grade I myxofibrosarcoma are characterized by distinct genetic alterations and specific composition of their extracellular matrix. J Cell Mol Med. 2009; 13:1291–1301.
28. Cates JM, Memoli VA, Gonzalez RS. Cell cycle and apoptosis regulatory proteins, proliferative markers, cell signaling molecules, CD209, and decorin immunoreactivity in low-grade myxofibrosarcoma and myxoma. Virchows Arch. 2015; 467:211–216.
29. Idowu BD, Al-Adnani M, O’Donnell P, Yu L, Odell E, Diss T, Gale RE, Flanagan AM. A sensitive mutation-specific screening technique for GNAS1 mutations in cases of fibrous dysplasia: the first report of a codon 227 mutation in bone. Histopathology. 2007; 50:691–704.
30. Wilson CH, McIntyre RE, Arends MJ, Adams DJ. The activating mutation R201C in GNAS promotes intestinal tumourigenesis in Apc(Min/+) mice through activation of Wnt and ERK1/2 MAPK pathways. Oncogene. 2010; 29:4567–4575.
31. Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989; 340:692–696.
32. Vallar L. GTPase-inhibiting mutations activate the alpha-chain of Gs in human tumours. Biochem Soc Symp. 1990; 56:165–170.
33. Schaffer LG, McGowan-Jordan J, Schmid M. ISCN 2013: An International System for Human Cytogenetic Nomenclature (Basel: Karger). 2013.
34. Singhal H, Ren YR, Kern SE. Improved DNA electrophoresis in conditions favoring polyborates and lewis acid complexation. PloS one. 2010; 5:e11318.