
INTRODUCTION
The use of cell-free DNA (cfDNA) as a “liquid biopsy” to detect cancer biomarkers is an emerging and promising area of cancer research. Small amounts of circulating cfDNA can be detected in plasma from healthy individuals [1], is often highly elevated in cancer patients [1] and correlates with disease burden [2–4]. Advancements in technologies such as next generation sequencing (NGS) and droplet digital polymerase chain reaction (ddPCR) have facilitated detection of low-abundance cancer-specific genomic alterations in the blood. Compared to biopsy, cfDNA is minimally invasive, captures a more comprehensive representation of disease heterogeneity, and is more facile for monitoring therapy resistance-associated genetic alterations [5].
Somatic alterations in steroid hormone receptors such as estrogen receptor (ER)α, ESR1 and androgen receptor, AR have been identified following progression on targeted therapies [6–12]. The feasibility of using plasma derived cfDNA to determine receptor hormone status in patients with advanced cancer has been demonstrated recently [13–17]. Analysis of AR status in cfDNA in men with metastatic castration resistant prostate cancer (mCRPC) showed that AR gene aberrations such as gene amplification or ligand binding domain (LBD) mutations are associated with worse outcomes following next generation therapies that inhibit the androgen/AR axis such as abiraterone and enzalutamide [14, 15, 17]. While validation is still needed, these early studies highlight the clinical potential for using plasma cfDNA to detect AR status as a biomarker for therapy resistance.
Translation of preclinical findings using cfDNA into a robust clinical test will require further technical considerations. Discovery of low-abundance somatic mutations among normal cfDNA poses challenges for rigorous clinical application. AR mutations detected by deep sequencing of plasma-derived cfDNA have been reported at allelic fractions as low as 0.11% [14]. Analytical optimization will be necessary to ensure specificity as deep sequencing has the potential for low level error that may be comparable to genuine low-abundant mutations. Cross platform analysis of low-abundant mutations using technologies such as ddPCR would offer insights to the validity of these low-abundant mutations, but secondary analysis is often limited due to sample depletion. Herein, we aimed to validate low-abundant AR mutations discovered by deep sequencing cfDNA from patients with mCRPC by ddPCR.
RESULTS
Patient cohort
We prospectively enrolled 11 patients treated at Johns Hopkins Hospital (Baltimore, MD) and Sibley Memorial Hospital (Washington D.C.) for mCRPC. Patients had histologically confirmed prostate adenocarcinoma, progressive disease despite androgen deprivation therapy, and documented metastatic disease by computed tomography (CT) or bone scan with technetium-99m-labeled methylene diphosphonate. Plasma-derived cfDNA was isolated from all patients prior to initiation of enzalutamide therapy. Patient characteristics at plasma collection are summarized in Table 1. Most patients had multiple therapies prior to collection and bone disease at collection. Patient response to enzalutamide is summarized in Supplementary Figure 2A-2D.
Table 1: Patient Characteristics
Characteristics |
(n=11) |
|---|---|
Age, years |
|
Median (range) |
71 (41-90) |
Race |
|
White |
11 |
Black |
0 |
Prior Treatment for Prostate Cancer, n (%) |
|
Radical Prostatectomy |
2 (18) |
Primary Radiation |
2 (18) |
ADT |
7 (64) |
Gleason Sum, n (%) |
|
≤7 |
0 (0) |
≥8 |
9 (81.8) |
Not Available |
2 (18.2) |
Prior Treatment for Metastatic Prostate Cancer, n (%) |
|
ADT Alone |
4 (36.4) |
ADT and Abiraterone |
1 (9.1) |
ADT, Abiraterone, and Chemotherapy |
1 (9.1) |
ADT and Chemotherapy |
2 (18.2) |
ADT, Chemotherapy, and Radiation |
3 (27.3) |
PSA, ng/ml |
|
Median (range) |
46.6 (0.9-183.7) |
Site of Metastases, n (%) |
|
Bone Metastases |
9 (81.8) |
Visceral Metastases |
1 (9.1) |
Lymph Lode Only |
1 (9.1) |
PSA Progression Free Survival, days |
|
Median (range) |
168 (35-466) |
Deep NGS of AR from plasma-derived cfDNA
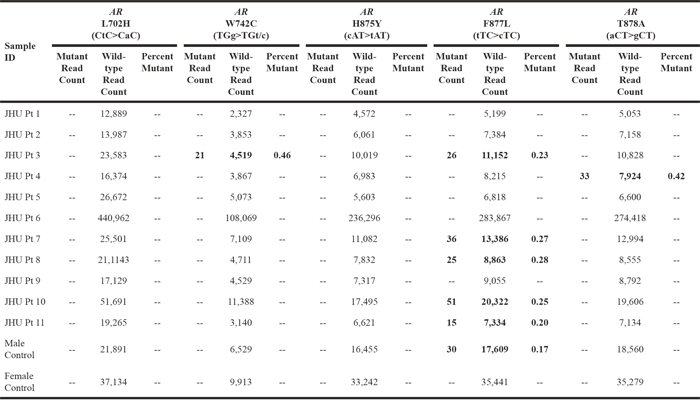
Plasma derived cfDNA from patients with mCRPC was amplified using the Qiagen Prostate targeted panel with Qiagen HotStarTaq and Qiagen HiFi Taq and then deep sequenced for AR mutations on the Illumina HiSeq. Plasma derived cfDNA from two healthy donors (one male and one female) was also amplified and sequenced. The AR (TGg>TGt/c) W742C hotspot mutation was detected in one patient at an allelic frequency of 0.46% (Table 2). The AR (aCT>gCT) T878A hotspot mutation was detected in one patient at an allelic frequency of 0.42% (Table 2). These mutations were not detected in cfDNA from the healthy controls (Table 2). Unexpectedly, the AR (tTC>cTC) F877L hotspot mutation was detected in approximately 45% of the patients (n=5/11) at allelic fractions between 0.20-0.28% (Table 2). While these allelic fractions were within previously reported ranges for AR hotspot mutations identified using similar technologies in comparable patients, the frequency of the AR F877L mutation in this cohort was over ten-fold higher than in previously published studies [14, 18, 19]. In addition, this mutation was also detected at a lower level in the healthy male control (Table 2). Other AR hotspot mutations, AR (CtC>CaC) L702H and AR (cAT>tAT) H875Y were not detected in any of the patient samples or controls (Table 2).
Table 2: NGS of AR using Qiagen Library Amplification Phred Quality Score ≥ 25
Validation of AR mutations from cfDNA by ddPCR
The unprecedented frequency of the AR F877L mutation in this cohort as well as its overall low allelic fraction and its occurrence in a healthy male control collectively indicate these mutations to be false positives. As the NGS workflow utilized in this study was designed to limit DNA contamination and the NGS data did not show evidence of sample to sample contamination, the AR F877L mutations were most likely not from contaminating DNA. Error can occur due to Taq polymerase infidelity during targeted library amplification prior to sequencing or during NGS and subsequent variant calling. As AR F877L mutations have been reported at low allelic frequencies and AR F877L was not detected in all patient samples or controls, we sought to query these samples by an alternate platform to determine if any were genuine mutations that were masked by low level sequencing error. We additionally sought to use an alternate platform to determine if this site is commonly prone to amplification mediated error. To do this, we examined samples by ddPCR with prior preamplification using high fidelity polymerases, NEB Phusion® or Invitrogen™ Platinum™ SuperFi™ (Table 3). Patient cfDNA, wild-type control genomic DNA, and a no template control were preamplified using Phusion® prior to ddPCR. Due to limited sample amount, the five positive samples by NGS for the AR F877L mutation, the one positive sample by NGS for the AR T878A mutation, wild-type genomic DNA, and a no template control were amplified by Platinum SuperFi™. Preamplified samples and controls and non-amplified controls were then examined for wild-type AR and the AR F877L hotspot mutation by ddPCR. Ten of the eleven samples preamplified by Phusion® were positive for the AR F877L (tTC>cTC) mutation by ddPCR at very low allelic fractions ranging from 0.007 to 0.033% (Table 4). Notably, Phusion® preamplified wild-type genomic DNA was also positive for the AR F877L (tTC>cTC) mutation by ddPCR while non-amplified wild-type genomic DNA was negative suggesting that the AR F877L mutation was introduced prior to ddPCR during preamplification with Phusion® (Table 4). DNA contamination is not likely the source of the AR F877L mutation as both the preamplified and the non-amplified no template controls were negative. In contrast to Phusion® preamplification, none of the Platinum SuperFi™ preamplified patient samples or wild-type control genomic DNA were positive for the AR F877L mutation (Table 4). Control genome equivalents assayed were comparable between the two polymerases suggesting that fidelity differences were not due to under representation or sensitivity. These data suggest that this specific AR locus is prone to PCR based error and that under these conditions at this locus, Platinum SuperFi™ has greater fidelity than Phusion®.
Table 3: Company Reported Polymerase Fidelity
Qiagen HotStarTaq |
NEB Phusion® High-Fidelity DNA Polymerase |
Invitrogen™ Platinum™ SuperFi™ DNA Polymerase |
|
|---|---|---|---|
Components |
Modified recombinant 94 kDa Taq DNA polymerase originally isolated from Thermus aquaticus |
Pyrococcus-like proofreading enzyme fused with a processivity-enhancing domain. |
Chemically engineered Pyrococcus-like enzyme |
Company Reported Fidelity |
Error Rate of 2 x 10-5 |
Error Rate of 4.4 x 10-7 |
Greater than 100X Taq fidelity |
Table 4: Comparison of NEB Phusion® and Invitrogen™ Platinum™ SuperFi™ Preamplification of AR by ddPCR for AR F877L
Sample ID |
AR |
AR |
||||
|---|---|---|---|---|---|---|
Mutant Droplet Count |
Wild-type |
Percent |
Mutant |
Wild-type |
Percent |
|
JHU Pt 1 |
5 |
18298 |
0.027 |
-- |
-- |
-- |
JHU Pt 2 |
8 |
31108 |
0.026 |
-- |
-- |
-- |
JHU Pt 3 |
5 |
27672 |
0.018 |
0 |
14824 |
0.000 |
JHU Pt 4 |
5 |
35181 |
0.014 |
0 |
14164 |
0.000 |
JHU Pt 5 |
11 |
32975 |
0.033 |
-- |
-- |
-- |
JHU Pt 6 |
0 |
41833 |
0.000 |
-- |
-- |
-- |
JHU Pt 7 |
7 |
68073 |
0.010 |
0 |
40588 |
0.000 |
JHU Pt 8 |
10 |
36587 |
0.027 |
0 |
10163 |
0.000 |
JHU Pt 9 |
6 |
25642 |
0.023 |
-- |
-- |
-- |
JHU Pt 10 |
4 |
59673 |
0.007 |
0 |
16317 |
0.000 |
JHU Pt 11 |
8 |
37478 |
0.021 |
0 |
17926 |
0.000 |
Preamplified |
11 |
30348 |
0.036 |
0 |
44104 |
0.000 |
Preamplified No Template Control |
0 |
0 |
0.000 |
0 |
0 |
0.000 |
Non-Amplified |
0 |
36018 |
0.000 |
0 |
7761 |
0.000 |
Non-Amplified |
964 |
0 |
100 |
810 |
0 |
100 |
Non-Amplified No Template Control |
0 |
0 |
0.000 |
0 |
0 |
0.000 |
As Phusion® has been used for preamplification of cfDNA prior to ddPCR without reported false positives [13], we sought to determine if the AR locus encompassing the F877 codon was particularly susceptible to error during amplification or if preamplification with Phusion® broadly introduced a low level of error under these conditions. To do this, we examined both the adjacent codon on exon 8 for the AR T878A hotspot mutation and an upstream codon on exon 5 for the hotspot mutation AR W742C by ddPCR of Phusion® and Platinum SuperFi™ preamplified wild-type genomic DNA and patient cfDNA. Due to limited sample amount, we selected seven of the eleven patient samples including the five that were positive for the AR F877L mutation and the one that was positive for the AR T878A mutation by deep sequencing and wild-type genomic DNA for preamplification by Phusion® and Platinum SuperFi™ prior to ddPCR for wild-type AR and the AR T878A mutation. Control cfDNA from a patient with an AR T878A mutation was included to further validate the efficacy of the AR T878A ddPCR probe. Five of the seven Phusion® preamplified patient samples as well as the wild-type genomic DNA were also positive for the AR T878A mutation, but at significantly (P=0.02) lower allelic frequencies (0.004 to 0.011%) than the AR F877L mutation (Table 5). Decreasing preamplification cycle number from 22 to 12 did not eliminate the introduction of either the AR F877L or AR T878A mutations when wild-type genomic DNA was preamplified by Phusion® (Table 6). In contrast to Phusion® preamplification, all Platinum SuperFi™ preamplified JHU patient samples and wild-type genomic DNA were negative for the AR T878A mutation (Table 5). Interestingly, Patient 4, who was positive for the AR T878A mutation by NGS, was negative by ddPCR while the AR T878A positive control cfDNA was positive for the AR T878A mutation. Collectively, this suggests that the AR T878A mutation detected by NGS in JHU patient 4 was a false positive. Again, genome equivalents were comparable between assays suggesting that differences were not due to under representation or sensitivity.
Table 5: Comparison of NEB Phusion® and Invitrogen™ Platinum™ SuperFi™ Preamplification of AR by ddPCR for AR T878A
Sample ID |
AR |
AR |
||||
|---|---|---|---|---|---|---|
Mutant |
Wild-type |
Percent |
Mutant |
Wild-type |
Percent |
|
JHU Pt 1 |
1 |
11707 |
0.009 |
0 |
41931 |
0.000 |
JHU Pt 3 |
1 |
15126 |
0.007 |
0 |
26745 |
0.000 |
JHU Pt 4 |
3 |
26983 |
0.011 |
0 |
30737 |
0.000 |
JHU Pt 7 |
0 |
12156 |
0.000 |
0 |
35931 |
0.000 |
JHU Pt 8 |
1 |
24205 |
0.004 |
0 |
27729 |
0.000 |
JHU Pt 10 |
0 |
51299 |
0.000 |
0 |
20279 |
0.000 |
JHU Pt 11 |
1 |
27082 |
0.004 |
0 |
32648 |
0.000 |
Preamplified |
2 |
36329 |
0.006 |
0 |
21511 |
0.000 |
Preamplified |
0 |
0 |
0.000 |
0 |
0 |
0.000 |
Preamplified cfDNA from a patient with an AR T878A mutation detected by NGS |
--- |
--- |
--- |
233 |
11890 |
1.960 |
Non-Amplified |
0 |
16132 |
0.000 |
0 |
24915 |
0.000 |
Non-Amplified |
122 |
0 |
100 |
7502 |
0 |
100 |
Non-Amplified |
0 |
0 |
0.000 |
0 |
0 |
0.000 |
Table 6: Comparison of NEB Phusion® and Invitrogen™ Platinum™ SuperFi™ Preamplification of AR by ddPCR for AR Hotspot Mutations
Wild-type |
NEB Phusion® |
NEB Phusion® |
Invitrogen™ Platinum™ SuperFi™ |
||||||
|---|---|---|---|---|---|---|---|---|---|
Mutant |
Wild-type |
Percent |
Mutant Read Count |
Wild-type |
Percent Mutant |
Mutant Read Count |
Wild-type |
Percent Mutant |
|
AR |
11 |
30348 |
0.036 |
1 |
8539 |
0.012 |
0 |
44104 |
0.000 |
AR |
2 |
36329 |
0.006 |
1 |
9617 |
0.010 |
0 |
21511 |
0.000 |
We next examined patient samples and wild-type genomic DNA preamplified by Phusion® and Platinum SuperFi™ for wild-type AR and the AR hotspot mutation W742C. Notably, Patient 7, who was negative for AR W742C by deep sequencing, and wild-type genomic DNA were both negative by ddPCR when preamplified by either Phusion® or Platinum SuperFi™ (Table 7). Patient 3, who was positive for the AR hotspot mutation W742C by deep sequencing, was also positive by ddPCR using either Phusion® or Platinum SuperFi™ preamplified cfDNA (Table 7). Collectively, this suggests that AR (tTC>cTC) F877L and to a lesser extent AR (aCT>gCT) T878A are prone to error during Phusion® mediated PCR amplification while other AR loci such as AR (TGf>TGt/c) W742C may not be.
Table 7: Comparison of NEB Phusion® and Invitrogen™ Platinum™ SuperFi™ Preamplification of AR by ddPCR for AR W742C
Sample ID |
AR |
AR |
||||
|---|---|---|---|---|---|---|
Mutant Droplet Count |
Wild-type |
Percent |
Mutant Droplet Count |
Wild-type |
Percent |
|
JHU Pt 3 |
4 |
25067 |
0.016 |
38 |
33611 |
0.113 |
JHU Pt 7 |
0 |
19753 |
0.000 |
0 |
15958 |
0.000 |
Preamplified |
0 |
17573 |
0.000 |
0 |
37069 |
0.000 |
Preamplified |
0 |
0 |
0.000 |
0 |
0 |
0.000 |
Non-Amplified |
0 |
31225 |
0.000 |
0 |
31225 |
0.000 |
Non-Amplified |
10602 |
0 |
100 |
10602 |
0 |
100 |
Non-Amplified |
0 |
0 |
0.000 |
0 |
0 |
0.000 |
Since prior pre-clinical and clinical studies [14, 19–22] support that AR F877L and AR T878A may mediate resistance to androgen-AR axis therapies such as enzalutamide, we examined for an association of AR gene aberrations including AR LBD hotspot mutations and AR amplification with enzalutamide response. AR amplification was determined by ddPCR (Supplementary Figure 1). Analyses of validated AR gene aberrations (AR hot spot mutations and AR amplification) support that AR gene aberrations trend, but not significantly, with a worse PSA response as measured as best PSA percent change (>0 versus ≤0) by Fisher’s exact test (Supplementary Figure 2A). Significance was further reduced by inclusion of the false positive AR F877L and AR T878A mutations detected by NGS (Supplementary Figure 2C). Kaplan-meier PSA progression-fee survival curves support that AR gene aberrations were significantly associated with a shorter time to PSA progression on enzalutamide by Log-rank (Mantel-Cox) tests (Supplementary Figure 2B). Significance was slightly decreased by inclusion of false positive AR F877L and AR T878A mutations detected by NGS (Supplementary Figure 2D). While the small cohort size limits robust conclusions pertaining to the association of AR gene aberrations with PSA response or PSA progression free survival, these findings are consistent with prior findings [14, 15, 17].
DISCUSSION
Mainstay treatment strategies for newly diagnosed metastatic prostate cancer exploit prostate cancer addiction to AR signaling by inhibiting the androgen/AR axis with androgen deprivation therapies (ADT). While ADT is initially effective in most men, prostate cancers almost universally recur after a variable amount of time leading to mCRPC. Despite androgen blockade, the majority of newly diagnosed mCRPC are thought to have continued dependence on androgen signaling. Similar to first line ADT, next generation therapies for mCRPC such as abiraterone and enzalutamide also function by inhibiting androgen/AR signaling. Somatic alterations in AR including splice variants [23–26] and genetic alterations such as amplification and LBD mutations [14, 15, 17] have been associated with resistance to these therapies. Somatic point mutations in AR have been shown to occur in 5-18% of patients with mCRPC [12, 27, 28] and account for nearly 60% of cases if combined with AR amplification [12]. Consequently, there is much enthusiasm in validating AR status as a biomarker to guide therapeutic decisions for men with mCRPC.
Evaluating and monitoring AR status in patients with mCRPC using traditional metastatic biopsies poses several clinical challenges including cost, patient discomfort, sample collection, and lack of ease for serial analyses. Single site biopsies may also not account for tumor heterogeneity. Due to these limitations, many rapidly developing technologies have focused on revolutionizing the detection of tumor specific genetic alterations in the blood as a “liquid biopsy”. Use of cfDNA as a tumor analyte combined with NGS as a platform for detection show great promise in the clinic as a means for mutation detection and monitoring. While these technologies have demonstrated proof of principle, further optimization and validation is necessary to define parameters for sensitivity and specificity. Concerns currently exist pertaining to the rate of false positives and the associated potential clinical ramifications. NGS of cfDNA for the detection of tumor specific mutations often involves polymerase based amplification which can introduce errors. While NGS approaches such as SAFESeqS [29] and Duplex sequencing [30] may dramatically limit sequencing based false positives, these technologies have yet to be widely implemented. Thus, rigorous optimization and standardization of these technologies will be needed prior to use for clinical decision making.
To begin to address issues pertaining to false positive rate and lower bound threshold for detection, we sought to identify mutations in AR by NGS of plasma-derived cfDNA from patients with mCRPC and then to cross-platform validate by ddPCR. Our findings suggest variability in polymerase fidelity at specific AR LBD hotspot loci. We demonstrate that some high fidelity polymerases used for preamplification prior to NGS and ddPCR can introduce low level mutations at AR LBD hotspot F877L and to a lesser extent at AR T878A. Interestingly, other AR LBD hotspots such as AR W742C did not show false positives by any of the tested polymerases. Sequence differences between these loci were not readily appreciable as they contain comparable GC content and single and dinucleotide repeats (Figure 1). The experimental error rates at the adjacent LBD hotspot loci, 877 and 878, far exceeded the reported overall error rate for these polymerases, thereby suggesting that AR F877L and AR T878A may be particularly prone to amplification error. These data support that previously unappreciable areas of the genome may be more susceptible to NGS preamplification based errors.

Figure 1: Genomic region surrounding loci of AR hotspot mutations.
Clinical data and laboratory based studies support that both the AR T878A and the AR F877L mutations have potential clinical significance. AR T878A was originally identified in the LNCaP prostate cancer cell line [31] while AR F877L was identified using an in vitro screen for resistance mechanisms to anti-androgens [21]. AR F877L and AR T878A have been detected in patients with mCRPC [14, 18, 19, 22]; however, the AR F877L mutation has only been detected by NGS of cfDNA from mCRPC patients [14, 18, 19] and has yet to be reported in a metastatic lesion by NGS [12, 27, 28]. Laboratory based studies support that both AR T878A [19] and AR F877L [18, 19, 22] may confer resistance to enzalutamide and ARN-509. Thus reliable detection of these and other AR LBD mutations may impact decisions in the clinic.
This study highlights the need for rigorous protocol optimization for the detection of mutations in cfDNA by NGS. We demonstrate that some loci may be more susceptible to polymerase based errors and thereby stress the need for loci specific polymerase optimization and standardization, use of proper controls, variant calling, and validation. While use of more stringent variant calling criteria would exclude the F877L false positives detected by NGS and improve specificity, it would consequently decrease sensitivity by also excluding genuine low abundant mutations such as that found in Patient 3. Currently, an accepted standard or platform for using cfDNA for mutation detection does not exist. These studies underscore the need to establish and to validate protocols for mutation detection and monitoring in cfDNA by NGS. By doing so, integration of these technologies will ultimately advance patient care.
MATERIALS AND METHODS
Patients and sample collection
Human subject research was approved by the Johns Hopkins Medicine Institutional Review Board (IRB). We prospectively enrolled patients diagnosed with metastatic castration resistant prostate cancer (mCRPC) (n=11) between September 2014 and April 2015 prior to initiation of enzalutamide. PSA progression was determined at the date of the first PSA rise that was followed by a subsequent PSA rise or at the date of physician determined change of therapy. All patients provided written and informed consent to participate in the study. Two healthy controls also participated in the study. Three 10 ml blood samples were collected in Streck BCT tubes prior to therapy initiation, stored at room temperature, and processed for plasma isolation within 24 hours. To optimize patient sample integrity and to limit DNA contamination, plasma was extracted in a bleach and UV cleaned hood specifically for plasma extraction in a room dedicated for blood processing, storage, and cell-free DNA isolation. Plasma was extracted from blood by centrifugation for 10 minutes at 1500 x g followed by a second centrifugation for 10 minutes at 3000 x g as previously described [13, 32, 33]. Plasma was stored at -80°C in 1mL aliquots. In a dedicated bleach and UV cleaned hood, cell-free DNA was extracted from plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen) per the manufacturer’s protocol. To limit contamination, only one patient sample was processed at a time. Cell-free DNA was quantified using the Quant-iT PicoGreen dsDNA assay kit (Invitrogen) as per the manufacturer’s protocol.
Deep next generation sequencing
GeneRead™ DNAseq Targeted Panel V2 (Qiagen) was used to prepare libraries from cfDNA for NGS as per the manufacturer’s protocol. NGS libraries were prepared in dedicated bleach and UV cleaned hood. Samples were PCR amplified using Qiagen HotStar Taq as per the manufacturer’s protocol. Libraries were amplified using Qiagen HiFi PCR Master Mix for 4 cycles. NGS was performed on the Illumina Hi-Seq with an average depth of coverage of 10,000x. Data was aligned to hg19 using bwa-0.7.7 (mem function). SAMtools was used to filter reads by quality (Phred ≥ 25 and mapQ >18 were included). The threshold for non-reference reads was 15 and single strand variant calls, non-reference reads less than or equal to 0.15%, non-reference reads less than twice the next highest non-reference frequency, variants in more than 50% of the samples, and variants over 50% of the allelic fraction were excluded. AR hotspot variants were additionally examined using the Integrative Genomics Viewer (IGV) http://www.broadinstitute.org/igv/. Variants that passed these filters were then validated by ddPCR. Variant calls were made independent/blinded to outcome data.
Droplet digital PCR
Control genomic DNA (Promega) was digested with MseI (NEB). Genomic DNA, patient cfDNA, and water (no template control) were PCR amplified using Phusion® with Phusion® HF buffer (NEB) or Platinum SuperFi™ (Invitrogen) at loci surrounding AR amino acid 742 and AR amino acid 877/878 for either 12 or 22 cycles. PCR amplification primer sequences are located in Supplementary Table 1. Three independent preamplification PCR reactions of 1 ng DNA each were pooled for each sample and control. Similarly, three independent preamplification PCR reactions were pooled for each no template control. Preamplified samples and controls were purified prior to ddPCR using the MinElute PCR Purification kit (Qiagen) and eluted in RNAase free water according to the manufacturer’s instructions. Non-amplified AR W742C, AR F877L, and AR T878A mutant control DNA for ddPCR were double-stranded purified DNA gBlocks®Gene Fragments (IDT) of approximately 400 base pairs that were reconstituted in RNAase free water. The non-amplified no template control had RNAase free water without template.
Dual labeled (FAM or HEX) fluorescent-quencher hydrolysis probes (IDT) were designed for AR hotspot mutations (W742C, F877L, and T878A) and their respective wild-type loci. Sequences for ddPCR primers and probes are located in Supplementary Table 1. Wild-type control genomic female DNA (Promega) digested with MseI and mutant DNA gBlocks®Gene Fragments (IDT) were used to optimize primer/probe conditions. Probes to AR (ABI Hs04121925_cn FAM), ZXDB (ABI Hs02220689_cn FAM), and NSUN3 (Bio-Rad dHsaCP2506682) were used to determine AR copy number. Samples with AR copy number greater than or equal to 1.9 were defined as having increased AR copy number. Wild-type control genomic female and male DNA (Promega) digested with MseI for AR or HaeIII for ZXDB were used for AR copy number controls. Cell-free DNA from healthy male and female donors was also used as a control. Droplet digital PCR (Bio-Rad) was performed in a dedicated UV equipped hood and according to the manufacturer’s protocol. Total WT and mutant DNA molecules were quantified by the QX200 Droplet Reader software. Results for each mutation analysis were recorded as the summation of four or more replicates.
Statistical analysis
Statistical Analyses were performed using GraphPad Prism software. All statistical tests were two-sided and P values less than 0.05 were considered statistically significant.
ACKNOWLEDGMENTS
We thank our patients who consented to partake in this study.
CONFLICTS OF INTEREST
The authors do not have conflicts of interest to disclose.
GRANT SUPPORT
This work was supported by the Department of Defense CDMRP Prostate Cancer Research Program PC130991, W81XWH-14-1-0284 (PJH, BHP, BT, AG), The Patrick C. Walsh Foundation (BHP, PJH), and The Prostate Cancer Foundation Young Investigator Award (ESA).
REFERENCES
1. Shapiro B, Chakrabarty M, Cohn EM, Leon SA. Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer. 1983; 51: 2116-20. doi: 10.1002/1097-0142(19830601)51:11<2116::AID-CNCR2820511127>3.0.CO;2-S.
2. Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, Rajan S, Humphray S, Becq J, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013; 368: 1199-209. doi: 10.1056/NEJMoa1213261.
3. Rago C, Huso DL, Diehl F, Karim B, Liu G, Papadopoulos N, Samuels Y, Velculescu VE, Vogelstein B, Kinzler KW, Diaz LA, Jr. Serial assessment of human tumor burdens in mice by the analysis of circulating DNA. Cancer Res. 2007; 67: 9364-70. doi: 10.1158/0008-5472.CAN-07-0605.
4. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA, Jr. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008; 14: 985-90. doi: 10.1038/nm.1789.
5. Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013; 10: 472-84. doi: 10.1038/nrclinonc.2013.110.
6. Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, He X, Liu S, Hoog J, Lu C, Ding L, Griffith OL, Miller C, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013; 4: 1116-30. doi: 10.1016/j.celrep.2013.08.022.
7. Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, Yelensky R, Brown M, Miller VA, Sarid D, Rizel S, Klein B, Rubinek T, et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013; 73: 6856-64. doi: 10.1158/0008-5472.CAN-13-1197.
8. Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, Kalyana-Sundaram S, Wang R, Ning Y, Hodges L, Gursky A, Siddiqui J, Tomlins SA, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013; 45: 1446-51. doi: 10.1038/ng.2823.
9. Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, Hudis C, Chen D, Taran T, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013; 45: 1439-45. doi: 10.1038/ng.2822.
10. Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, Ferrer-Lozano J, Perez-Fidalgo JA, Cristofanilli M, Gomez H, Arteaga CL, Giltnane J, Balko JM, et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014; 20: 1757-67. doi: 10.1158/1078-0432.CCR-13-2332.
11. Chen EJ, Sowalsky AG, Gao S, Cai C, Voznesensky O, Schaefer R, Loda M, True LD, Ye H, Troncoso P, Lis RL, Kantoff PW, Montgomery RB, et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin Cancer Res. 2015; 21: 1273-80. doi: 10.1158/1078-0432.CCR-14-1220.
12. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015; 161: 1215-28. doi: 10.1016/j.cell.2015.05.001.
13. Chu D, Paoletti C, Gersch C, VanDenBerg DA, Zabransky DJ, Cochran RL, Wong HY, Toro PV, Cidado J, Croessmann S, Erlanger B, Cravero K, Kyker-Snowman K, et al. ESR1 Mutations in Circulating Plasma Tumor DNA from Metastatic Breast Cancer Patients. Clin Cancer Res. 2016; 22: 993-9. doi: 10.1158/1078-0432.CCR-15-0943.
14. Azad AA, Volik SV, Wyatt AW, Haegert A, Le Bihan S, Bell RH, Anderson SA, McConeghy B, Shukin R, Bazov J, Youngren J, Paris P, Thomas G, et al. Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clin Cancer Res. 2015; 21: 2315-24. doi: 10.1158/1078-0432.CCR-14-2666.
15. Romanel A, Gasi Tandefelt D, Conteduca V, Jayaram A, Casiraghi N, Wetterskog D, Salvi S, Amadori D, Zafeiriou Z, Rescigno P, Bianchini D, Gurioli G, Casadio V, et al. Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med. 2015; 7: 312re10. doi: 10.1126/scitranslmed.aac9511.
16. Salvi S, Casadio V, Conteduca V, Burgio SL, Menna C, Bianchi E, Rossi L, Carretta E, Masini C, Amadori D, Calistri D, Attard G, De Giorgi U. Circulating cell-free AR and CYP17A1 copy number variations may associate with outcome of metastatic castration-resistant prostate cancer patients treated with abiraterone. Br J Cancer. 2015; 112: 1717-24. doi: 10.1038/bjc.2015.128.
17. Salvi S, Casadio V, Conteduca V, Lolli C, Gurioli G, Martignano F, Schepisi G, Testoni S, Scarpi E, Amadori D, Calistri D, Attard G, De Giorgi U. Circulating AR copy number and outcome to enzalutamide in docetaxel-treated metastatic castration-resistant prostate cancer. Oncotarget. 2016; 7:37839-37845. doi: 10.18632/oncotarget.9341.
18. Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B, Hager JH. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013; 3: 1020-9. doi: 10.1158/2159-8290.CD-13-0226.
19. Lallous N, Volik SV, Awrey S, Leblanc E, Tse R, Murillo J, Singh K, Azad AA, Wyatt AW, LeBihan S, Chi KN, Gleave ME, Rennie PS, et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biol. 2016; 17: 10. doi: 10.1186/s13059-015-0864-1.
20. Wyatt AW, Azad AA, Volik SV, Annala M, Beja K, McConeghy B, Haegert A, Warner EW, Mo F, Brahmbhatt S, Shukin R, Le Bihan S, Gleave ME, et al. Genomic Alterations in Cell-Free DNA and Enzalutamide Resistance in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016. doi: 10.1001/jamaoncol.2016.0494.
21. Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, Chen Y, Greene GL, Shen Y, Sawyers CL. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife. 2013; 2: e00499. doi: 10.7554/eLife.00499.
22. Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, Yuan J, Kovats SG, Kim S, Cooke VG, Monahan JE, Stegmeier F, Roberts TM, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013; 3: 1030-43. doi: 10.1158/2159-8290.CD-13-0142.
23. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, Lotan TL, Zheng Q, De Marzo AM, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014; 371: 1028-38. doi: 10.1056/NEJMoa1315815.
24. Qu F, Xie W, Nakabayashi M, Zhang H, Jeong SH, Wang X, Komura K, Sweeney CJ, Sartor O, Lee GM, Kantoff PW. Association of AR-V7 and prostate specific antigen RNA levels in blood with efficacy of abiraterone acetate and enzalutamide treatment in men with prostate cancer. Clin Cancer Res. 2016. doi: 10.1158/1078-0432.CCR-16-1070.
25. Todenhofer T, Azad A, Stewart C, Gao J, Eigl BJ, Gleave ME, Joshua AM, Black PC, Chi KN. AR-V7 transcripts in whole blood RNA of patients with metastatic castration resistant prostate cancer correlate with response to Abiraterone acetate. J Urol. 2016. doi: 10.1016/j.juro.2016.06.094.
26. Scher HI, Lu D, Schreiber NA, Louw J, Graf RP, Vargas HA, Johnson A, Jendrisak A, Bambury R, Danila D, McLaughlin B, Wahl J, Greene SB, et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016. doi: 10.1001/jamaoncol.2016.1828.
27. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012; 487: 239-43. doi: 10.1038/nature11125.
28. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV, Varambally S, Tomlins SA, Nanus DM, Tagawa ST, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016; 22: 298-305. doi: 10.1038/nm.4045.
29. Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A. 2011; 108: 9530-5. doi: 10.1073/pnas.1105422108.
30. Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci U S A. 2012; 109: 14508-13. doi: 10.1073/pnas.1208715109.
31. Veldscholte J, Ris-Stalpers C, Kuiper GG, Jenster G, Berrevoets C, Claassen E, van Rooij HC, Trapman J, Brinkmann AO, Mulder E. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun. 1990; 173: 534-40. doi: 10.1016/S0006-291X(05)80067-1.
32. Higgins MJ, Jelovac D, Barnathan E, Blair B, Slater S, Powers P, Zorzi J, Jeter SC, Oliver GR, Fetting J, Emens L, Riley C, Stearns V, et al. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin Cancer Res. 2012; 18: 3462-9. doi: 10.1158/1078-0432.CCR-11-2696.
33. Beaver JA, Jelovac D, Balukrishna S, Cochran RL, Croessmann S, Zabransky DJ, Wong HY, Valda Toro P, Cidado J, Blair BG, Chu D, Burns T, Higgins MJ, et al. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin Cancer Res. 2014; 20: 2643-50. doi: 10.1158/1078-0432.CCR-13-2933.