
INTRODUCTION
Colorectal cancer (CRC) is a major worldwide health problem owing to its high prevalence and mortality rates [1]. Immune surveillance and responses have been shown to prevent and inhibit the initiation and progression of CRC [2]. However, Immune escape is still an intriguing problem involved in CRC pathogenesis and metastasis [3]. Thorough understanding of anti-CRC immunity will shed new lights on developing effective therapeutics for CRC.
Infiltration of immune cells into tumors has been associated with destruction of tumor cells, reduction of tumor burden, and better clinical prognosis [4]. Among various immune cells, T cells are especially important for suppressing and eliminating CRC cells. Tumor antigen-specific T cells can be detected in the blood and bone marrow of 30% to 40% of patients with CRC [5, 6]. CRC patients with more T cell infiltration have better outcomes [4, 7, 8]. In particular, TNF-α and IFN-γ are both critical for T cells to protect CRC patients through inhibition of cancer cell proliferation and induction of apoptosis [9–17]. However, how T cell function is regulated in CRC sites has not been completely understood.
In this study, we examined the effect of CRC cell-derived Wnt proteins on intratumoral T cell function in a murine CRC implantation model. We found that CRC cell lines expressed an array of Wnt proteins, while intratumoral T cells increased various Frizzled (FZD) proteins as Wnt receptors. Meanwhile, both active β-catenin and total β-catenin were elevated in intratumoral T cells in comparison with splenic T cells. We further found that CRC cell-derived Wnt proteins suppressed IFN-γ expression and increased IL-17a expression in activated CD4+ T cells. However, the cytotoxic activity of CD8+ T cells was not altered by CRC cells. Enforced expression of β-catenin in intratumoral CD4+ T cells increased IL-17a expression. Moreover, enforced expression of β-catenin in intratumoral CD4+ T cells enhanced proliferation and inhibited apoptosis of CRC cells. Taken together, our study disclosed a new mechanism by which CRC impairs T cell immunity.
RESULTS
CRC cell lines express various Wnt proteins
Elevated expression of Wnt3, Wnt3a and Wnt10a in CRC has been disclosed by previous studies [18–20]. We firstly identify the expression of Wnt3, Wnt3a, Wnt5a and Wnt10a in several CRC cell lines. As shown in Figure 1A, in comparison with normal mouse colon tissue, expression of Wnt3, Wnt3a and Wnt10a was significantly increased in CT26.CL25, HT-29, SW480 and HCT-116 cells, whereas Wnt5a expression was only moderately increased in CT26.CL25 and SW480 cells. Among these cell lines, CT26.CL25 expressed high levels of Wnt3, Wnt3a and Wnt10a. Therefore, CT26.CL25 cells were utilized in the following experiments. To determine the in vivo expression of Wnt proteins in CT26.CL25 cells, CT26.CL25 cells were subcutaneously inoculated into Rag1−/− mice to form tumor grafts. Western blot analysis showed that Wnt3, Wnt3a and Wnt10a were indeed expressed in the tumor grafts, and their expression levels were higher than those in normal subcutaneous tissue and normal mouse colon (Figure 1B). To determine the expression of Wnts in non-CRC cell types in the tumor grafts, we sorted host-derived endothelial cells, fibroblasts, macrophages and dendritic cells and tested mRNA levels of Wnt3, Wnt3a, Wnt5a and Wnt10a in these cells. In comparison with implanted CT26.CL25 cells, non-CRC cell types expressed very low or no Wnt3, Wnt3a, Wnt5a and Wnt10a, suggesting that implanted CRC cells were the major source of these Wnts (Supplementary Figure 1). In addition, other Wnts that were reported to be present in both normal colon tissue and CRC, such as Wnt2b, Wnt4 and Wnt7b [21], were all expressed in these CRC cell lines, although at different levels (Figure 1C).
Intratumoral T cells express FZD proteins
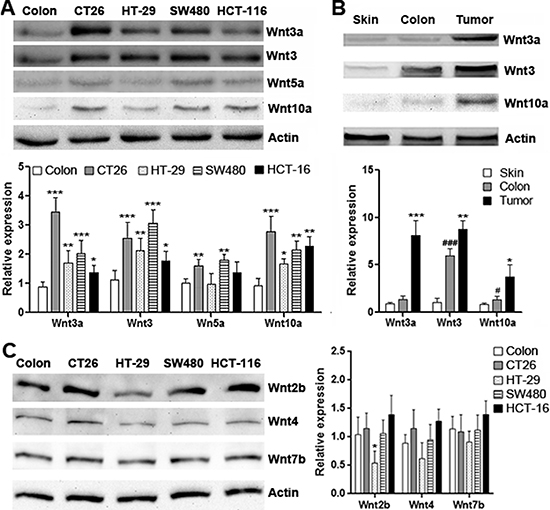
Figure 1: Expression of Wnt proteins in CRC cell lines. (A) Expression of Wnt3a, Wnt3, Wnt5a and Wnt10a in mouse and human CRC cell lines. Upper panel: representative Western blot images. Lower panel: statistical analysis for expression of each Wnt protein. N = 6 per group. *p < 0.05; **p < 0.01; ***p < 0.001 compared with normal colon tissue. (B) Expression of Wnt3a, Wnt3 and Wnt10a in normal tissues and CT26.CL25 tumor grafts. Upper panel: representative Western blot images. Lower panel: Statistical analysis for expression of each Wnt protein. Skin: normal skin tissue. Colon: normal colon tissue. Tumor: tumor grafts. N = 4 per group. *p < 0.05; **p < 0.01; ***p < 0.001 compared with normal colon tissue. ###p < 0.05; ###p < 0.001 compared with normal skin tissue. (C) Expression of Wnt2b, Wnt4 and Wnt7b. Left panel: representative Western blot images. Right panel: Statistical analysis for expression of each Wnt protein. N = 3 per group. *p < 0.05 compared with normal colon tissue.
A previous research has outlined expression of FZD proteins in resting and effector T cells in vitro [22]. So we also checked expression of these FZD proteins in intratumoral T cells to characterize the potential Wnt signaling in anti-tumor T cells. To do so, splenic CD3+ T cells were enriched by flow cytometry from BALB/C mice and were transferred into tumor-bearing Rag1−/−mice. Three weeks after transfer, T cells were localized in proximity to CRC cells in tumor grafts (Supplementary Figure 2). FZD proteins were determined in T cells isolated from spleens and tumor grafts. As shown in Figure 2A, TCR+CD4+ and CD8+ donor-derived T cells were present in both spleens and tumor grafts. Flow cytometry and Western blot analysis indicated that splenic T cells expressed very low levels of these FZD proteins except for FZD-6. However, FZD-3, FZD-5 and FZD-7 were increased in intratumoral CD4+ and CD8+ T cells in comparison with splenic counterparts, whereas FZD-6 was only slightly increased in intratumoral CD4+ T cells. FZD-4 was just increased in a small subpopulation of either CD4+ or CD8+ T cells (Figure 2B and 2C). Taken together, our data suggested that intratumoral T cells have higher expression of FZD proteins than splenic T cells.
β-catenin is activated in intratumoral T cells
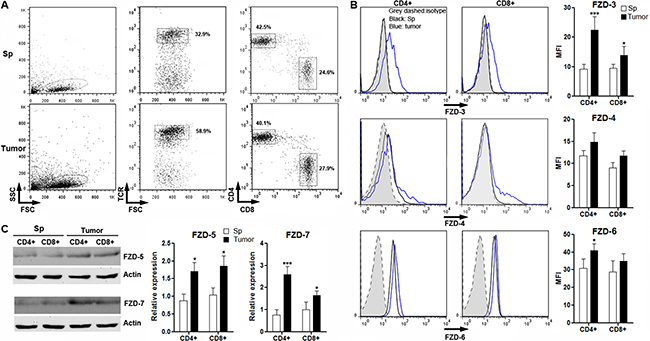
Figure 2: Expression of FZD protein in intratumoral T cells. (A) Gating strategy for splenic and intratumoral CD4+ and CD8+ T cells. Numbers in the plots are the percentages of gated populations. Sp: spleen. Tumor: tumor grafts. This is a representative of three independent experiments. (B) Expression of FZD-3, FZD-4 and FZD-6 on T cells were determined by flow cytometry. Left panel: representative histograms. Right panel: statistical analysis for mean fluorescence intensity of each FZD protein. (C) Expression of FZD-5 and FZD-7 in T cells were determined by Western blot. Left panel: representative image. Right panel: statistical analysis for each FZD protein. Sp: spleen. Tumor: tumor grafts. N = 3 per group. *p < 0.05; ***p < 0.001 compared with splenic T cell subsets.
To further define the activation status of canonical Wnt signaling in T cells, both active β-catenin and total β-catenin were detected in splenic and intratumoral T cells. We found that intratumoral CD4+ and CD8+ T cells expressed remarkably higher active β-catenin and total β-catenin than splenic T cell subsets (Figure 3A and 3B), suggesting β-catenin activation in intratumoral T cells. It was also noted that β-catenin expression was very weak in splenic T cells, which was consistent with previous reports [23, 24]. The increase of active β-catenin in intratumoral T cells was also confirmed by flow cytometry analysis (Figure 3C).
CT26.CL25 cells induce activation of β-catenin in activated T cells
To identify the role of CRC cells in inducing β-catenin activation in T cells, we co-cultured CT26.CL25 cells with T cells in the presence or absence of agonistic antibodies in Transwell plates (Figure 4A), followed by detection of active β-catenin in T cells. As shown in Figure 4B, either agonistic antibodies or tumor cells alone was unable to induce up-regulation of active β-catenin in T cells, while co-existence of agonistic antibodies and tumor cells increased expression of active β-catenin, suggesting that both T cell activation and tumor cell-derived soluble factors are indispensable for efficient Wnt signaling in T cells. The up-regulation of active β-catenin was further confirmed by flow cytometry analysis (Figure 4C). Moreover, co-culture with tumor cells had no remarkable impact on T cell apoptosis (Figure 4D), suggesting that potential changes of T cell functions might not be resulted from alteration of T cell viability.
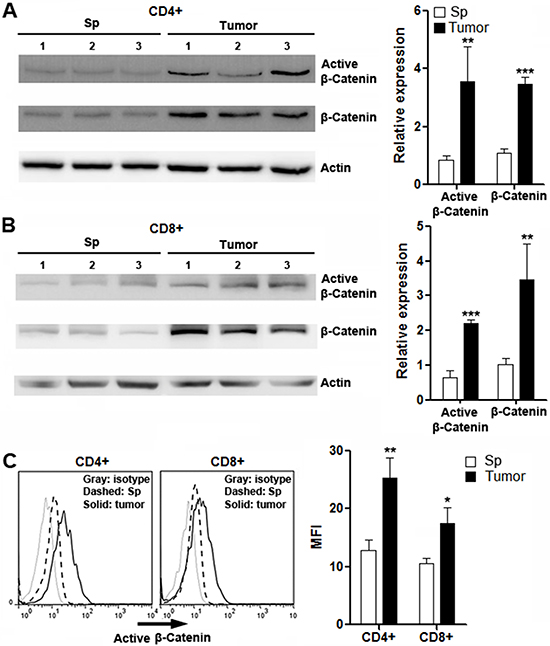
Figure 3: Expression of active β-catenin and total β-catenin in intratumoral T cells. (A and B) Expression of active β-catenin and total β-catenin in splenic and intratumoral CD4+ (A) and CD8+ (B) T cells were determined by Western blot. Left panels: representative Western blot images. Right panels: statistical analysis. (C) Expression of active β-catenin in splenic and intratumoral CD4+ and CD8+ T cells were determined by flow cytometry. Left panel: representative histograms. Right panel: statistical analysis for mean fluorescence intensity of active β-catenin. Sp: spleen. Tumor: tumor grafts. N = 3 per group. *p < 0.05; **p < 0.01; ***p < 0.001 compared with splenic T cell counterparts.
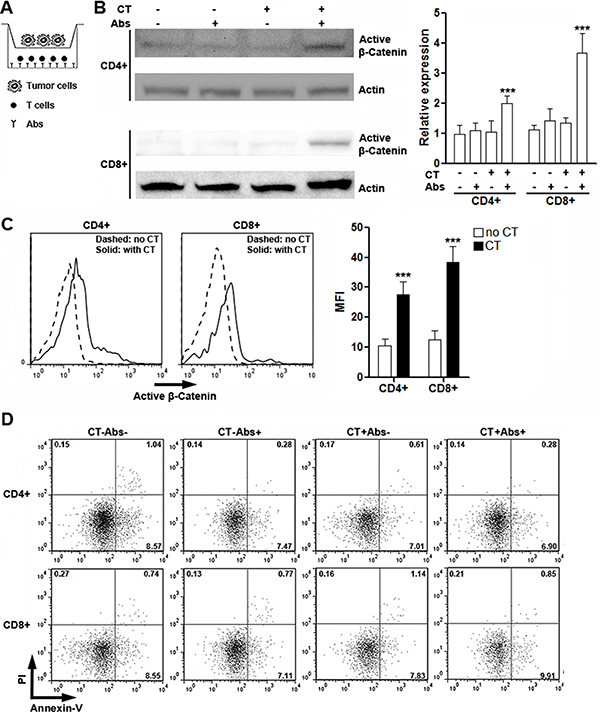
Figure 4: CT26.CL25 cells induce activation of β-catenin in activated T cells. (A) Illustration of co-culture system. (B) Expression of active β-catenin in cultured splenic CD4+ and CD8+ T cells was determined by Western blot. Left panels: representative Western blot images. Right panels: statistical analysis for active β-catenin. CT: CT26.CL25 cells. Abs: agonistic antibodies. N = 6 per group. ***p < 0.001 compared with T cell culture without CT26.CL25 cells and agonistic antibodies. (C) Expression of active β-catenin in activated splenic CD4+ and CD8+ T cells was determined by flow cytometry. Note that T cells shown here are all stimulated with agonistic antibodies. Left panel: representative histograms. Right panel: statistical analysis. No CT: without CT26.CL25 cells. CT: co-culture with CT26.CL25 cells. N = 6 per group. ***p < 0.001 compared with “No CT” group. (D) Flow cytometry analysis of T cell apoptosis in culture. Numbers in the quadrants are percentages of corresponding gated populations. This is a representative of two independent experiments.
CT26.CL25 cells bias CD4+ T cell polarization towards Th17 via Wnt signaling
To determine the influences of CRC cells on T cell immunity, we co-cultured CT26.CL25 cells with T cells in the presence or absence of agonistic antibodies in Transwell plates before detection of cytokine production and master regulator expression. For CD4+ T cells without agonistic antibody stimulation, co-culture with CT26.CL25 cells could not their change cytokine production and master regulator expression, suggesting that CT26.CL25 cells could not induce activation of resting CD4+ T cells (Figure 5A and 5B). In the presence of agonistic antibodies, CT26.CL25 cells inhibited expression of IL-2 and IFN-γ but increased IL-17a production in CD4+ T cells (Figure 5A). Consistently, in the presence of agonistic antibodies, CT26.CL25 cells lower the expression of T-bet while up-regulating RORγt (Figure 5B). Expression of IL-4 or Gata-3 was not altered (Figure 5A and 5B). For CD8+ T cells, no significant changes were observed in the presence of CT26.CL25 cells (Figure 5C). Our findings suggested that CT26.CL25 cells might predominantly influence CD4+ T cell function. So in the following experiments we focused on CD4+ T cells only.
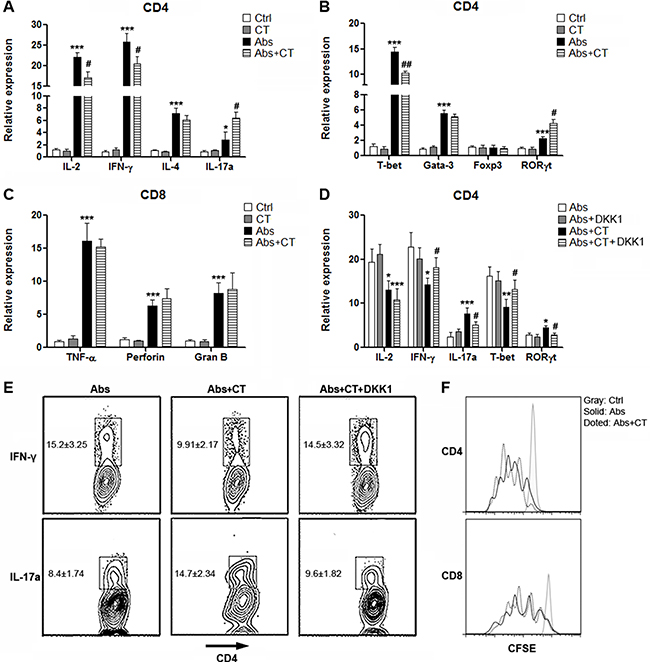
Figure 5: CT26.CL25 cells bias CD4+ T cell polarization towards Th17 via Wnt signaling. (A and B) Expression of cytokines (A) and master regulators (B) in cultured CD4+ T cells were determined by real-time PCR. Ctrl: untreated T cells. CT: T cells co-cultured with CT26.CL25 cells. Abs: agonistic antibody-stimulated T cells. Abs+CT: agonistic antibody-stimulated T cells co-cultured with CT26.CL25 cells. (C) Expression of cytotoxic mediators in cultured CD8+ T cells were determined by real-time PCR. (D) Expression of cytokines and master regulators in cultured CD4+ T cells were determined by real-time PCR. Abs: agonistic antibody-stimulated T cells. Abs+DKK1: agonistic antibody-stimulated T cells in the presence of DKK1. Abs+CT: agonistic antibody-stimulated T cells co-cultured with CT26.CL25 cells. Abs+CT+DKK1: agonistic antibody-stimulated T cells co-cultured with CT26.CL25 cells and DKK1. (E) Expression of IFN-γ and IL-17a were measured by flow cytometry analysis. Numbers in the plots are percentages of gated populations. (F) CFSE dilution in proliferative CD4+ T cells. N = 6 per group. For A to C, *p < 0.05; **p < 0.01; ***p < 0.001 compared with Ctrl group. #p < 0.05; ##p < 0.01 compared with Abs group. For D, *p < 0.05; **p < 0.01; ***p < 0.001 compared with Abs group. #p < 0.05 compared with Abs+CT group.
To examine the role of Wnt signaling in CRC cell-induced alterations of CD4+ T cells, we added Wnt antagonist DKK1 into the co-culture system. As shown in Figure 5D, in the absence of CT26.CL25 cells, DKK1 itself did not change the expression of cytokines or master regulators, suggesting that Wnt signaling did not function during CD4+ T cell activation. However, DKK1 abolished the effects of CT26.CL25 cells on CD4+ T cell activation, demonstrated by restored expression of IFN-γ and T-bet, as well as down-regulation of IL-17a and RORγt. The changes of IFN-γ and IL-17a were also confirmed by flow cytometry analysis (Figure 5E). Furthermore, CT26.CL25 cells did not alter the proliferation of activated CD4+ T cells (Figure 5F).
Enforced expression of β-catenin in CD4+ T cells through lentiviral transduction
To clarify the role of Wnt signaling in T cell immunity in vivo, expression of β-catenin was enforced in CD4+ T cells. These CD4+ T cells contained CRC-reactive T cells, because they were sorted from the draining lymph nodes of BALB/C mice which were subcutaneously inoculated with CT26.CL25 cells. Transduction of β-catenin Lentiviral Activation Particles (Thereinafter Lenti-β) prompted robust expression of β-catenin as well as active β-catenin (Figure 6A and 6B). T cell apoptosis was not influenced by lentirivral transduction or β-catenin up-regulation (Figure 6C). Expression of two Wnt signaling target genes, Tcf-1 and LEF-1, was notably increased in Lenti-β-transduced T cells, further confirming the activation of canonical Wnt signaling (Figure 6D). To measure the transmigration of T cells into the colon cancer site, T cells transduced with control lentivirus (Thereinafter Lenti-C) were labeled with CFSE before adoptive transfer with T cells transduced with Lenti-β into tumor-bearing Rag1−/− mice. One week after transfer, the proportions of CFSE+ and CFSE− T cells were detected in intratumoral T cells (Figure 6E). As shown in Figure 6E, almost equal percentages of CFSE+ and CFSE− T cells were found in intratumoral T cells, suggesting that β-catenin expression did not interfere with T cell migration. Furthermore, active β-catenin expression level was markedly higher in Lenti-β-transduced T cells in both spleens and tumor grafts (Figure 6F to Figure 6G).
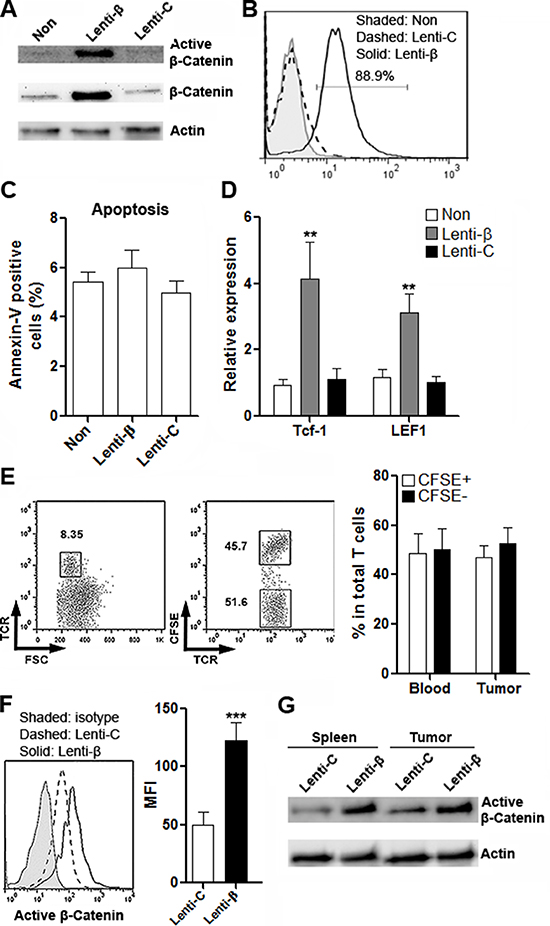
Figure 6: Enforced expression of β-catenin in CD4+ T cells through lentiviral transduction. (A) Expression of active β-catenin and total β-catenin in CD4+ T cells after lentiviral transduction was determined by Western blot. This is a representative of two independent experiments. Non: non-transduced T cells. Lenti-β: T cells transduced with β-catenin Lentiviral Activation Particles. Lenti-C: T cells transduced with control lentiviral particles. (B) Expression of active β-catenin in T cells was detected by flow cytometry. This is a representative of two independent experiments. (C) T cells apoptosis after lentiviral transduction. N = 4 per group. (D) Expression of Tcf-1 and LEF1 in T cells were evaluated by real-time PCR after lentiviral transduction. N = 6 per group. **p < 0.01 compared with “Non” group. (E) Proportions of CFSE- and CFSE+ T cells in tumor grafts were evaluated by flow cytometry. Left panel: representative dot plots. Right panel: statistics. Numbers in the plots are proportions of corresponding gated populations. N = 3 per group. (F) Expression of active β-catenin in donor-derived intratumoral T cells was determined by flow cytometry. Left panel: representative histograms. Right panel: statistics of MFI. N = 3 per group. ***p < 0.001. (G) Expression of active β-catenin in donor-derived splenic and intratumoral T cells was determined by Western blot. This is a representative of two independent experiments.
Wnt signaling in CD4+ T cells favors CRC graft growth
To determine the CD4+ T cell reaction against CRC, CD4+ T cells were transduced with Lenti-C or Lenti-β and were then adoptively transferred into tumor-bearing Rag1−/− mice. Cytokine production and master regulator expression in intratumoral T cells was measured two weeks after transfer. As shown in Figure 7A, in comparison with Lenti-C-transduced T cells, Lenti-β-transduced T cells showed lower IFN-γ and higher IL-17a production, while IL-2 and IL-4 were not altered. Consistently, reduced T-bet expression and increased RORγt were found in Lenti-β-transduced T cells (Figure 7B). These results were consistent with our in vitro data mentioned above. To evaluate the anti-CRC immune reaction in CRC grafts, cytokine production in the whole CRC graft tissue was determined two weeks after transfer. We found that the expression of IFN-γ was decreased while the expression of IL-17a was increased in the whole CRC graft tissue in mice receiving Lenti-β-transduced T cells (Figure 7C). To evaluate CRC graft growth, CD45-H-2Kd+ CRC cells were detected for further characterization (Figure 7D). Ki67 staining of CRC cells indicated that Lenti-C-transduced T cells inhibited cancer cell growth, whereas Lenti-β-transduced T cells were weaker in doing so (Figure 7E and 7F and Supplementary Figure 3A). Apoptosis assay demonstrated that Lenti-β-transduced T cells were less capable of inducing cancer cell apoptosis than Lenti-C-transduced T cells (Figure 7G and 7H and Supplementary Figure 3B). Moreover, measurement of CRC graft size showed larger tumor volume in mice receiving Lenti-β-transduced T cells, compared with mice receiving Lenti-C-transduced T cells (Figure 7I). In general, our data indicated that excessive Wnt signaling weakened anti-cancer reaction of CD4+ T cells, and favored CRC graft growth.
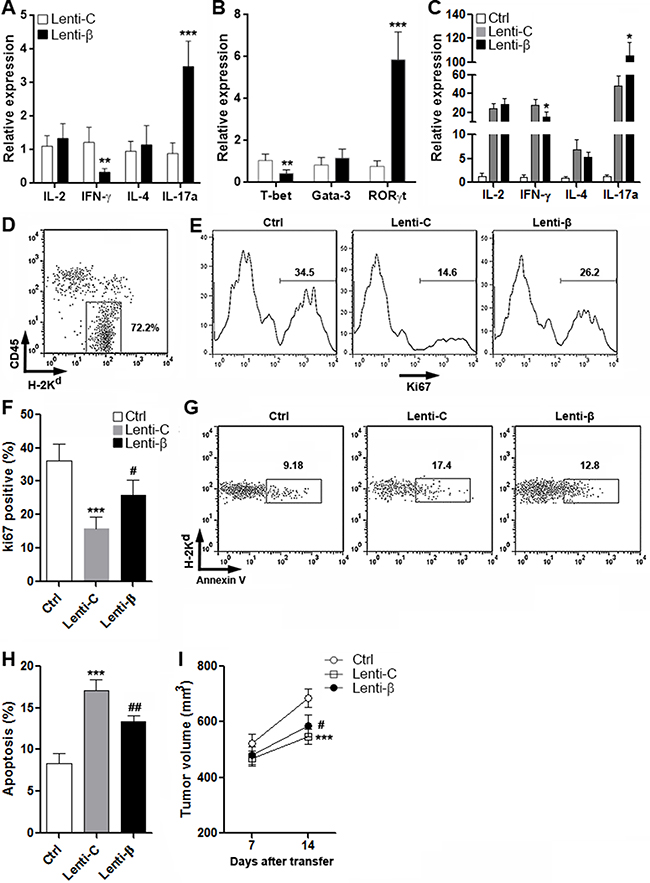
Figure 7: Enforced expression of β-catenin in CD4+ T cells favors growth of CRC grafts. (A and B) Expression of cytokines (A) and master regulators (B) in donor-derived intratumoral CD4+ T cells were assessed by real-time PCR. Lenti-β: T cells transduced with β-catenin Lentiviral Activation Particles. Lenti-C: T cells transduced with control lentiviral particles. N = 6 per group. **p < 0.01; ***p < 0.001 compared with Lenti-C group. (C) Expression of indicated cytokines in CRC grafts. Ctrl: mice receiving PBS. Lenti-C: mice receiving T cells transduced with control lentiviral particles. Lenti-β: mice receiving T cells transduced with β-catenin Lentiviral Activation Particles. N = 6 per group. *p < 0.05 compared with Lenti-C group. (D) Gating strategy for CT26.CL25 cell detection in CRC grafts. (E and F) Proliferation of implanted CT26.CL25 cells. Histograms of Ki67 staining were shown in (E), and statistics was shown in (F). N = 6 per group. (G and H) Apoptosis of implanted CT26.CL25 cells was measured by flow cytometry. Representative dot plots of Annexin V staining were shown in (G), and statistics was shown in (H). N=6 per group. (I) Tumor volume after adoptive transfer. N = 10 per group. For F, H and I, ***p < 0.001 compared with Ctrl group. #p < 0.05, ##p < 0.01 compared with Lenti-C group.
DISCUSSION
Wnts are crucial for cell proliferation, survival, migration and polarity, specification of cell fate, and self-renewal of stem cells [25]. The canonical Wnt pathway is a central mechanism in cancer biology, either beneficial or detrimental to the prognosis of cancer patients [26]. In CRC, activation of Wnt pathway has also been linked to carcinogenesis [27]. For example, expression of Wnt3, Wnt3a and Wnt10a are increased in CRC [18–20]. Wnt2 and Wnt5a expression are also elevated in colon cancer compared to normal colon and during the progression from adenoma to carcinoma [28, 29]. In contrast, the expression of other Wnts, including Wnt2b, 4, 7b, and 10b, does not change, as they remain strongly expressed in both normal colon and colon cancer [21]. Our data was consistent with above reports, showing higher Wnt3, Wnt3a, Wnt5a and Wnt10a in most cell lines tested. The CT26.CL25 cell-derived subcutaneous tumor grafts also expressed significant Wnt proteins.
Some immune cells expressing Wnt receptors could be target cells of Wnt proteins. Indeed, Wnt proteins induce changes of immune cell viability, differentiation and functions [30]. Due to the critical role of T cells in tumor immunity, Wnt signaling in T cells was focused in our research. Our study was inspired by a recent report demonstrating elevated β-catenin expression in CRC-infiltrating T cells [31]. We speculated that CRC cells could be the source of various Wnt ligands which activate canonical Wnt signaling in T cells. Based on a previous study [22], we found that expression of almost all FZDs in splenic naïve T cells were very weak except for FZD-6, and intratumoral T cells markedly increased FZD-3, FZD-5 and FZD-7. Hence, our data suggests higher FZD expression in CRC-reactive intratumoral T cells. However, future study will be necessary to test the antigen specificity of intratumoral T cells. Furthermore, other FZDs that have not been tested here, such as FZD-1, FZD-2 and FZD-8, should be evaluated in future.
We also found that intratumoral T cells had high levels of active β-catenin and total β-catenin, suggesting activation of Wnt signaling. However, due to insufficient information about engagement of Wnt ligands with specific FZD receptors, it is difficult for us to speculate which Wnt ligand or which FZD is predominant for suppressing T cell function. Probably different Wnt ligands simultaneously bind to different FZDs to synergistically induce Wnt signaling.
Our in vitro study suggests that CRC cells indeed stimulate canonical Wnt signaling in activated T cells. Interestingly, although TCR signaling was reported to stabilize β-catenin in T cells [32], our study did not show the same result. In our study, agonistic antibodies could not accumulate β-catenin, in accordance with a previous report indicating constitutive degradation of β-catenin in both resting and CD3/CD28 antibody-primed T cells [23]. Hence, it seems that activation of Wnt signaling unlikely resulted from TCR or CD3 signaling.
Studies indicate that Wnt/β-catenin signaling is a key regulator of T cell development at various stages of thymocyte differentiation [30, 33]. However, the exact significance of Wnt/β-catenin signaling for mature T cell immunity is still controversial. Inconsistent or even contradictory results have been generated in the past decades [24, 34–39], reflecting the subtle role of Wnt signaling in regulating T cell function. It is very likely that the effect of Wnt signaling is differential in distinct subpopulations of T cells. Regardless the previous inconsistent data, the inhibitory effect of Wnt signaling on T cell function has been repeatedly observed [23, 31, 39]. We also showed that CRC predominantly impair Th1 function. Interestingly, CRC-induced Wnt signaling increased IL-17a and RORγt expression in CD4+ T cells, which was consistent with two previous research [36, 40] but in contrast to to another two studies [41, 42]. It is possible that the comprehensive effect of CRC-derived Wnt ligands favors Th17 differentiation. It is also likely that CRC cells produce other cytokines and soluble mediators which work with Wnt ligands to induce Th17 polarization. Further research on the significance of Wnt signaling in Th17 polarization is necessary.
Our in vivo study using enforced β-catenin expression in CD4+ T cells further confirmed the in vitro data. The lower IFN-γ production in β-catenin-overexpressed T cells was accompanied by higher proliferation and less apoptosis of CRC cells, suggesting that Wnt signaling impaired CD4+ T cell-mediated anti-CRC immunity. In addition, IL-17a was reported to facilitate colon cancer development, promoting cancer-elicited inflammation and preventing cancer cells from immune surveillance [43–45]. Therefore, Wnt signaling not only inhibits Th1 cell-mediated CRC cell death, but also enhances Th17 cell-mediated CRC growth. We did not use CD8+ T cells for in vivo study because we did not see substantial functional changes of CD8+ T cells in vitro. However, we plan to test them in a similar system in the near future.
It should be noted that Wnt5a, a protein triggering non-canonical Wnt signaling, is also expressed in CRC cell lines, although it seems that its expression level is relatively weaker than other Wnt proteins. Non-canonical Wnt signaling has been shown to play roles in T cell development [46–48] but its role in mature T cell function is not clear. In addition, non-canonical Wnt signaling might play a critical role in CRC carcinogenesis. In particular, Wnt5a can induce an antagonistic signaling against canonical Wnt signaling [49] so it could inhibit CRC initiation or progression. However, due to its relatively low expression and the complexity of Wnt signaling, it is still hard to speculate the exact role of Wnt5a in CRC. More carefully designed experiments are needed in future.
Taken together, we are the first to indicate that CRC-derived Wnt ligands can influence T cell immunity in the tumor microenvironment. Our study shed new lights on the study of enhancing anti-CRC T cell immunity and might be beneficial for developing novel therapeutic interventions.
MATERIALS AND METHODS
CRC cell line culture
CRC cell line CT26.CL25 (mouse), HT-29, SW480 and HCT-116 cells (human) were obtained from the American type culture collection. Cells were cultured in Dulbecco-modified essential media (DMEM, Gibco) containing 4.5 g/l glucose supplemented with 10% fetal bovine serum (FBS, Hyclone), 3.44 mg/ml L-glutamine and 100 mg/ml of penicillin-streptomycin.
Mouse CRC cell implantation model
All animal experiments were conducted in compliance with institutional guidelines and Jilin University Guidelines for the Use of Animals. All animal procedures were approved by Jilin University Animal Care and Use Committee. Eight-to-ten week old male BALB/C and Rag1−/− mice (C57BL/6J background) were obtained from the Weitonglihua(Beijing) Laboratory Animal Technology Co., Ltd.. Rag1−/− mice were subcutaneously (s.c.) inoculated with 0.5 × 106 CT26.CL25 cells in 0.2 ml phosphate-buffered saline (PBS) at the lower left flank. Four weeks after inoculation, the mice were euthanized by inhalation of carbon dioxide for an average of 5 min. Tumor volume was measured according to the standard formula 1/2 × L × W2.
Adoptive transfer of T cells
CD4+ and CD8+ T cells were sorted from BALB/C mouse axillary and inguinal lymph nodes using EasySep CD4+ T cell isolation Kit and CD8+ T cell isolation Kit (both from Miltenyi Biotec), respectively, according to the manufacturer’s manual. In some experiments, the donor BALB/C mice were inoculated with 1 × 106 CT26.CL25 cells at the left flank, and T cells were sorted two weeks after inoculation followed by in vitro expansion and lentiviral transduction as mentioned below. 1 × 107 T cells (total T cells or CD4+ T cells) were retro-orbitally injected into tumor-bearing Rag1−/− mice two weeks after inoculation of CT26.CL25 cells. The adoptive transfer was performed every 3 days for a week.
Isolation of T cells and CT26.CL25 cells from implanted tumors
The tumor-bearing mice were anesthetized with isoflurane before perfusion with 10 ml ice-cold PBS from the heart. Fresh tumor tissues were cut into about 1 mm3 pieces and digested at 37°C for 30 min in 2 ml of RPMI 1640 supplemented with 0.05% collagenase Type IV (Sigma-Aldrich), 0.002% DNase I (Roche) and 20% FBS (Hyclone). Digested tumor tissues were then pressed through a 70-μm mesh to prepare single cell suspensions. For intratumoral T cell isolation or detection, the single cell suspension was carefully layered onto equal volume of Ficoll-Paque reagent (GE Healthcare) followed by centrifugation at 400 g for 30 min at room temperature. Mononuclear cells were then collected from the interface, washed in 3 volumes of PBS, and were incubated in 3 ml of Tris-NH4Cl solution for 3 min to lyse residual red blood cells. Cells were washed with PBS twice before sorting or detection T cells using flow cytometry. For CT26.CL25 cell detection, the single cell suspensions were directly stained with indicated antibodies or markers followed by flow cytometry analysis.
Western blot
The cells, normal mouse colon tissue and normal mouse skin tissue were lysed in RIPA buffer containing protease inhibitor cocktail (Sigma-Aldrich). The following antibodies were used: anti-β-actin (ab8226), anti-Wnt2b (ab178418), anti-Wnt3 (ab32249), anti-Wnt3a (ab28472), anti-Wnt4 (ab91226), anti-Wnt5a (ab72583), anti-Wnt7b (ab94915), anti-Wnt10a (ab106522), anti-FZD-5 (ab75234), and anti-FZD-7 (ab64635) were purchased from Abcam. Anti-active β-catenin (D13A1) and anti-total β-catenin (D10A8) were purchased from Cell Signaling Technology. Membranes were developed with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and the optical density was analyzed using a Biospetrum 500 imaging system.
RNA extraction, cDNA synthesis and real-time PCR
Total RNA was extracted from cells or tissues using the RNeasy Mini Kit (Qiagen). One microgram of total RNA from each sample was transcribed into cDNA using SuperScript® III First-Strand Synthesis System (Invitrogen) following the manufacturers’ instruction. qRT-PCR was performed using Fast SYBR® Green Master Mix (Invitrogen) on a 7300 qRT-PCR System (Invitrogen). Data was analyzed with 7300 system software. Primer sequences for each gene are as follows: β-actin (5′-AGAGGGAAATCGTGCGTGAC-3′ and 5′-CAATAGTGATGACCTGGCCGT-3′). T-bet (5′-ACCGCTTATATGTCCACCCA-3′ and 5′-GAGAGACTGCAGGACGATCA-3′). Gata3 (5′-CCCATTACCACCTATCCGCC-3′ and 5′- GTTCACACACTCCCTGCCTT-3′). Foxp3(5′- TTGGCCAGCGCCATCTT-3′ and 5′-TGCCTCCTCCAGAGAGAAGTG-3′). RORc (5′-CACGGCCCTGGTTCTCAT-3′ and 5′-GCAGATGTTCCACTCTCCTCTTCT-3′). IL-2 (5′-CCTGAGCAGGATGGAGAATTACA-3′ and 5′-TCCAGAACATGCCGCAGAG-3′). IL-4 (5′-CTTTCGGGCTTTTCGATGCC-3′ and 5′-TCAGTGATGTGGACTTGGACTC-3′). IL-17a (5′-AGTCCAGGGAGAGCTTCATCT-3′ and 5′-CACGCTGAGCTTTGAGGGAT-3′). IFN-γ (5′-TGAACGCTACACACTGCATCTTGG-3′ and 5′-CGACTCCTTTTCCGCTTCCTGAG-3′). Perforin(5′-CTGGCAGGGACGATGACCT-3′ and 5′-GGGAACCAGACTTGG GAGC-3′). Granzyme B (5′-ATCAAGGATCAGCAGCCTGA-3′ and 5′-TG ATGTCATTGGAGAATGTCT-3′). TNF-α (5′-GCCTCTTCTCATTCCTGCT
TG-3′ and 5′-CTGATGAG AGGGAGGCCATT-3′). PCR conditions used for all primer sets were as follows: 95°C hot start for 10 min, followed by 40 amplification cycles of 95°C for 30 s (denaturing), 60°C for 1 min (annealing, extension and detection). Relative abundance of RNA was analyzed using 2−ΔΔCt method.
Flow cytometry analysis
The following anti-mouse antibodies or reagents were used for detection and sorting of T cells and CT26.CL25 cells: APC anti-CD45 (30-F11), FITC anti- H-2Kd (SF1-1.1), PE anti-TCRβ (H57-597), APC-Cy7 anti-CD4 (GK1.5), PE-Cy7 anti-CD8a (53-6.7), FITC anti-IFN-γ (MP6-XT22) and Alexa Fluor® 488 anti-IL-17a (TC11-18H10) were purchased from BD Pharmingen. Anti-FZD-3, anti-FZD-4 and anti-FZD-6 were purchased from R&D Systems. Anti-active β-catenin (D13A1) was purchased from Cell Signaling Technology. PE anti-Ki67 was purchased from Biolegend. PE Annexin V and carboxyfluorescein diacetate succinimidyl ester (CFSE) were purchased from BD Pharmingen.
For cell surface staining, cells were incubated with corresponding antibodies in PBS for 30 min on ice before analysis on a BD LSRII flow cytometer. Dead cells that were stained by propidium iodide (PI, 5 μg/ml) were excluded. For intracellular cytokine staining, cells were fixed and permeabilized with BD Cytofix/perm and Perm/wash buffer according to the manufacturer’s protocol, respectively. Then cells were stained at room temperature for 30 min with FITC anti-IFN-γ or Alexa Fluor® 488 anti-IL-17a. For active β-catenin staining, cells were fixed for 10 min at 37°C with BD Cytofix/perm buffer followed by permeabilization with 90% methanol for 30 min on ice. Then cells were stained for 30 min at room temperature with anti-active β-catenin antibody (1:50). Cells were washed with PBS once before incubation with FITC goat anti-rabbit IgG (BD Pharmigen) for 0 min at room temperature. For cell apoptosis assay, cells were stained with 2 μg/ml PI and PE Annexin V (BD Pharmigen) according to the manufacturer’s instruction. All samples were detected on a BD LSRII flow cytometer (BD Bioscience). All flow cytometry data was analyzed with Flowjo 7.6.1 software. Cell sorting was performed on a BD FACSAria cell sorter based on cell surface marker staining.
Co-culture of T cells with CT26.CL25 cells
T cells were treated in the presence or absence of agonistic antibodies before co-culture. Briefly, a 96-well plate was pre-coated with 5 μg/ml anti-CD3 monoclonal antibody (17A2, eBiscience). 1 × 105 sorted CD4+ or CD8+ T cells were seeded into each pre-coated well and incubated for two days in the presence of 2 μg/ml anti-CD28 antibody (37.51, eBioscience). Then these T cells were transferred into a 24-well plate pre-coated with 5 μg/ml anti-CD3 monoclonal antibody. Meanwhile, 2.5 × 105 CT26.CL25 cells were seeded into each Transwell insert (0.4 μm pore. Corning Costar), and the inserts were placed in the T cell-containing 24-well plate. After 48 h co-culture, T cells were collected and subject to further processing. In some experiments, recombinant mouse Dickkopf 1 (Dkk1, R&D Systems), an inhibitor of Wnt signalling, was added at 400 ng/ml in the co-culture.
For T cell proliferation assay, T cells were labelled with 2 μM CFSE before stimulation with agonistic antibodies and co-culture as mentioned above. CFSE dilution in T cells were then evaluated by flow cytometry.
For intracellular cytokine staining, the cancer cell-containing Transwell inserts were removed after co-culture. T cells were re-stimulated with 20 ng/ml phorbol myristate acetate (PMA) and 1 μM Ionomycin in the presence of 5 μg/ml brefedin A and 5 μg/ml monensin for 4 h before flow cytometry analysis.
Lentiviral transduction
β-catenin Lentiviral Activation Particles were purchased from Santa Cruz Biotechnology. 1.5 × 106 CD4+ T cells were expanded in vitro for 7 days in 6-well plates with plate-bound anti-CD3 antibody and soluble anti-CD28 antibody in the presence of 100 U/ml recombinant mouse IL-2 (R&D systems). At day 5 after expansion, polybrene was added into the medium to 5 μg/ml. Then T cells were transduced with lentiviral particles at an MOI of 20 overnight. Cells were washed and expanded for additional 2 days. Then T cells were washed with PBS and were subject to further experiments.
Statistics
Most data was presented as mean ± SEM and analyzed by statistical software (Prism 6.0, GraphPad Software). Student’s t test or one-way ANOVA were used for comparison of mean between the groups. P values < 0.05 were considered significant.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by National Natural Science Foundation of China grant (No. 81372295 & No. 81402374).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Deschoolmeester V, Baay M, Lardon F, Pauwels P, Peeters M. Immune Cells in Colorectal Cancer: Prognostic Relevance and Role of MSI. Cancer Microenviron. 2011; 4:377–392.
2. Jacobs J, Smits E, Lardon F, Pauwels P, Deschoolmeester V. Immune Checkpoint Modulation in Colorectal Cancer: What’s New and What to Expect. J Immunol Res. 2015; 2015:158038.
3. Pancione M, Giordano G, Remo A, Febbraro A, Sabatino L, Manfrin E, Ceccarelli M, Colantuoni V. Immune escape mechanisms in colorectal cancer pathogenesis and liver metastasis. J Immunol Res. 2014; 2014:686879.
4. Prall F, Duhrkop T, Weirich V, Ostwald C, Lenz P, Nizze H, Barten M. Prognostic role of CD8+ tumor-infiltrating lymphocytes in stage III colorectal cancer with and without microsatellite instability. Hum Pathol. 2004; 35:808–816.
5. Koch M, Beckhove P, Op den Winkel J, Autenrieth D, Wagner P, Nummer D, Specht S, Antolovic D, Galindo L, Schmitz-Winnenthal FH, Schirrmacher V, Buchler MW, Weitz J. Tumor infiltrating T lymphocytes in colorectal cancer: Tumor-selective activation and cytotoxic activity in situ. Ann Surg. 2006; 244:986–992; discussion 992–983.
6. Bonertz A, Weitz J, Pietsch DH, Rahbari NN, Schlude C, Ge Y, Juenger S, Vlodavsky I, Khazaie K, Jaeger D, Reissfelder C, Antolovic D, Aigner M, et al. Antigen-specific Tregs control T cell responses against a limited repertoire of tumor antigens in patients with colorectal carcinoma. J Clin Invest. 2009; 119:3311–3321.
7. Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, Meatchi T, Bruneval P, Cugnenc PH, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005; 353:2654–2666.
8. Pages F, Galon J, Fridman WH. The essential role of the in situ immune reaction in human colorectal cancer. J Leukoc Biol. 2008; 84:981–987.
9. Reissfelder C, Stamova S, Gossmann C, Braun M, Bonertz A, Walliczek U, Grimm M, Rahbari NN, Koch M, Saadati M, Benner A, Buchler MW, Jager D, et al. Tumor-specific cytotoxic T lymphocyte activity determines colorectal cancer patient prognosis. J Clin Invest. 2015; 125:739–751.
10. Wang L, Wang Y, Song Z, Chu J, Qu X. Deficiency of interferon-gamma or its receptor promotes colorectal cancer development. J Interferon Cytokine Res. 2015; 35:273–280.
11. Slattery ML, Lundgreen A, Bondurant KL, Wolff RK. Interferon-signaling pathway: associations with colon and rectal cancer risk and subsequent survival. Carcinogenesis. 2011; 32:1660–1667.
12. Mori S, Sawada T, Okada T, Kubota K. Anti-proliferative effect of interferon-gamma is enhanced by iron chelation in colon cancer cell lines in vitro. Hepatogastroenterology. 2008; 55:1274–1279.
13. Langaas V, Shahzidi S, Johnsen JI, Smedsrod B, Sveinbjornsson B. Interferon-gamma modulates TRAIL-mediated apoptosis in human colon carcinoma cells. Anticancer Res. 2001; 21:3733–3738.
14. Ni C, Wu P, Zhu X, Ye J, Zhang Z, Chen Z, Zhang T, Wang K, Wu D, Qiu F, Huang J. IFN-gamma selectively exerts pro-apoptotic effects on tumor-initiating label-retaining colon cancer cells. Cancer Lett. 2013; 336:174–184.
15. Zins K, Abraham D, Sioud M, Aharinejad S. Colon cancer cell-derived tumor necrosis factor-alpha mediates the tumor growth-promoting response in macrophages by up-regulating the colony-stimulating factor-1 pathway. Cancer Res. 2007; 67:1038–1045.
16. Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, Oshima M, Fujii C, Mukaida N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008; 118:560–570.
17. Wang X, Lin Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol Sin. 2008; 29:1275–1288.
18. Voloshanenko O, Erdmann G, Dubash TD, Augustin I, Metzig M, Moffa G, Hundsrucker C, Kerr G, Sandmann T, Anchang B, Demir K, Boehm C, Leible S, et al. Wnt secretion is required to maintain high levels of Wnt activity in colon cancer cells. Nat Commun. 2013; 4:2610.
19. Qi L, Sun B, Liu Z, Cheng R, Li Y, Zhao X. Wnt3a expression is associated with epithelial-mesenchymal transition and promotes colon cancer progression. J Exp Clin Cancer Res. 2014; 33:107.
20. Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, Boisguerin V, Bukur T, Sorn P, Paret C, Diken M, Kreiter S, Tureci O, et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics. 2014; 15:190.
21. Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, Pretlow TP, Yang B, Akiyama Y, Van Engeland M, Toyota M, Tokino T, Hinoda Y, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004; 36:417–422.
22. Wu B, Crampton SP, Hughes CC. Wnt signaling induces matrix metalloproteinase expression and regulates T cell transmigration. Immunity. 2007; 26:227–239.
23. Driessens G, Zheng Y, Locke F, Cannon JL, Gounari F, Gajewski TF. Beta-catenin inhibits T cell activation by selective interference with linker for activation of T cells-phospholipase C-gamma1 phosphorylation. J Immunol. 2011; 186:784–790.
24. Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM, Paulos CM, Muranski P, Restifo NP. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009; 15:808–813.
25. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013; 13:11–26.
26. Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012; 4.
27. Najdi R, Holcombe RF, Waterman ML. Wnt signaling and colon carcinogenesis: beyond APC. J Carcinog. 2011; 10:5.
28. Holcombe RF, Marsh JL, Waterman ML, Lin F, Milovanovic T, Truong T. Expression of Wnt ligands and Frizzled receptors in colonic mucosa and in colon carcinoma. Mol Pathol. 2002; 55:220–226.
29. Park JK, Song JH, He TC, Nam SW, Lee JY, Park WS. Overexpression of Wnt-2 in colorectal cancers. Neoplasma. 2009; 56:119–123.
30. Staal FJ, Luis TC, Tiemessen MM. WNT signalling in the immune system: WNT is spreading its wings. Nat Rev Immunol. 2008; 8:581–593.
31. Keerthivasan S, Aghajani K, Dose M, Molinero L, Khan MW, Venkateswaran V, Weber C, Emmanuel AO, Sun T, Bentrem DJ, Mulcahy M, Keshavarzian A, Ramos EM, et al. beta-Catenin promotes colitis and colon cancer through imprinting of proinflammatory properties in T cells. Sci Transl Med. 2014; 6:225ra228.
32. Lovatt M, Bijlmakers MJ. Stabilisation of beta-catenin downstream of T cell receptor signalling. PLoS One. 2010; 5.
33. Ioannidis V, Beermann F, Clevers H, Held W. The beta-catenin—TCF-1 pathway ensures CD4(+)CD8(+) thymocyte survival. Nat Immunol. 2001; 2:691–697.
34. Driessens G, Zheng Y, Gajewski TF. Beta-catenin does not regulate memory T cell phenotype. Nat Med. 2010; 16:513–514; author reply 514–515.
35. Yu Q, Sharma A, Oh SY, Moon HG, Hossain MZ, Salay TM, Leeds KE, Du H, Wu B, Waterman ML, Zhu Z, Sen JM. T cell factor 1 initiates the T helper type 2 fate by inducing the transcription factor GATA-3 and repressing interferon-gamma. Nat Immunol. 2009; 10:992–999.
36. Notani D, Gottimukkala KP, Jayani RS, Limaye AS, Damle MV, Mehta S, Purbey PK, Joseph J, Galande S. Global regulator SATB1 recruits beta-catenin and regulates T(H)2 differentiation in Wnt-dependent manner. PLoS Biol. 2010; 8:e1000296.
37. Prlic M, Bevan MJ. Cutting edge: beta-catenin is dispensable for T cell effector differentiation, memory formation, and recall responses. J Immunol. 2011; 187:1542–1546.
38. Ding Y, Shen S, Lino AC, Curotto de Lafaille MA, Lafaille JJ. Beta-catenin stabilization extends regulatory T cell survival and induces anergy in nonregulatory T cells. Nat Med. 2008; 14:162–169.
39. van Loosdregt J, Fleskens V, Tiemessen MM, Mokry M, van Boxtel R, Meerding J, Pals CE, Kurek D, Baert MR, Delemarre EM, Grone A, Koerkamp MJ, Sijts AJ, et al. Canonical Wnt signaling negatively modulates regulatory T cell function. Immunity. 2013; 39:298–310.
40. Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, Sukumar M, Reger RN, Yu Z, Kern SJ, Roychoudhuri R, Ferreyra GA, Shen W, et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity. 2011; 35:972–985.
41. Dai W, Liu F, Li C, Lu Y, Lu X, Du S, Chen Y, Weng D, Chen J. Blockade of Wnt/beta-Catenin Pathway Aggravated Silica-Induced Lung Inflammation through Tregs Regulation on Th Immune Responses. Mediators Inflamm. 2016; 2016:6235614.
42. Lee YS, Lee KA, Yoon HB, Yoo SA, Park YW, Chung Y, Kim WU, Kang CY. The Wnt inhibitor secreted Frizzled-Related Protein 1 (sFRP1) promotes human Th17 differentiation. Eur J Immunol. 2012; 42:2564–2573.
43. Ernst M, Putoczki T. IL-17 cuts to the chase in colon cancer. Immunity. 2014; 41:880–882.
44. Wang K, Kim MK, Di Caro G, Wong J, Shalapour S, Wan J, Zhang W, Zhong Z, Sanchez-Lopez E, Wu LW, Taniguchi K, Feng Y, Fearon E, et al. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity. 2014; 41:1052–1063.
45. Wu D, Wu P, Huang Q, Liu Y, Ye J, Huang J. Interleukin-17: a promoter in colorectal cancer progression. Clin Dev Immunol. 2013; 2013:436307.
46. Liang H, Coles AH, Zhu Z, Zayas J, Jurecic R, Kang J, Jones SN. Noncanonical Wnt signaling promotes apoptosis in thymocyte development. J Exp Med. 2007; 204:3077–3084.
47. Louis I, Heinonen KM, Chagraoui J, Vainio S, Sauvageau G, Perreault C. The signaling protein Wnt4 enhances thymopoiesis and expands multipotent hematopoietic progenitors through beta-catenin-independent signaling. Immunity. 2008; 29:57–67.
48. Famili F, Naber BA, Vloemans S, de Haas EF, Tiemessen MM, Staal FJ. Discrete roles of canonical and non-canonical Wnt signaling in hematopoiesis and lymphopoiesis. Cell Death Dis. 2015; 6:e1981.
49. Ishitani T, Kishida S, Hyodo-Miura J, Ueno N, Yasuda J, Waterman M, Shibuya H, Moon RT, Ninomiya-Tsuji J, Matsumoto K. The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca(2+) pathway to antagonize Wnt/beta-catenin signaling. Mol Cell Biol. 2003; 23:131–139.