
INTRODUCION
Gastrokine 1 (GKN1) is a tissue-specific 18 kDa protein that is highly expressed in the gastric mucosa of many mammalian species [1, 2]. Its biological function is poorly understood, but it is thought to be involved in the replenishment of the surface lumen epithelial cell layer, in maintaining mucosal integrity, and in cell proliferation and differentiation [3–6]. GKN1 is down-regulated in samples from Helicobacter pyilori-infected gastric mucosa but is absent in gastric adenocarcinoma tissues [1, 2, 7]. We previously showed that GKN1 down-regulation is one of the leading causes of gastric cancer (GC) development [8, 9]. Its over-expression in gastric adenocarcinoma cell lines AGS and MKN28 activated the expression of Fas receptor, while treatment with an anti-Fas antibody significantly increased apoptosis [10]. Moreover, treatment of tumor cells with recombinant human GKN1 reduced the proliferation of AGS cells compared with human embryonic kidney cells (HEK 293) and non-gastric cancer cells (H1355) [11]. These data suggest that GKN1 functions as a tumor suppressor and a modulator of apoptotic signals in GC. GKN1 could also be considered a biomarker for GC because individuals with a lower expression of the protein have an increased risk of developing gastric diseases [12].
GKN1 (CA11, accession number: BK0017373) is located in a 6 kb region of chromosome 2p13 and contains six exons. The mechanism by which GKN1 is silenced in GC and the role of epigenetic changes in this is unknown. Recently, Yoon et al. 2011 investigated this aspect in a sample group of 81 gastric carcinomas and 40 gastric adenomas [13]. No mutation was detected in gastric tumors and hyper-methylation of the GKN1 promoter was only observed in two tumors, whereas DNA copy number and GKN1 mRNA levels were significantly decreased in all GC samples. More recently, the Epstein–Barr nuclear antigen 1 (EBNA1) protein was reported to directly bind GKN1 and GKN2 promoters [14]. Treatment of AGS-Epstein–Barr virus (EBV) and AGS-EBNA1 cell lines with 5’ azacytidine showed that GKN1 and GKN2 were transcriptionally silenced by DNA methylation, and that latent EBV infection further reduced GKN1 and GKN2 expression in AGS cells. EBNA1 depletion by small interfering RNA partially alleviated this repression. However, the ectopic expression of EBNA1 slightly increased GKN1 and GKN2 basal mRNA levels, but reduced their responsiveness to demethylating agents. These findings indicated that EBNA1 contributes to the transcriptional complex and epigenetic deregulation of GKN1 and GKN2 tumor suppressor genes in EBV-positive GC.
Although these studies suggest that epigenetic modifications are involved in the deregulation of GKN1 in GC, no studies have yet investigated histone modifications or the recruitment of histone-modifying enzymes and GKN1 co-repressors in GC. Therefore, in the present study, we attempted to clarify whether epigenetic mechanisms are associated with GKN1 silencing in GC and to determine whether this event might be involved in the development and progression of GC.
RESULTS
GKN1 expression levels in non-tumoral and tumoral tissues
We first analyzed the expression levels of GKN1 in six gastric tissue specimens from our collection of paired samples of non-tumoral (N1–N6) and tumoral (T1–T6) gastric tissues from the same patients. Tissue T1 showed a well-differentiated adenocarcinoma of intestinal type, T2 showed a severe dysplasia grade associated with a small area of intraglandular adenocarcinoma that was moderately differentiated, T3 and T6 showed a poorly differentiated adenocarcinoma of diffuse type, T4 showed a poorly differentiated adenocarcinoma of intestinal type, and T5 showed a moderately differentiated adenocarcinoma of intestinal type. The clinicopathologic characteristics of GC patients are summarized in Table 1.
Table 1: Characteristics of Gastric Cancer Patients
Variable |
Gastric Cancer |
|---|---|
Age at surgery (Y) |
|
Mean |
67 ± 13 |
Range |
45 – 78 |
Sex ratio (M/F) |
3/3 |
Tumor type |
Intestinal 3, |
Grade of differentiation |
Well 1, |
The peritumoral areas of intestinal type GC showed a variable degree of gastric atrophy with diffuse intestinal metaplasia, while the peritumoral areas of diffuse type GC showed a variable degree of non-dysplastic inflammation. Figure 1 (panels A and C) shows the expression profiles of GKN1 in the six paired non-tumoral and tumoral tissues as evaluated by western blotting. Compared with non-tumoral tissues, all tumoral samples showed a down-regulation or an almost total absence of GKN1, using the GAPDH expression profile as a control (panels B and D) and based on the densitometric analysis of GKN1 expression (Figure 1E). Figure 1C shows the GKN1 expression profiles of samples C1 and C2 taken from healthy individuals undergoing sleeve gastrectomy as positive controls.
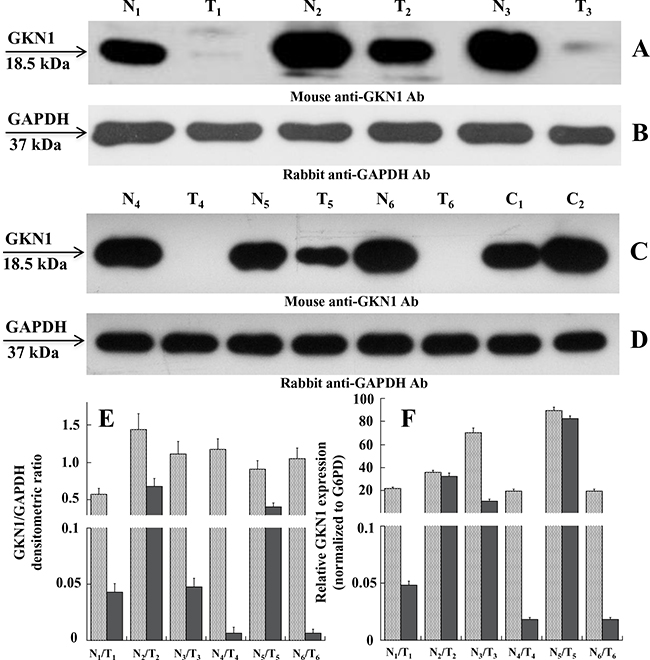
Figure 1: Expression levels of GKN1 in human gastric tissues. A and C. Western blot of tissue extracts analyzed in paired non-tumoral (N1-N6) and tumoral (T1-T6) human gastric samples, respectively, using mouse anti-GKN1 antibody (Ab). E. Expression levels of GKN1 protein in non-tumoral (N1-N6) and tumoral (T1-T6) paired samples evaluated from the densitometry of GKN1 bands normalized towards the corresponding densitometry of GAPDH bands B and D. F. qRT-PCR analysis. Total RNA was prepared from gastric tissues and analyzed by qRT-PCR for GKN1 mRNA level compared to G6PD mRNA as reference sample. Data from three experiments are reported as mean values ± SD.
To determine if the observed down-regulation also occurred at the transcriptional level, we performed quantitative reverse transcription (qRT)-PCR on total RNA isolated from paired gastric non-tumoral and tumoral tissues. Figure 1F shows a decrease of GKN1 mRNA levels in tumoral tissues compared with non-tumoral tissues, which supports the western blot findings.
GKN1 down-regulation in GC is associated with trimethylation of histone 3 at lysine 9 on the GKN1 promoter
To investigate the possible causes of GKN1 inactivation, chromatin immunoprecipitation (ChIP) assays for the repressive trimethylation of histone 3 at lysine 9 (H3K9triMe) were performed. A 600 bp promoter region of GKN1 (identified by the UCSC Genome Browser) including the 5’-untranslated region (UTR) was divided into three different segments (A, B, and C) of about 160 bp, and corresponding PCR primers were designed (Figure 2A). ChIP assays performed on these three DNA segments revealed a significant increase in H3K9triMe modification in tumoral tissues compared with non-tumoral tissues. Figure 3 shows the results of an average of six independent experiments performed on six paired non-tumoral (N1–N6) and tumoral (T1–T6) specimens.
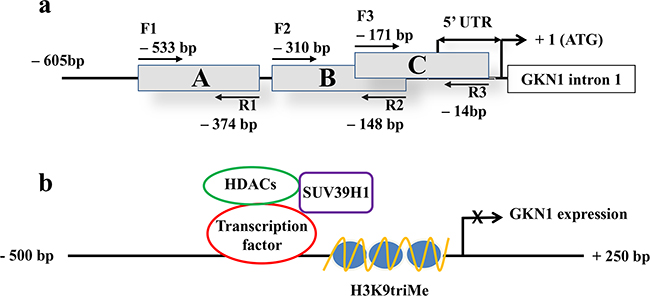
Figure 2: 5’-region of GKN1 gene analyzed by ChIP assay and proposed gene expression regulation. a. The A, B and C regions of GKN1 gene promoter delimited by the corresponding primer pairs (F1-R1, F2-R2, F3-R3) are boxed. The length of the 5’ UTR of GKN1 mRNA is indicated by double arrows. +1 indicates the position of the start codon. b. Scheme showing the proposed mechanism of histone/SUV39H1 actions on GKN1 gene promoter.
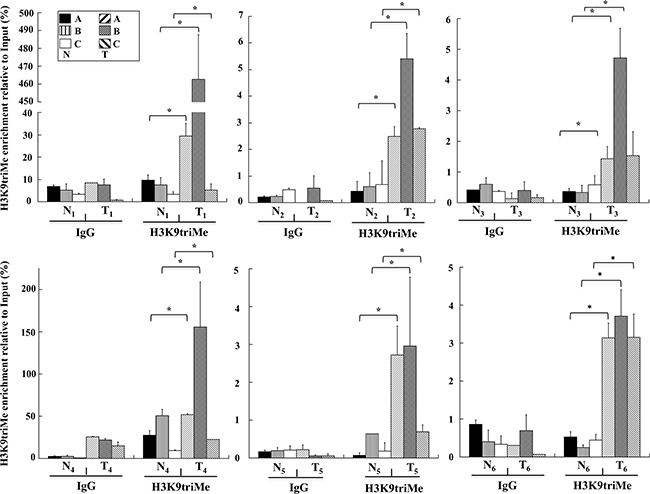
Figure 3: H3K9triMe levels on human GKN1 gene promoter. ChIP assays performed on human non-tumoral (N1-N6) and tumoral (T1-T6) human gastric samples, respectively. H3K9triMe enrichment relative to input is reported as 2ΔCt × 100, where ΔCt is the difference between CtInput and CtIP. All quantitative ChIP data were derived from three independent experiments, and for each experiment qPCR was performed in triplicate. * p<0.05, compared to corresponding control.
H3K9triMe is associated with the recruitment and/or activation of a histone-lysine N-methyltransferase on the GKN1 promoter
Next, we examined the control of the H3K9triMe modification on the GKN1 promoter. Previous work suggested that the H3K9triMe modification mainly occurs by a specific histone methyltransferase, histone-lysine N-methyltransferase SUV39H1, that trimethylates Lys-9 of histone H3 using mono-methylated H3 Lys-9 as a substrate [15]. Therefore, we used ChIP assays to verify the presence of this enzyme in the GKN1 promoter region. Figure 4 clearly shows a significant increase in the binding of SUV39H1 to the GKN1 promoter region in tumoral samples compared with non-tumoral tissues.
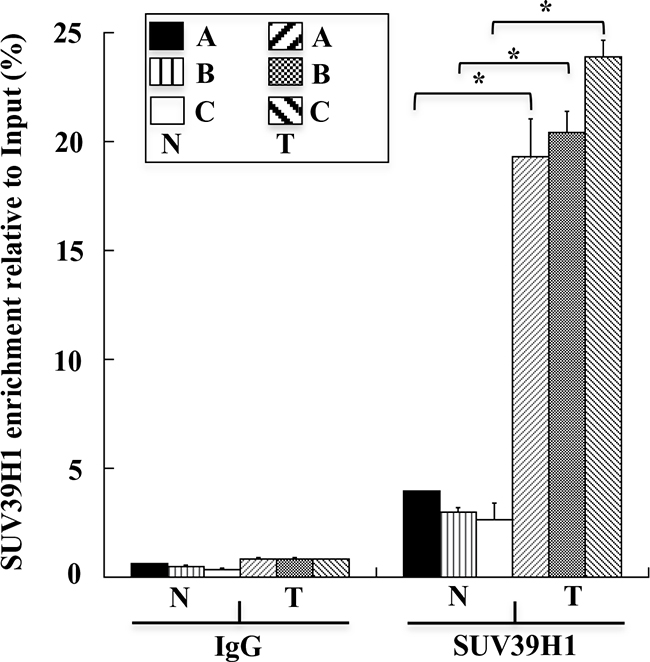
Figure 4: SUV39H1 levels on human GKN1 gene promoter. ChIP assays performed on human non-tumoral N1-N6(N) and tumoral T1-T6(T) human gastric samples, respectively. SUV39H1 enrichment relative to input is reported as 2ΔCt× 100, where ΔCt is the difference between CtInput and CtIP. All quantitative ChIP data were derived from three independent experiments, and for each experiment qPCR was performed in triplicate. * p<0.05, compared to corresponding control.
The expression of SUV39H1, histone deacetylase 1, and H3K9triMe in GC tissues and cell lines
We next investigated the relationship between the expression of GKN1 and that of SUV39H1, histone deacetylase 1 (HDAC1), and H3K9triMe. As shown in Figure 5 (panels A and D), we observed an increase in the expression of SUV39H1 in tumoral tissues (T1–T6) compared with non-tumoral tissues (N1–N6), based on the corresponding GAPDH western blot band intensity (Figure 5, panels C and F). Similar expression profiles for HDAC1 were seen in paired non-tumoral and tumoral gastric tissues (Figure 5, panels B and E). We were only able to analyze H3K9triMe expression in the nuclear extracts of one paired non-tumoral (N6) and tumoral (T6) sample. As shown in Figure 5G, tumoral tissue showed higher level of H3K9triMe expression compared with non-tumoral tissue, as determined by the western blot band intensity ratio with respect to that of lamin A/C (Figure 5H).

Figure 5: SUV39H1, HDAC1 and H3K9triMe expression in human gastric tissues and cell lines. SUV39H1, HDAC1 expression in human non-tumoral (N) and tumoral (T) cell extracts (panels A-F) and H3K9triMe expression in nuclear extracts (panels G-H) was assessed by Western blot with the specific antibodies. Relative expression of SUV39H1, HDAC1 in sample tissues is reported as band intensity ratio with that of the corresponding GAPDH whereas that of H3K9triMe is shown as band intensity ratio with that of the corresponding lamin A/C. Protein expression in gastric cancer cell lines (panels I-L) was assessed by Western blot with the specific antibodies on nuclear extracts of AGS cells (lane 1), AGS cells transfected with flGKN1 (lane 2) and NCI-N87 cells (lane 3). Relative expression of SUV39H1, HDAC1 and H3K9triMe is reported as band intensity ratio with that of the corresponding lamin A/C.
The expression levels of SUV39H1 and HDAC1 were next evaluated in healthy sleeve gastrectomy specimens (C1 and C2) (Figure 5, panels D and E). In these cases, expression levels appeared similar to those of non-tumoral tissues (N4–N6).
Lastly, we evaluated the expression levels of SUV39H1, HDAC1, and H3K9triMe proteins in GC cell lines. Because of the lack of a non-tumoral gastric cell line, we analyzed protein expression in GKN1-transfected and non-transfected GC cells (AGS) and in an additional non-transfected gastric cancer cell line (NCI-N87). As reported in Figure 5, AGS cells transfected with GKN1 demonstrated lower levels of SUV39H1 (Figure 5I), HDAC1 (Figure 5J), and H3K9triMe (Figure 5K) expression compared with non-transfected cells, as determined by the western blot band intensity ratio with respect to that of lamin A/C (Figure 5L).
Treatment of GC cells with trichostatin A induces the up-regulation of GKN1 mRNA
To further understand the effect of epigenetic modifications on GKN1 expression, we tested the possible role of histone acetylation. MKN28, AGS, and KATO III GC cell lines were treated with trichostatin A (TSA), an inhibitor of histone deacetylases (HDACs), and GKN1 mRNA levels were evaluated by qRT-PCR. The treatment of AGS cells with TSA for 24 h led to an increase in GKN1 mRNA expression of about 28-fold compared with untreated cells, whereas no effect was observed in MKN28 and KATO III cells. TSA treatment for 48 h increased GKN1 mRNA expression by around 50-, 160- and 110-fold in MKN28, AGS, and KATO III cells, respectively (Figure 6). However, these results were not associated with protein re-expression, as evaluated by western blotting (data not shown).
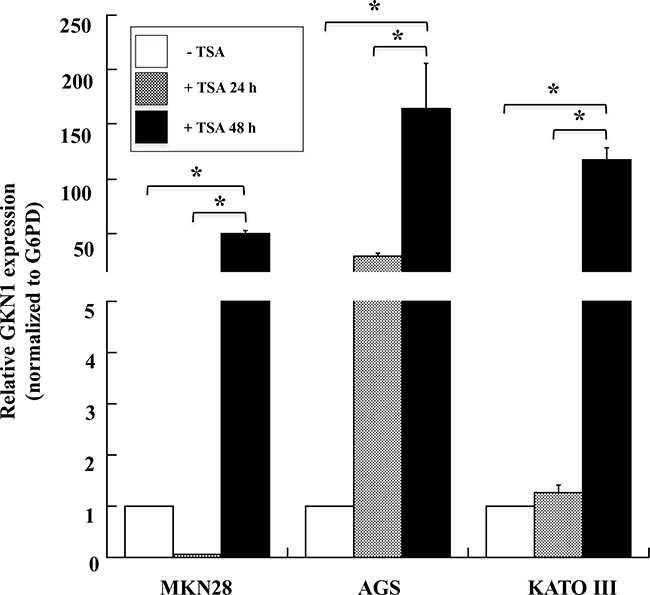
Figure 6: TSA induces the expression of GKN1 mRNA in gastric cancer cell lines. qRT-PCR analysis of GKN1 mRNA in MKN28, AGS and KATO III gastric cancer cell lines after TSA treatment of the cells for 24 and 48 hours. G6PD was used as internal standard for normalization The relative expression of GKN1 was evaluated using as control cells treated with DMSO. Data from a representative experiment are reported as mean values ± SD. * p<0.05.
The up-regulation of GKN1 mRNA prompted us to use the TSA-treated AGS cell line as a positive control to confirm the results obtained in human gastric tissues in a cellular gastric model. As shown in Figure 7, a ChIP assay showed that GKN1 up-regulation in AGS cells after TSA treatment was associated with a reduction of the H3K9triMe repressive modification (Figure 7A). This confirmed our findings in gastric tissues, and revealed an increase of H3-acetylation-activating modification in the same three regions of the GKN1 promoter analyzed earlier (Figure 7B).
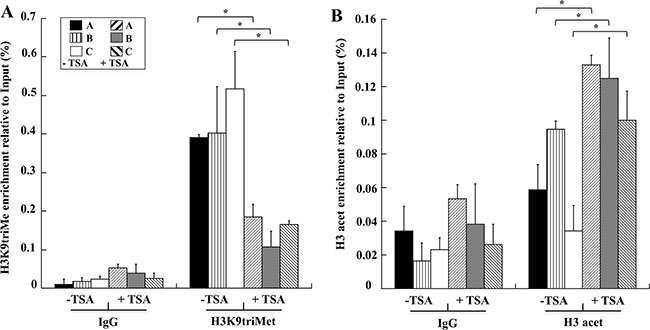
Figure 7: TSA induces in AGS cells decreased levels of H3K9triMe and increased levels of H3 acetylation. Levels of H3K9triMe A. and H3 acetylation B. determined by ChIP assays on AGS cells not treated (-TSA) and treated with TSA (+TSA). H3K9triMe and H3 acetylation enrichment relative to input are reported as 2ΔCt × 100, where ΔCt is the difference between CtInput and CtIP. All quantitative ChIP data were derived from three independent experiments, and for each experiment, qRT-PCR was performed in triplicate. * p<0.05, compared to corresponding control.
DISCUSSION
Inflammation is a key risk factor in the development of many types of cancers, and chronic inflammation of the stomach initiates the histopathologic progression of chronic gastritis through gastric atrophy, intestinal metaplasia, dysplasia, and finally GC [16]. Indeed, H. pylori infection of the gastric mucosa and consequent chronic inflammation is a major step in the initiation and development of GC [17]. Promoter hypermethylation in tumor-related genes is often detected in premalignant gastric lesions [18], suggesting a relationship with the induction and or promotion of GC [19, 20]. Moreover, aberrant DNA methylation represents one of the most important inactivating mechanisms of tumor suppressor genes often associated with H. pylori infection [21, 22].
GKN1 expression is known to decrease throughout the progressive stages of neoplastic transformation. Previous investigations revealed no CpG hyper-methylation of the GKN1 promoter in GC tissues [13] and altered GKN1 expression associated with the severity of gastritis and DNA methylation in non-neoplastic gastric mucosa [23]. Moreover, GKN1 expression in AGS cells induced endogenous micro RNA (miR)-185 that directly targeted the epigenetic effectors DNA (cytosine-5)-methyltransferase 1 (DNMT1) and histone-lysine N-methyltransferase enzyme (EZH2). The histone acetyl transferase Tip60 was shown to be up-regulated and HDAC1 to be down-regulated in an miR-185-independent manner, thus inducing cell cycle arrest by regulating cell cycle proteins in GC cells [24]. However, the precise relationship between gastritis and GKN1 has not been evaluated.
In the present study, we investigated the possible causes of GKN1 inactivation by evaluating whether other epigenetic mechanisms could be involved in this process. Because DNA methylation, histone deacetylation, and methylation of histone H3 at lysine 9 are the three best-characterized covalent modifications associated with a repressed chromatin state, we focused our attention on these modifications [25, 26]. We show for the first time that a mechanism comprising histone modifications appears to be involved in the dysregulation of GKN1 transcription in GC. By comparing human non-tumoral tissues with corresponding tumoral ones using ChIP assays, we revealed an increase of the repressive histone modification H3K9triMe (Figure 3) accomplished by recruitment of the specific histone methyltransferase SUV39H1 (Figure 4; Figure 5A, 5C) in tumoral tissues. Upregulation of SUV39H1 and H3K9triMe both at transcriptional and translational level has been demonstrated in several cancers therefore, SUV39H1 and H3K9triMe have important roles in cancer development and progression and the pharmacological inhibition of SUV39H1 may be a promising therapeutic approach for cancer treatment [27, 28]. Our present study documents the overexpression of Suv39H1 and histone tri-methylated H3K9 in gastric carcinoma.
It is interesting to note that although the enrichment levels of H3K9triMe modification observed in the six samples appeared quantitatively different, the GKN1 promoter region where this modification was mostly enriched (region B) was the same in all samples. Additionally, no significant difference in H3K9triMe in non-tumoral gastric tissues was observed compared with tumoral ones, suggesting a correlation with the GKN1 protein expression shown in Figure 1. In fact, the H3K9triMe enrichment observed by ChIP (Figure 3) was associated with tumoral samples in which strong down-regulation of GKN1 expression was observed. The prominent role of H3K9triMe in GC was also confirmed by the finding that H3K9 trimethylation positively correlates with tumor stage, lymphovascular invasion, and cancer recurrence in gastric carcinoma whereas higher levels of H3K9 trimethylation correlate with poor survival [29].
Histone acetylation is one of the main determinants of chromatin structure [30], and it can be regulated dynamically through the involvement of transactivating factors with intrinsic histone acetylase activity or through the recruitment of deacetylase complexes that repress gene expression [31]. Indeed, several reports indicated that HDAC1 is up-regulated in many cancer cell lines and tissues [32], including GC, at both the transcriptional and translational levels [33]. Our findings are in agreement with this (Figure 5) and imply that increased HDAC1 expression causes histone hypoacetylation and the silencing of several tumor suppressor genes in GC. Therefore, we investigated whether underacetylation might contribute to GKN1 transcriptional inhibition using TSA to increase general histone acetylation in an attempt to bypass the inhibitory effects of DNA methylation. Because TSA can arrest the cell cycle, induce apoptosis, regulate cell differentiation, and inhibit cell migration in the absence of cytotoxicity [34–36], we used milder experimental conditions (TSA, 90 ng/ml; time of treatment, 24 and 48 h) to reduce the possible inhibition of cell proliferation [37]. Under these same experimental conditions, TSA was previously shown to reduce AGS cell viability by less than 10% [38], and to dose-dependently inhibit MKN28 cell growth up to a concentration of 500 ng/ml [39]. Treatment of GC cell lines MKN28, AGS, and KATO III with TSA in the present study strongly increased GKN1 mRNA expression (Figure 6), suggesting that histone deacetylation represents an important mediator of GKN1 repression associated with DNA methylation. In fact, the TSA treatment of AGS cells led to a reduction of H3K9triMe and an increase of histone acetylation (Figure 6). Because histone acetylation is a fundamental regulatory mechanism for controlling gene accessibility, our results indicate that histone methylation is a unique mechanism for establishing local histone deacetylation, and generating maintainable epigenetic chromosomal states. However, it must be pointed out that even in this condition, the GKN1 mRNA level was still very low because the cycle numbers required for its amplification were about 10-fold lower than those required for the amplification of housekeeping glucose-6-phosphate dehydrogenase (data not shown). In any case, no GKN1 protein re-expression was observed under these conditions by western blotting. This finding could be due to proteosome-mediated degradation of GKN1. To ascertain this possibility, we treated AGS cells with proteosome inhibitor (MG132). No GKN1 expression was observed (not shown). This suggests the presence of further regulation at the translational level, perhaps by mechanisms mediated by miRNAs, resulting in translational repression and gene silencing. For example, miRNA-544 directly targets the 3’-UTR of the newly-identified tumor suppressor gene IRX1, whose hypermethylation decreases expression of the protein in GC [40]. Therefore, miRNAs and promoter hypermethylation are important epigenetic mechanisms for transcriptional inactivation of tumor suppressors.
Recently, Yoon et al. 2015, showed that NKX6.3, considered a possible tumor suppressor for GC, is a transcriptional factor for GKN1. They showed that NKX6.3 is strongly down-regulated in GC cells; however, its over-expression in AGS and MKN1 GC cells induced the re-expression of GKN1 protein [41]. Therefore, it is possible that the strong increase in GKN1 mRNA transcribed by over-expressed NKX6.3 in AGS cells escapes the post-transcriptional mechanism that regulates GKN1 mRNA translation. With this in mind, the effects of GKN1 on SUV39H1, HDAC1, and H3K9triMe expression observed in transfected AGS cells in the present study (Figure 5) suggest that GKN1 functions as a direct or indirect modulator of the epigenetic factors involved in gene silencing during gastric carcinogenesis [24].
We propose the following model describing the mechanisms which H3K9triMe by SUV39H1 acts on the GKN1 promotor (Figure 2B). A transcription factor functions as a negative regulator by recruiting SUV39H1 and HDACs to the GKN1 promoter to induce histone deacetylation and methylation, thus resulting in GKN1 repression. This model is in agreement with recent findings showing that restoration of GKN1 protein suppressed GC cell growth through an miRNA-mediated mechanism for DNA epigenetic modification [24]. Therefore, the loss of GKN1 function contributes to malignant transformation and the proliferation of gastric epithelial cells in gastric carcinogenesis.
CONCLUSIONS
In conclusion, utilizing several specimens from patients with gastric carcinoma, we found that GKN1 expression is significantly reduced in dysplasia and tumor gastric mucosa, and is inversely correlated with the recruitment of H3K9triMe and Suv39H1 to the GKN1 promoter. Tumoral GKN1 expression also appeared to be associated with an overall increase of the expression profiles of H3K9triMe, Suv39H1, and HDAC1 proteins. These findings provide evidence that epigenetic mechanisms leading to the inactivation of GKN1 play a key role in the multi-step process of gastric carcinogenesis. While our results are relevant and reliable given that they were obtained in vivo using human specimens, we nevertheless aim to confirm them in a larger number of samples. Moreover, it will be also interesting to evaluate the role of miRNAs as regulators of GKN1 expression in GC. This will enable us to obtain a greater understanding of these mechanisms to determine whether they are involved in the development and progression of GC.
MATERIALS AND METHODS
Materials
Dulbecco’s modified Eagle’s medium (DMEM-F12) and fetal bovine serum (FBS) were purchased from Cambrex (Rutherford, NJ, USA). Mouse GKN1 monoclonal antibody (M01), clone 2E5, was purchased from Abnova (Taipei, Taiwan). Rabbit monoclonal to Histone H3 (tri methyl K9) [ab8898] and to HDAC1 [ab109411] antibodies were from Abcam (Cambridge, MA, USA), mouse SUV39H1 (clone MG44) [05–615] and rabbit acetyl-Histone H3 [06–599] antibodies were from Millipore (Temecula, CA, USA). DMSO and Trichostatin A (TSA) were from Sigma (Milan, Italy). Rabbit GAPDH monoclonal (glyceraldehyde-3-phosphate dehydrogenase) and rabbit Lamin A/C polyclonal antibodies were from Santa Cruz Biotecnology (Dallas, TX, USA).
Cell cultures, transfection, human tissues and Western blotting
Human gastric adenocarcinoma cell lines (AGS, MKN28, KATO III, NCI-N87) were grown in DMEM-F12 supplemented with heat inactivated FBS, 1% penicillin/streptomycin and 1% L-glutamine at 37°C in a 5% CO2 atmosphere. AGS were transfected with 4 μg of vector pcDNA3.1-flGKN1(His)6 encoding the full length GKN1 (flGKN1, containing the first 20 amino acids leader peptide and His6-Tag sequence at the C-terminal) as already described [10]. The efficiency of transfection of gastric cancer cells with flGKN1 was always evaluated by a parallel transfection using EGFP vector as control. In general, after transfection, the average value of the ratio between number of green fluorescent cells/total number of cells was 0.5 ± 0.1.
Human gastric tissues were from patients with GC recruited at Hospital A. Cardarelli, Naples, Italy. All patients were interviewed regarding smoking habit, alcohol intake and chronic use of drugs. Hospital Pathologist performed the macro dissection of tumor and non-tumor tissues of GC patients during surgery. Gastric cancer was staged and graded according to the American Joint Committee on Cancer criteria [42]. The characterization of non-tumoral gastric mucosa was based on macroscopic aspects of normal compared with tumoral tissue as evaluated by the hospital pathologist [43], and from our previous work showing that GKN1 was highly expressed in gastric non-tumoral tissues but down-regulated or totally absent in GC tissues [3]. The study reported in the manuscript has been carried out in the frame of a research protocol entitled “Role of gastrokine 1 in gastric cancer” that has the approval from the Ethic Committee of the University of Naples Federico II (Comitato Etico Università Federico II). The assigned protocol number of the study was 34/15 [43].
Proteins from cell extracts (about 20 μg) were analyzed by Western blotting using mouse anti-GKN1 at 1:500, rabbit anti-Histone H3 (tri methyl K9) at 1:1000, rabbit anti-HDAC1 at 1:1000, mouse anti-SUV39H1 at 1:500, anti-GAPDH at 1:1000 and rabbit anti-Lamin A/C at 1:1000 dilution. Detection was performed using the enhanced chemiluminescence detection kit (SuperSignal West Pico) following manufacturer’s instructions. Western blot band intensity was measured with ImageJ 1.46r software.
mRNA isolation and qRT-PCR
Total RNA was extracted from normal and cancer human tissues or from DMSO or TSA treated cells using TRIzol reagent solution (Invitrogen) according to the manufacturer’s protocol. cDNA was synthesized using the reverse transcription kit from Roche Molecular Systems (Roche, Penzberg, Germany) according to the manufacturer’s protocol. GKN1 cDNA was amplified by qRT-PCR (forward and reverse primers 5’-ctttctagctcctgccctagc-3’ and 5’-tggttgcagcaaagccattt-3’, respectively) using the housekeepingglucose 6-phosphate dehydrogenase (G6PD) mRNA as an internal standard for normalization, according to standard procedures (Applied Biosystems, Foster City, CA, USA). qRT-PCR was performed with the SYBR Green PCR MasterMix (Applied Biosystems) under the following conditions: 10 minutes at 95°C, followed by 40 cycles (15 seconds at 95°C and 1 minute at 60°C). Each reaction was performed in triplicate. We used the 2–ΔΔCT method to calculate the relative expression levels [44].
Chromatin immunoprecipitation assay
Samples from normal and cancer human tissues were processed for chromatin immunoprecipitation (ChIP) assay. Cellular sospension was collected by centrifugation at 2000 rpm at 4°C for 10 minutes and then resuspended in 6× volume of cell lysis buffer [5 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) pH 8.0, 85 mM KCl, 0.5% NP-40] plus phenylmethylsulfonyl fluoride (PMSF) (1 mM) and trypsin inhibitor (10 μg/ mL) as protease inhibitors. Cells were then incubated on ice for 15 minutes and lysed using a dounce several times. Nuclei were collected at 5000 rpm at 4°C for 10 minutes and the pellet was resuspend in 5× volume of nuclei lysis buffer (50 mM TrisHCl pH 8.1, 10 mM EDTA, 1% SDS) plus the same protease inhibitors as the cell lysis buffer. The solution was incubate on ice for 20 minutes and subsequently freezed and thawed in liquid nitrogen 2 times to aid in nuclear lysis. After centrifugation at 5000 rpm at 4°C for 10 minutes, the obtained chromatin was sonicated according to the procedure described by Federico et al. 2009 [45]. Samples were subjected to IP with the following specific antibodies against histone modification anti-tri methyl K9-Histone3, anti-acetyl H3 and the specific histone methyltransferase anti-SUV39H1. For qRT-PCR, 2 μl aliquot of IP DNA (150 μl) were amplified with a set of three primers pairs (all primers are listed in the 5’ to 3’ direction); region A: F1, ggggtaggtttgg tgggagttgc, R1, atcacagctgaaaagccacgtgta; region B: F2, cgcccacagctttgactgggt, R2, tgccatgagccagtgtaccagga; region C: F3, tcctggtacactggctcatggca, R3, agcagtggacag aggagtaggca. GAPDH promoter amplicon was used as a negative control in all experiments (data not shown). IgGs were used as nonspecific controls, and input DNA values were used to normalize the values from quantitative ChIP samples.
ChIP assay from AGS cells treated with dimethyl sulfoxide (DMSO) or TSA were processed as above reported. For each assay, about 5×106 AGS cells were used for chromatin preparation and IP.
Treatment of gastric cancer cell lines with TSA
AGS, MKN28 and KATO III cells were plated in 10 cm culture dishes and grown for 24 hours before drug treatment. The next day, about 8.8 × 106 cells were incubated in fresh culture medium containing a TSA/DMSO solution up to a final concentration of 300 nM. Control cells were treated with an equivalent volume of DMSO. After 24 or 48 hours, cells were harvested and used either to evaluate the GKN1 mRNA expression by qRT-PCR or for ChIP assays.
Statistical analysis
Statistical analysis was performed by two-tailed paired Student’s t-test using KaleidaGraph 4.1.1 software. Western blot band intensity was evaluated with ImageJ 1.41o software. Data were reported as means ± standard deviation (SD). The significance was accepted at the level of p < 0.05.
Abbreviations
ChIP, chromatin immunoprecipitation; DMSO, dimethyl sulfoxide; DNMT1, DNA (cytosine-5)-methyltransferase 1; EZH2, enhancer of zeste homolog 2; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GC, gastric cancer; GKN1, gastrokine 1; G6PD, glucose-6-phosphate dehydrogenase; EBNA1, Epstein-Barr nuclear antigen 1; EBV, Epstein-Barr virus; HDAC, histone deacetylase; H3K9triMe, trimethylation of Histone 3 at lysine 9; IM, intestinal metaplasia; PIPES, 1,4-piperazinediethanesulfonic acid; PMSF, phenylmethylsulfonyl fluoride; Tip60, Histone acetyltransferase; TSA, trichostatin A; SUV39H1, Histone-lysine N-methyltransferase
ACKNOWLEDGMENTS
We thank Dr. G. de Dominicis, Hospital Cardarelli, Naples, Italy, for tissue histopathological classification. We thank also Valentina Villano for her technical support in tissue culture.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
FUNDINGS
This work was supported by funds from Programmi di Ricerca Scientifica di Rilevante Interesse Nazionale (2012CK5RPF_004), PON Ricerca e Competitività 2007-2013 (PON01_02782) and POR Campania FSE 2007-2013, Project CRÈME.
Authors’ contributions
FA and CSDS were involved in Chip assays, AF performed qRT-PCR analyses and statistical evaluation, GM was involved in TSA cell treatments and RNA preparation, MDP take care of patient recruitment and informed consent, ER take care of cell tissue culture, cell growth and transfecions and RNA preparation, PA was involved in the preparation of a draft manuscript and in the reading and approval of the final manuscript.
REFERENCES
1. Martin TE, Powell CT, Wang Z, Bhattacharyya S, Walsh- Reitz MM, Agarwal K, Toback FG. A novel mitogenic protein that is highly expressed in cells of the gastric antrum mucosa, Am. J. Physiol. Gastrointest. Liver Physiol. 2003; 285:G332-G343.
2. Oien KA, McGregor F, Butler S, Ferrier RK, Downie I, Bryce S, Burns S, Keith WN. Gastrokine 1 is abundantly and specifically expressed in superficial gastric epithelium, down-regulated in gastric carcinoma, and shows high evolutionary conservation. J. Pathol. 2004; 203:789-797.
3. Lacy ER, Morris GP, Cohen MM. Rapid repair of the surface epithelium in human gastric mucosa after acute superficial injury. J Clin Gastroenterol. 1993; 17:S125-S135.
4. Podolsky DK. Healing the epithelium: Solving the problem from two sides. J Gastroenterol. 1997; 32:122-126.
5. Toback FG, Walsh-Reitz MM, Musch MW, Chang EB, Del Valle J, Ren H, Huang E, Martin TE. Peptide fragments of AMP-18, a novel secreted gastric antrum mucosal protein, are mitogenic and motogenic. Am J Physiol Gastroint Liver Physiol. 2003; 285:G344-G353.
6. Walsh-Reitz MM, Huang EF, Musch MW, Chang EB, Martin TE, Kartha S, Toback FG. AMP-18 protects barrier function of colonic epithelial cells: Role of tight junction proteins. Am J Physiol Gastroint Liver Physiol. 2005; 289:G163-G171.
7. Nardone G, Martin G, Rocco A, Rippa E, La Monica G, Caruso F, Arcari P. Molecular expression of gastyrokine 1 in normal mucosa and in Helicobacter pylori-related pre-neoplastic and neoplastic lesions. Cancer Biol. Ther. 2008; 7:1890-1895.
8. Nardone G, Rippa E, Martin G, Rocco A, Siciliano RA, Fiengo A, Cacace G, Malorni A, Budillon G, Arcari P. Gastrokine 1 expression in patients with and without Helicobacter pylori infection. Dig. Liver Dis. 2007; 39:122- 129.
9. Rippa E, Martin G, Rocco A, La Monica G, Fiengo A, Siciliano RA, Cacace G, Malori A, Nardone G, Arcari P. Changes of protein expression in Helicobacter pylori-infected human gastric mucosa. Current Topics in Peptide & Protein Research. 2007; 8:35-43.
10. Rippa E, La Monica G, Allocca R, Romano MF, De Palma M, Arcari P. Overexpression of gastrokine 1 in gastric cancer cells induces fas-mediated apoptosis. Journal of Cellular Physiology. 2011; 226:2571-2578.
11. Pavone LM, Del Vecchio P, Mallardo P, Altieri F, De Pasquale V, Rea S, Martucci NM, Di Stadio CS, Pucci P, Flagiello A, Masullo M, Arcari P, Rippa E. Structural characterization and biological properties of human gastrokine 1. Mol Biosyst. 2013; 9:412-421.
12. Shiozaki K, Nakamori S, Tsujie M, Okami J, Yamamoto H, Nagano H, Dono K, Umeshita K, Sakon M, Furukawa H, Hiratsuka M, Kasugai T, Ishiguro S, Monden M. Human stomach-specific gene, CA11, is down-regulated in gastric cancer. Int J Oncol. 2001; 19:701-707.
13. Yoon JH, Song JH, Zhang C, Jin M, Kang YH, Nam SW, Lee JY, Park WS. Inactivation of the Gastrokine 1 gene in gastric adenomas and carcinomas. Journal of Pathology. 2011; 223:618-625.
14. Lu F, Tempera I, Lee HT, Dewispelaere K, Lieberman PM. EBNA1 binding and epigenetic regulation of gastrokine tumor suppressor genes in gastric carcinoma cells. Virol. J. 2014; 11:12.
15. Müller MM, Fierz B, Bittova L, Liszczak G, Muir TW. A two-state activation mechanism controls the histone methyltransferase Suv39h1. Nat. Chem. Biol. 2016 [Epub ahead of print]
16. Fox JG, Wang TC. Helicobacter pylori-not a good bug after all! N. Engl. J. Med. 2001; 345:829-832.
17. Wang F, Meng W, Wang B, Qiao L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer. Lett. 2013 [Epub ahead of print].
18. Park SY, Yoo EJ, Cho NY, Kim N, Kang GH. Comparison of CpG island hypermethylation and repetitive DNA hypomethylation in premalignant stages of gastric cancer, stratified for Helicobacter pylori infection. J. Pathol. 2009; 219:410-416.
19. Leung SY, Yuen ST, Chung LP, Chu KM, Chan AS, Ho JC. hMLH1 promoter methylation and lack of hMLH1 expression in sporadic gastric carcinomas with high-frequency microsatellite instability. Cancer. Res. 1999; 59:159-164.
20. Suzuki H, Itoh F, Toyota M, Kikuchi T, Kakiuchi H, Hinoda Y, Imai K. Distinct methylation pattern and microsatellite instability in sporadic gastric cancer. Int. J. Cancer 1999; 83:309-313.
21. Niwa T, Tsukamoto T, Toyoda T, Mori A, Tanaka H, Maekita T, Ichinose M, Tatematsu M, Ushijima T. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer. Res. 2010; 70:1430-1440.
22. Chen HY, Zhu BH, Zhang CH, Yang DJ, Peng JJ, Chen JH, Liu FK, He YL. DNA methylation of multiple genes in gastric cancer: association with CpG island methylator phenotype and Helicobacter pylori infection. J. Korean Gastric Cancer Assoc. 2006; 6:227-236.
23. Choi WS, Seo HS, Song KY, Yoon JH, Kim O, Nam SW, Lee JY, Park WS. Gastrokine 1 expression in the human gastric mucosa is closely associated with the degree of gastritis and DNA methylation. J Gastric Cancer. 2013; 13:232-241.
24. Yoon JH, Choi YJ, Choi WS, Ashktorab H, Smoot DT, Nam SW, Lee JY, Park WS. GKN1-miR-185-DNMT1 axis suppresses gastric carcinogenesis through regulation of epigenetic alteration and cell cycle. Clin Cancer Res. 2013; 19:4599-4610.
25. Tariq M, Paszkowski J. DNA and histone methylation in plants. Trends Genet. 2004; 20:244-251.
26. Freitag M, Selker EU. Controlling DNA methylation: many roads to one modification. Curr Opin Genet Dev. 2005; 15:191-199.
27. Chiba T, Saito T, Yuki K, Zen Y, Koide S, Kanogawa N, Motoyama T, Ogasawara S, Suzuki E, Ooka Y, Tawada A, Otsuka M, Miyazaki M, Iwama A, Yokosuka O. Histone lysine methyltransferase SUV39H1 is a potent target for epigenetic therapy of hepatocellular carcinoma. Int J Cancer. 2015; 136:289-298.
28. Kang MY, Lee BB, Kim YH, Chang DK, Kyu Park S, Chun HK, Song SY, Park J, Kim DH. Association of the SUV39H1 histone methyltransferase with the DNA methyltransferase 1 at mRNA expression level in primary colorectal cancer. Int J Cancer. 2007; 121:2192-2197.
29. Park YS, Jin MY, Kim YJ, Yook JH, Kim BS, Jang SJ. The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Ann Surg Oncol. 2008; 15:1968-1976.
30. Hebbes TR, Clayton AL, Thorne AW, Crane-Robinson C. Core histone hyperacetylation co-maps with generalized DNase I sensitivity in the chicken beta-globin chromosomal domain. EMBO J. 1994; 13:1823-1830.
31. Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997; 389:348–352.
32. Nakagawa M, Oda Y, Eguchi T, Aishima S, Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M, Tsuneyoshi M. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep. 2007; 18:769-774.
33. Choi JH1, Kwon HJ, Yoon BI, Kim JH, Han SU, Joo HJ, Kim DY. Expression Profile of Histone Deacetylase 1 in Gastric Cancer Tissues. Jpn J Cancer Res. 2001; 92:1300-1304.
34. Takai N, Ueda T, Nishida M, Nasu K, Narahara H. A novel histone deacetylase inhibitor, Scriptaid, induces growth inhibition, cell cycle arrest and apoptosis in human endometrial cancer and ovarian cancer cells. Int J Mol Med 2006; 17:323-329.
35. Chang J, Varghese DS, Gillam MC, Peyton M, Modi B, Schiltz RL, Girard L, Martinez ED. Differential response of cancer cells to HDAC inhibitors trichostatin A and depsipeptide. Br J Cancer 2012; 106:116-125.
36. Agudelo M, Gandhi N, Saiyed Z, Pichili V, Thangavel S, Khatavkar P, Yndart-Arias A, Nair M. Effects of alcohol on histone deacetylase 2 (HDAC2) and the neuroprotective role of trichostatin A (TSA). Alcohol Clin Exp Res 2011; 35:1550-1556.
37. Wang YG, Wang N, Li GM, Fang WL, Wei J, Ma JL, Wang T, Shi M. Mechanisms of trichostatin A inhibiting AGS proliferation and identification of lysine-acetylated proteins. World J Gastroenterol. 2013; 19:3226-3240.
38. Li L, Fan B, Zhang LH, Xing XF, Cheng XJ, Wang XH, Guo T, Du H, Wen XZ, Ji JF. Trichostatin A potentiates TRAIL-induced antitumor effects via inhibition of ERK/FOXM1 pathway in gastric cancer. Tumour Biol. 2016; 37:10269-10278.
39. Suzuki T, Yokozaki H, Kuniyasu H, Hayashi K, Naka K, Ono S, Ishikawa T, Tahara E, Yasui W. Effect of trichostatin A on cell growth and expression of cell cycle- and apoptosis-related molecules in human gastric and oral carcinoma cell lines. Int J Cancer. 2000; 88:992-997.
40. Zhi Q, Guo X, Guo L, Zhang R, Jiang J, Ji J, Zhang J, Zhang J, Chen X, Cai Q, Li J, Liu B, Zhu Z, Yu Y. Oncogenic miR-544 is an important molecular target in gastric cancer. Anticancer Agents Med Chem. 2013; 13:270-275.
41. Yoon JH, Choi WS, Kim O, Choi SS, Lee EK, Nam SW, Lee JY, Park WS. NKX6.3 controls gastric differentiation and tumorigenesis. Oncotarget. 2015; 6:28425-28439. doi: 10.18632/oncotarget.4952.
42. American Joint Committee on Cancer: AJCC Cancer Staging Manual. 6th ed. Springer: 2002; p.99-106.
43. Di Stadio CS, Altieri F, Miselli G, Elce A, Severino V, Chambery A, Quagliariello, V, Villano V, de Dominicis G, Rippa E, Arcari P. AMP18 interacts with the anion exchanger SLC26A3 and enhances its expression in gastric cancer cells. Biochimie. 2016; 121:151-160.
44. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402-408.
45. Federico A, Pallante P, Bianco M, Ferraro A, Esposito F, Monti M, Cozzolino M, Keller S, Fedele M, Leone V, Troncone G, Chiariotti L, Pucci P, et al. Chromobox protein homologue 7 protein, with decreased expression in human carcinomas, positively regulates E-cadherin expression by interacting with the histone deacetylase 2 protein. Cancer Res. 2009; 69:7079-7087.