
INTRODUCTION
Oxidative stress is a crucial initiating factor in a variety of pathological conditions including osteoporosis, resulting from excessive generation of ROS or damage of the anti-oxidative stress defense system in impairment of osteoblastic bone formation [1]. Most of studies reported that the pathogenesis of osteoporosis is associated with oxidative stress in osteoblasts [2, 3]. Overproduction of ROS disrupts the balance between oxidation and antioxidant defense systems, leading to bone loss by promoting lipid peroxidation and lowering antioxidant enzymes, inducing apoptosis of osteoblasts and inhibiting bone formation [4–6]. In terms of the possible importance of ROS in bone metabolism, it is essential for cells to effectively up-regulate antioxidants, decrease ROS generation and scavenge free radical, which may contribute to bone loss and antagonize osteoporosis.
Nuclear factor-erythroid 2-related factor 2 (Nrf2), also known as NF-E2-like 2 (NFE2L2), plays an essential role in the cellular defense against various inflammatory and oxidative stress-induced diseases, including osteoporosis, which is a transcription factor that binds to antioxidant response element and regulates the production of multiple anti-oxidative enzymes [7–11]. Under stress conditions, Nrf2 dissociates from Kelch-like ECH-associated protein-1 (Keap1) and translocates into the nucleus to activate the expression of antioxidant-responsive genes and induction of phase II detoxifying enzymes, which is considered as a major regulator of oxidant resistance and involved in the maintenance of the cellular redox homeostasis, and elimination of reactive oxidants and electrophilic agents [8, 10, 11]. Heme oxygenase-1 (HO-1), a well-known intracellular inducible phase II enzyme, is involved in defense against various oxidative-inducing agents, including LPS [9], hydrogen peroxide [12], hypoxia [13] and subsequent lethality [14]. Upon oxidative stress, HO-1 is up-regulated and enhancement of HO-1 expression is potentially important in iron homeostasis, antioxidant defense, and anti-resorption of bone [15–17]. The HO-1 gene is primarily regulated at the transcriptional level, and transcriptional regulation of HO-1 protein expression is mediated by Nrf2 activation [18]. In particular, a non-cytotoxic pharmacological agent may maximize the intrinsic anti-oxidant potential of cells and represent a promising therapeutic intervention in treating a variety of disorders related to oxidative stress and inflammation via increasing HO-1 expression [18, 19]. Indeed, PI3K/Akt is a key survival signaling pathway in governing the cellular defense system against oxidative injury, and regulating cell proliferation survival and apoptosis associated with Nrf2 activation and HO-1 induction [20, 21]. Therefore, the PI3K/Akt pathway plays a vital role in the Nrf2-mediated antioxidant response, making it a potential target of drug therapy by modulating Nrf2 as an upstream signaling molecule.
So far, numerous polyphenols have been identified and proposed as the therapeutic agents to counteract oxidative stress-related diseases, which is involved in the balance between the cellular production and elimination of free radicals [22]. CGA, a type of polyphenolic compound formed by esterification of caffeic and quinic acids, is the major active ingredient found in many traditional Chinese medicines such as Flos Lonicerae Japonicae and Eucommia Ulmoides, and it is also abundant in some fruits, dietary vegetables, and daily beverages like coffee, pineapple, beans, strawberries and apples [23]. Previous reports have demonstrated that CGA exhibits a wide range of diverse pharmacological effects, including anti-inflammatory, anti-oxidative, and anti-carcinogenic activities [3, 24, 25]. However, the antioxidant effects and molecular mechanisms of CGA in osteoblasts have not been elucidated yet. Therefore, the present study established an in vitro experimental model of osteoblasts exposed to H2O2 to evaluate the effect of CGA on H2O2-induced oxidative stress of the osteoblast MC3T3-E1 cells, and investigate whether CGA conferred an antioxidant defense capacity via modulation of PI3K/Akt activity and activation of Nrf2 and HO-1 expressions.
RESULTS
H2O2 inhibits MC3T3-E1 cells proliferation and induces apoptosis
The results showed that the viability of MC3T3-E1 cells exposed to H2O2 was decreased in both time- and dose- dependent manners (Figure 1A). MC3T3-E1 cells were stained with DCFH-DA to assess the effect of H2O2 on the intracellular ROS production. Intracellular ROS production significantly increased with passage of time in H2O2-treated MC3T3-E1 cells (Figure 1B). Flow cytometric analysis demonstrated that the apoptotic osteoblasts increased with the increase of the dose of H2O2 (Figure 1C). As observed under microscope, treatment with H2O2 (400 μM) for 4 h resulted in significant cell shrinkage and a decrease in the rate of cellular attachment compared to the control group (Figure 1D). According to the results, the concentration (400 μM) of H2O2 for 4 h was chosen to be the model condition of oxidative stress in osteoblast cells for further research.
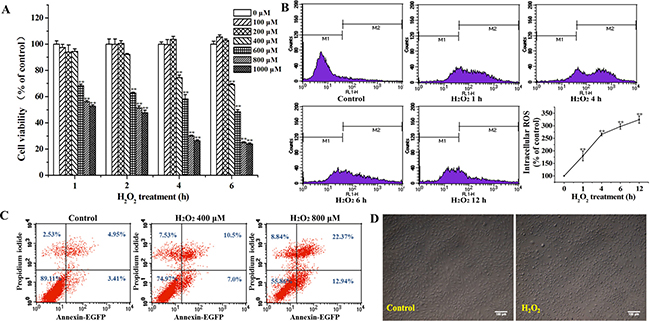
Figure 1: Effects of H2O2 on the apoptosis of MC3T3-E1 cells. (A) MC3T3-E1 cells were treated with various concentrations of H2O2 (0~1000 μM) for 1, 2, 4 and 6 h, and the cell viability was analyzed by MTT assay. (B) Cells were treated with 400 μM H2O2 for the indicated times (0, 1, 4, 6, and 12 h), and the cells were stained with DCFH-DA to detect the intracellular ROS production in different times by flow cytometry. (C) Cells were treated with 0, 400 and 800 μM H2O2 for 4 h, and apoptosis was determined by flow cytometry followed by Annexin V–PI double staining. (D) Cells were treated with 400 μM H2O2 for 4 h, and the cell morphology was observed using an inverted/phase-contrast microscope. Data represent means ± S.E.M of three independent experiments and differences between mean values were assessed by one-way ANOVA. **p < 0.01 indicates the significant difference compared with control group.
CGA promotes MC3T3-E1 cells proliferation
Cell viability was tested after being treated with different concentrations (0, 5, 25, 50, 100, 200, 400 μM) of CGA for 24 and 48 h. Various concentrations of CGA had a marked role in promoting cell proliferation without cytotoxicity, and this effect was in a time- and dose-dependent manner in the range of 25 to 400 μM (Figure 2).
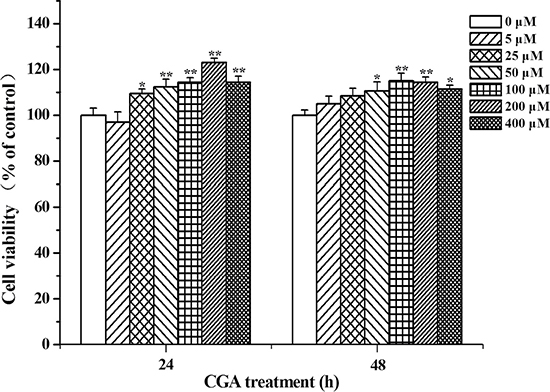
Figure 2: Effects of CGA on cell viability. Effect of CGA on the viability of MC3T3-E1 cells was measured using MTT assay. Cells were treated with various concentrations (0, 5, 25, 50, 100, 200, 400 μM) of CGA for 24, 48 h. Data represent means ± S.E.M of six separate experiments and differences between mean values were assessed by one-way ANOVA. *p < 0.05 and **p < 0.01 indicate the significant difference compared with control group.
CGA improves cell viability and reduces MC3T3-E1 apoptosis after H2O2 exposure
The results demonstrated that H2O2 exposure markedly reduced cell viability, which was attenuated by CGA treatment (Figure 3A). The results of cell apoptosis detection by flow cytometry using Annexin V/PI double staining showed that after exposure to H2O2 for 4 h, CGA lowered apoptosis in a dose-dependent manner (Figure 3B, 3C). Compared with the control group, exposure to 400 μM H2O2 for 4 h activated caspase-3 of the MC3T3-E1 cells. CGA treatment was found to reduce caspase-3 activity in a dose-dependent manner (Figure 3D). This result suggests that CGA inhibited caspase 3-mediated cell apoptosis induced by H2O2.
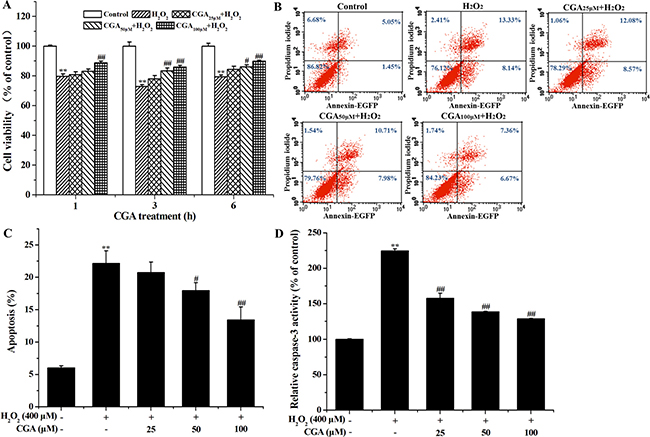
Figure 3: Protective effect of CGA on H2O2-induced cytotoxicity and inhibitory effect of CGA on H2O2-induced apoptosis in MC3T3-E1 cells. (A) Cells were pretreated with or without CGA at the indicated concentrations for 1, 3, 6 h and then incubated in the presence of H2O2 (400 μM). The cell viability was determined by MTT assay. (B) Cells were pretreated with or without CGA at the indicated concentrations for 3 h before treatment with H2O2 (400 μM). Apoptosis was measured by flow cytometry, followed by Annexin V-EGFP (FL 1 channels) and PI (FL 2 channels) double staining. (C) The percentage of apoptosis was counted including early apoptosis (in the lower-right quadrants) and late apoptosis (in the upper-right quadrant). (D) The activity of caspase-3 in cell lysates was measured using respective substrate peptide Ac-DEVD-ρNA. Data represent means ± S.E.M of three independent experiments and differences between mean values were assessed by one-way ANOVA. *p < 0.05 and **p < 0.01 indicate the significant difference compared with control group; #p < 0.05 and ##p < 0.01 indicate the significant difference compared with H2O2-treated group.
CGA inhibits H2O2-induced oxidative stress in MC3T3-E1 cells
Treatment of MC3T3-E1 cells with H2O2 alone clearly increased the levels of ROS, MDA and NO, and decreased GSH content. In contrast, pre-treatment with CGA significantly reversed the effect of H2O2-induced ROS, MDA, GSH (Figure 4A–4D), and NO level (Figure 4E) in a dose-dependent manner, respectively.
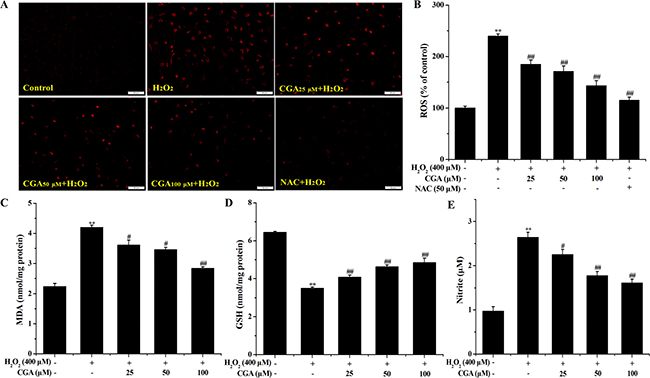
Figure 4: Effects of CGA on intracellular ROS, MDA, GSH and NO levels after H2O2 treatment. (A) and (B) MC3T3-E1 cells were pretreated with or without CGA at the indicated concentrations in the presence or absence of 50 μM NAC for 1 h before treatment with 400 μM H2O2, ROS generation was observed using fluorescence microscopy and the fluorescence intensity of ROS was measured by a fluorescence microplate reader. (C), (D) and (E) Cells were pretreated with or without CGA at the indicated concentrations for 3 h and incubated in the presence of H2O2 (400 μM). Data represent means ± S.E.M of three independent experiments and differences between mean values were assessed by one-way ANOVA. *p < 0.05 and **p < 0.01 indicate the significant difference compared with control group; #p < 0.05 and ##p < 0.01 indicate the significant difference compared with H2O2-treated group.
CGA increases HO-1 expression and it protects oxidative stress in MC3T3-E1 cells
Western blot analysis demonstrated that 100 μM CGA treatment significantly increased HO-1 protein level in a time-dependent manner (Figure 5A). Furthermore, cells were treated with various doses of CGA (0, 25, 50, 100 μM) for 3 h, and CGA concentration -dependently, increased HO-1 protein expression (Figure 5B). ZnPP IX, a HO-1 inhibitor, significantly reduced the expression of HO-1 induction by CGA (Figure 5C). The increased cell viability by CGA after exposure to H2O2 was significantly inhibited by ZnPP IX (Figure 5D), indicating that HO-1 induction is responsible for increased cell viability with CGA-pretreated cells under oxidative stress condition.
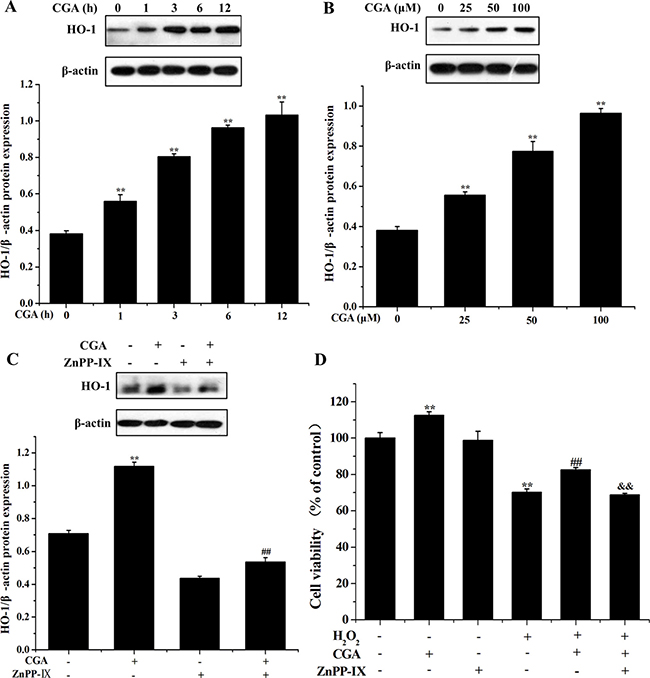
Figure 5: Effects of CGA on HO-1 protein induction. (A) Cells were treated with CGA (100 μM) for indicated time periods (0, 1, 3, 6 and 12 h). (B) Cells were treated with indicated concentrations (0, 25, 50 and 100 μM) of CGA for 3 h. Cell lysates were subjected to western blot analysis with anti-HO-1 antibody. (C) Cells were pretreated with ZnPP-IX (5 μM) for 1 h prior to incubation with CGA (100 μM) for 3 h and then proteins were extracted for western blot analysis using anti-HO-1 antibody. (D) Cells were pretreated with ZnPP-IX (5 μM) for 1 h, and then incubated with or without CGA (100 μM) for 3 h, followed by exposure of H2O2. Cell viability was measured using the MTT assay. **p < 0.01 indicates the significant difference compared with control group; ##p < 0.01 indicates the significant difference compared with H2O2-treated group; &&p < 0.01 indicates the significant difference compared with CGA-treated group.
CGA increases Nrf2 nuclear translocation and activates PI3K/Akt pathway in MC3T3-E1 cells
Western blot analysis showed that nuclear Nrf2 protein expression increased at 1 h, 3 h, and continued to rise up to 6 h, whereas Nrf2 protein in the cytoplasm decreased at 3 h, 6 h, and continued to rise up to 12 h compared with the control group (Figure 6A). CGA concentration-dependently induced translocation of Nrf2 from cytosol to nucleus (Figure 6B). Our data showed that CGA treatment notably enhanced Akt phosphorylation in a dose- and time-dependent manner, while no significant changes were found in the total Akt protein level (Figure 6C, 6D). These finding suggested that CGA-induced HO-1 expression and Nrf2 nuclear translocation, which may be associated with PI3K/Akt signaling pathway
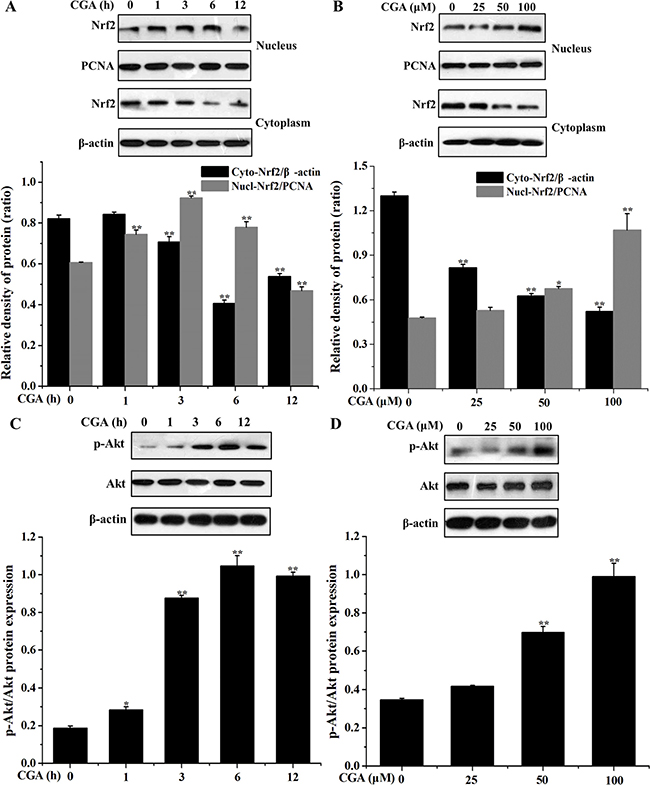
Figure 6: Effects of CGA on Nrf2 nuclear translocation and PI3K/Akt phosphorylation in MC3T3-E1 cells. (A) and (C) Cells were treated with CGA (100 μM) for indicated time points (0, 1, 3, 6 and 12 h). (B) and (D) Cells were treated with indicated concentrations (0, 25, 50 and 100 μM) of CGA for 3 h. (A) and (B) The nuclear and cytosolic levels of Nrf2 were determined by western blot analysis. PCNA was used as nuclear loading control. (C) and (D) Cell lysates were subjected to western blot analysis with anti-Akt or p-Akt antibodies. β-actin was used as loading control. **p < 0.01 indicates the significant difference compared with control group.
Role of PI3K/Akt on CGA-induced HO-1 expression and Nrf2 nuclear translocation in MC3T3-E1 cells
Our results revealed that inhibition of the PI3K/Akt pathway significantly suppressed Akt phosphorylation and the nuclear translocation of Nrf2 induced by CGA (Figure 7A, 7B). Similarly, pharmacological inhibition of PI3K/Akt significantly suppressed the protein level of HO-1 induced by CGA (Figure 7C).
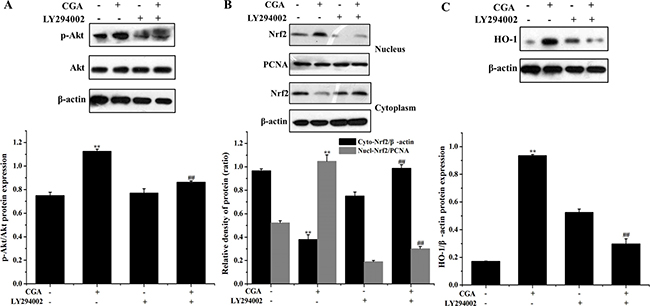
Figure 7: Role of the PI3K/Akt pathway in CGA-induced Nrf2 transcription and HO-1 expression. (A), (B) and (C) Cells were pretreated with 20 μM LY294002 for 1 h, and then treated with 100 μM CGA for 3 h. Nrf2, p-Akt and HO-1 levels were determined by western blot analysis. **p < 0.01 indicates the significant difference compared with control group; ##p < 0.01 indicates the significant difference compared with H2O2-treated group.
Role of PI3K/Akt pathway on cytoprotection induced by CGA in MC3T3-E1 cells
LY294002, an inhibitor of PI3K/Akt, abolished the protective effect of CGA against H2O2-induced decreased cell viability and increased apoptotic rate (Figure 8), indicating that the protective role of CGA was through the PI3K/Akt pathway, a vital upstream signaling pathway that mediated the Nrf2-related cytoprotective effect.
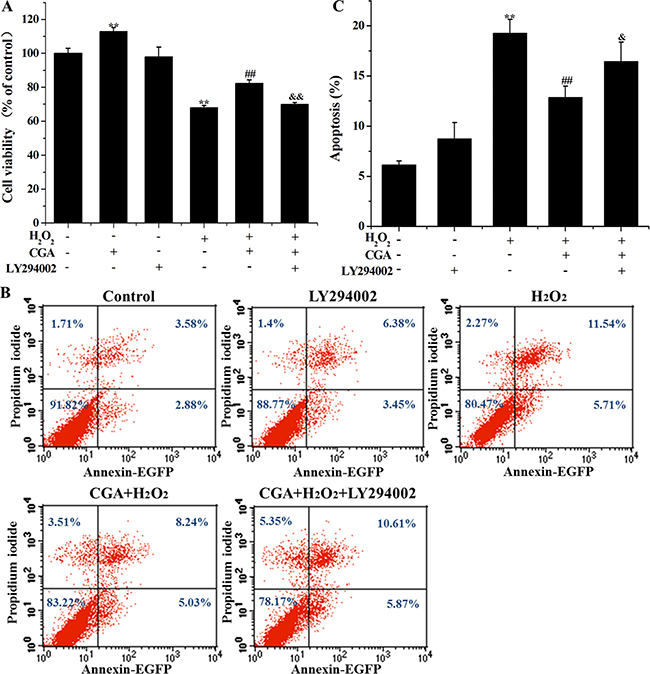
Figure 8: Effects of CGA and selective inhibitor LY294002 on H2O2-induced cytotoxicity and apoptosis in MC3T3-E1 cells. Cells were pretreated with 20 μM LY294002 for 1 h, and then incubated with or without CGA (100 μM) for 3 h, followed by exposure with H2O2. (A) Cell viability was measured using the MTT assay. (B) and (C) Cell apoptosis was determined by the Annexin V-EGFP/PI staining assay. **p < 0.01 indicates the significant difference compared with control group; ##p < 0.01 indicates the significant difference compared with H2O2-treated group; &p < 0.05 and &&p < 0.01 indicate the significant difference compared with CGA-treated group.
DISCUSSION
Several observational studies showed that ROS production reduces the bone formation of osteoblastic cells, decreases bone mass and alters bone micro-architecture in age-related osteoporosis or in secondary osteoporosis [1, 4, 15]. H2O2, as involvement of oxidative stress, may induce the self-generation of free radicals and produce a wide range of damages including apoptosis, necrosis or autophagy of various types of cells, including osteoblast cells [26–28]. In this study, MC3T3-E1 cells were exposed to H2O2 to perform a model of oxidative damage, we found that treating MC3T3-E1 cells with H2O2 resulted in reduction of cell viability, ROS production and apoptosis. Moreover, bone pathophysiology is highly sensitive to ROS production, contributing to define the cellular and molecular basis of the biological activities of ROS [29]. Therefore, it is tempting to suggest that exogenous administration of antioxidants should be investigated as a potential approach to protect cells against ROS for the treatment of osteoporosis. Previous studies have revealed the antioxidant effect of CGA, effectively scavenging free radicals and inhibiting lipid peroxidation [25, 30, 31]. Recent report demonstrated that CGA efficiently inhibits osteoblast apoptosis and ameliorates hormone-induced femoral head necrosis [32]. Therefore, the protective effects and underlying mechanisms of CGA against oxidative damage were explored in the current study. The MTT colorimetric assay showed that CGA can increase the proliferation capabilities of osteoblasts. Moreover, the results showed that CGA significantly attenuated H2O2-induced cell damage through increasing cell viability, decreasing apoptotic rate and caspase-3 activity in MC3T3-E1 cells.
MDA is frequently used to measure the degree of lipid peroxidation, which is closely related to cell damage [33]. GSH is a vital antioxidant that scavenges hydrogen peroxide and ROS [34]. When excessively produced, NO may react with superoxide anion radicals, leading to the strong oxidant, peroxynitrite, and destroying functional tissues [35]. In the current study, CGA significantly decreased ROS and NO production, and the content of MDA, while increasing the level of GSH in MC3T3-E1 cells. These findings indicated that blocking H2O2-induced apoptosis by CGA may be mediated by an increase in antioxidant capacity, and/or the prevention of oxidative stress.
HO-1, one of the pivotal cytoprotective enzymes in cellular defense, is a key protein that plays an important role in the cellular adaptation to oxidative stress inflicted by pathological events in a wide variety of cells [7, 8, 12, 36]. Many investigations have confirmed that upregulation of HO-1 contributes to the cellular defense mechanism in response to oxidative insults [20, 34, 37]. The present study showed that CGA treatment significantly upregulated HO-1 protein expression in a dose- and time-dependent manner, indicating that CGA exhibited antioxidant activity by upregulation HO-1 expression. Moreover, inhibition of HO-1 can partially reverse the increased cell viability induced by CGA in the presence of H2O2. These data clearly demonstrated that the increased HO-1 protein expression in MC3T3-E1 cells is responsible for CGA-induced protective effect against H2O2-induced cytotoxicity, implicating that CGA confers protection against oxidative stress by increasing HO-1 levels.
Nrf2 is known as an upstream mediator of ARE-dependent phase II enzyme expression [38]. The Nrf2 translocation has been further demonstrated to up-regulate the expression of antioxidant genes [7, 36, 37], which plays an essential role in the induction of HO-1 and may provide a useful therapeutic approach to combating oxidative stress. Furthermore, current study reveals that the induction of HO-1 expression is mediated by Nrf2 activation in MC3T3-E1 cells [18]. Western blot analysis depicted that CGA treatment significantly increased Nrf2 protein level in the nucleus from 1 h to 6 h and in a dose range of 50 μM to 100 μM, which suggested that CGA may up-regulate HO-1 protein expression by activating Nrf2 signaling pathway. When MC3T3-E1 cells were treated with CGA for a long period time (12 h), nuclear Nrf2 dramatically decreased, which indicated that Nrf2 anti-oxidative stress pathway just adaptively responded to CGA treatment in a short time.
Furthermore, PI3K/Akt signaling pathway is commonly involved in HO-1 expression and in Nrf2-dependent transcription in diverse cell types in response to oxidative stress and different stimuli [7, 33, 37, 39]. Therefore, the current experiments were designed to determine a role for PI3K/Akt as a possible regulator of cell survival in CGA-induced HO-1 expression. In the present findings, CGA was found to increase the phosphorylation level of Akt protein, suggesting that enhancement of Akt protein phosphorylation may contribute to the CGA-induced HO-1 expression. However, LY294002, a specific PI3K/Akt inhibitor, significantly suppressed the nuclear localization of Nrf2 and markedly blocked HO-1 protein expression in the presence of CGA. Moreover, inhibition of PI3K/Akt signaling pathway partially reverse the increased cell viability and decreased apoptotic rate induced by CGA in the presence of H2O2, it indicating that PI3K/Akt pathway plays a central role in the mechanism of CGA-induced Nrf2/HO-1 activation.
Our findings demonstrated that CGA activates nuclear translocation of Nrf2, induces the expression of HO-1, and reduces H2O2-induced cell death in vitro via activating PI3K/Akt pathway in MC3T3-E1 cells as shown in Figure 9. However, it has to be emphasized that whether there is the same effect of CGA in vivo or in other cell lines. Moreover, further investigations will be carried out in the future to reveal that whether other redox-sensitive transcription factors and targets, such as activator protein-1 (AP-1) and nuclear factor-κB (NF-κB) [8, 16], and other possible pathways, such as mitogen-activated protein kinase (MAPK) and AMP- activated protein kinase (AMPK) [8, 34, 40, 41], may participate in the process of CGA-mediated antioxidant effect.
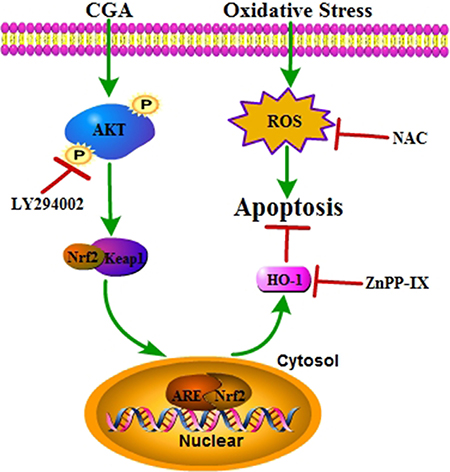
Figure 9: A proposed signaling pathway involved in CGA against H2O2-induced oxidative damage. Schematic diagram shows that CGA induces Nrf2 –mediated cytoprotective protein via activation of PI3K/Akt signaling, which protects against oxidative stress of osteoblast cells. H2O2 induced ROS generation resulting in cell apoptosis. Meanwhile, CGA activates Nrf2/HO-1 through the PI3K/Akt pathway. Activation of Nrf2/HO-1 protects against H2O2 –induced osteoblast apoptosis. Green arrow indicates stimulation and red bar indicates inhibition.
MATERIALS AND METHODS
Reagents and antibodies
CGA [> 96% high-performance liquid chromatography (HPLC) purity] was purchased from National Institutes for Food and Drug Control (Beijing, China). 3- (4, 5-dimethyl-thiazol-2-yl)-2,5-dipheny l tetrazolium bromide (MTT), dimethyl sulfoxide (DMSO) and Zinc protoporphyrin IX (ZnPP) were purchased from Sigma-Aldrich Chemical (Sigma, USA). Minimum essential medium (α-MEM) and fetal bovine serum (FBS) were purchased from Gibco (Grand island, NY, USA). Primary antibodies to HO-1 (Cat # ab13248), Nrf2 (Cat # ab62352) and corresponding secondary antibodies (Cat # ab6721, Cat # ab6728) were obtained from Abcam (Abcam, USA). Antibodies for β-actin (Cat # AC004) and proliferating cell nuclear antigen (PCNA, Cat # A0264) were purchased from ABclonal Technology (Wuhan, China). LY294002 (Cat # 9901) and antibodies against Akt (Cat # 4685), and p-Akt (Cat # 4060) were purchased from Cell Signaling Technology (Danvers, MA, USA).
Cell culture and treatment
The mouse osteoblastic cell line MC3T3-E1 was purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). Cells were cultured in α-MEM medium supplemented with 10% FBS and 1% antibiotics (100 U/mL penicillin and 100 U/mL streptomycin) at 37°C in a humidified atmosphere of 5% CO2. All cells were washed with phosphate buffer saline (PBS, PH 7.2–7.4) before incubation with CGA or H2O2. After reaching 85% confluence, the cells were treated with medium containing different concentrations of CGA (0, 25, 50, 100 μM) for 1, 3, 6 h before treatment with H2O2 (400 μM) for 4 h. ZnPP IX (5 μM) or LY294002 (20 μM) was treated for 1 h before CGA treatment. CGA, ZnPP IX or LY294002 were dissolved in DMSO, and the final DMSO concentration was ≤ 0.1% (v/v).
Cell viability assay
Cell viability was determined by MTT reduction assay. In brief, MC3T3-E1 cells were pre-incubated with α-MEM containing 10% FBS overnight in 96-well plates at a density of 5 × 104 cells per well. According to the experimental design, treated cells in each well were added with α-MEM solution containing 10% MTT. The cells were incubated at 37°C for 4 h, the supernatants were removed, and the formazan crystals were dissolved in 150 μl DMSO. Absorbance was recorded at a wavelength of 490 nm and reference wavelength of 630 nm using a microplate reader (Bio-Rad, Foster, California, USA).
Assay of Annexin V-EGFP/PI apoptosis
Cellular apoptosis was determined using the Annexin V-EGFP/PI Cell Apoptosis Detection Kit (KeyGEN, Nanjing, China). Briefly, cells were plated into 6-well plates and treated with LY294002, CGA or H2O2 according to the experimental design. Following each specific treatment, cells were harvested, then washed twice with ice-cold PBS and centrifuged at 1000 rpm for 10 min. Cells were resuspended in 100 μl of 1× binding buffer and transferred to sterile flow cytometry glass tubes 5 μl of Annexin V-EGFP and 5 μl of propidium iodide were added and incubated at room temperature (25°C) in dark conditions for 15 min. Finally, detection was performed using flow cytometric analysis according to manufacturer’s instructions (Beckman, Fullerton, California, USA).
Measurement of ROS production
The level of ROS was detected by measuring intracellular ROS formation using a ROS detection kit (Beyotime, Haimen, China). Briefly, MC3T3-E1 cells, cultured in 6-well plates for 24 h before treatment, were pretreated with CGA (0, 25, 50, 100 μM) and then exposed with H2O2 (400 μM) for 1 h to induce ROS production. Cells were washed twice with PBS and then incubated with 5μM 2′, 7′- dichlorofluorescein diacetate (DCFH-DA) for 30 min. Fluorescence staining was visualized using a fluorescence microscope (Olympus, IX71), and fluorescence assays were measured and quantified using a fluorescence microplate reader (Tecan, Sunrise) at excitation/emission 525/610 nm and a flow cytometer (Beckman, Fullerton, California, USA) at excitation/emission 488/525 nm.
Determination of caspase-3 activity
MC3T3-E1 cells were cultured in 6-well plates and then treated with CGA (0, 25, 50, 100 μM) for 3 h prior to 400 μM H2O2 treatment for 4 h. In brief, treated cells were extracted using lysis buffer (Beyotime, Haimen, China), and then the supernatants were collected and incubated with the substrate Ac-DEVD-pNA (acetyl-Asp-Glu-Val-Asp p-nitroanilide), which produces yellow pNA. Absorbance was measured in a microplate reader (Bio-Rad, Foster, California, USA) at a wavelength of 405 nm.
Analysis of MDA, GSH and NO
MC3T3-E1 cells were treated with different concentration of CGA (0, 25, 50, 100 μM) for 3 h, after which H2O2 (400 μM) was added for 4 h. The cells in each group were lysed and prepared to determine MDA and GSH contents using commercial colorimetric assay kits (Beyotime, Haimen, China) according to manufacturer’s instructions. In addition, culture supernatants were collected and assayed to measure NO release using a NO assay kit (Beyotime, Haimen, China). The results were calculated with reference to a standard curve and expressed as nM/mg protein for MDA and GSH and μM for NO, respectively.
Western blot analysis
MC3T3-E1 cells were treated with the indicated agents according to the experimental design. Total cellular protein of MC3T3-E1 cells was extracted using a total protein extraction kit (KeyGEN, Nanjing, China), and nuclear and cytoplasmic extractions were prepared using a nuclear and cytoplasmic extraction kit (KeyGEN, Nanjing, China), respectively. The protein content was quantified using a BCA protein assay kit (Beyotime, Haimen, China) according to the manufacturer’s instructions. Proteins were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a poly-vinylidine difluoride membrane (PVDF, Millipore, MA, USA). After being blocked, the membranes were incubated over night at 4°C with specific primary antibodies. Then membranes were probed with HRP-conjugated secondary antibody for 1 h at room temperature and detection was performed by an enhanced chemiluminescence system (ECL, Beyotime, Haimen, China). The results were normalized to β-actin or PCNA and analyzed using Image J (National Institutes of Mental Health, Bethesda, MD, USA).
Statistical analysis
Results from a representative of three independent experiments were expressed as means ± standard error of the mean (SEM), and assessed by the one-way analysis of variance (ANOVA) followed by least significant difference (LSD) test for multiple comparisons, and post hoc test using SPSS 19.0 (SPSS, Inc., Chicago, IL, USA). A value of p < 0.05 was considered statistically significant.
CONCLUSIONS
In summary, our results showed that CGA protected MC3T3-E1 cells against H2O2-induced caspase 3-dependent cell death by decreasing ROS, MDA and NO production, while increasing GSH content, it also demonstrated that CGA augments the cellular antioxidant defense capacity by enhancing Nrf2 nuclear translocation and upregulating HO-1 induction, and that the PI3K/Akt signaling pathway plays a central role in this mechanism, thereby protecting osteoblasts from oxidative damage.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by grants from the National Natural Science Foundation of China (NO. 30571362, NO. 30972230 and NO. 31272623).
CONFLICTS OF INTEREST
The author declares that there is no conflict of interest regarding the publication of this paper.
REFERENCES
1. Baek KH, Oh KW, Lee WY, Lee SS, Kim MK, Kwon HS, Rhee EJ, Han JH, Song KH, Cha BY, Lee KW, Kang MI. Association of oxidative stress with postmenopausal osteoporosis and the effects of hydrogen peroxide on osteoclast formation in human bone marrow cell cultures. Calcif Tissue Int. 2010; 87:226–235.
2. Cervellati C, Bergamini CM. Oxidative damage and the pathogenesis of menopause related disturbances and diseases. Clin Chem Lab Med. 2015; 54: 739–753.
3. Liu YJ, Zhou CY, Qiu CH, Lu XM, Wang YT. Chlorogenic acid induced apoptosis and inhibition of proliferation in human acute promyelocytic leukemia HL60 cells. Mol Med Rep. 2013; 8:1106–1110.
4. Arai M, Shibata Y, Pugdee K, Abiko Y, Ogata K. Effects of reactive oxygen species (ROS) on antioxidant system and osteoblastic differentiation in MC3T3-E1 cells. IUBMB Life. 2007; 59:27–33.
5. Pisoschi AM, Pop A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur J Med Chem. 2015; 97:55–74.
6. Wauquier F, Leotoing L, Coxam V, Guicheux J, Wittrant Y. Oxidative stress in bone remodelling and disease. Trends Mol Med. 2009; 15:468–477.
7. Hwang YP, Jeong HG. Ginsenoside Rb1 protects against 6-hydroxydopamine-induced oxidative stress by increasing heme oxygenase-1 expression through an estrogen receptor-related PI3K/Akt/Nrf2-dependent pathway in human dopaminergic cells. Toxicol Appl Pharmacol. 2012; 242:18–28.
8. Lidia I, Ferrándiz ML, Brines R, Guede D, Cuadrado A, Alcaraz MJ. Effects of Nrf2 deficiency on bone microarchitecture in an experimental model of osteoporosis. Oxid. Med. Cell Longev. 2014; 2014:390–392.
9. Lv HM, Yu ZX, Zheng YW, Wang LD, Qin XF, Cheng GH, Ci XX. Isovitexin exerts anti-inflammatory and anti-oxidant activities on lipopolysaccharide-induced acute lung injury by inhibiting MAPK and NF-kappaB and activating HO-1/Nrf2 pathways. Int J Biol Sci. 2016; 12:72–86.
10. Sun YX, Xu AH, Yang Y, Li JL. Role of Nrf2 in bone metabolism. J Biomed Sci. 2015; 22: 1–7.
11. Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009; 284:13291–13295.
12. Chen YC, Chow JM, Lin CW, Shen SC. Baicalein inhibition of oxidative-stress-induced apoptosis via modulation of ERKs activation and induction of HO-1 gene expression in rat glioma cells C6. Toxicol Appl Pharmacol. 2006; 216:263–273.
13. Reddy PH, Reddy TC, Marreddy R, Reddy NM, Noel S, Rabb H, Reddy SP. Nrf2-Akt interactions regulate heme oxygenase 1 expression in kidney epithelia during hypoxia and hypoxia-reoxygenation. Am J Physiol Renal Physiol. 2016; doi: 10.1152/ajprenal.00362.
14. Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006; 86:583–650.
15. Lee D, Kook SH, Ji H, Lee SA, Choi KC, Lee KY, Lee JC. N-acetyl cysteine inhibits H2O2-mediated reduction in the mineralization of MC3T3-E1 cells by down-regulating Nrf2/HO-1 pathway. BMB Rep. 2015; 48:636–641.
16. Min KJ, Lee JT, Joe EH, Kwon TK. An IκBα phosphorylation inhibitor induces heme oxygenase-1(HO-1) expression through the activation of reactive oxygen species (ROS)-Nrf2-ARE signaling and ROS-PI3K/Akt signaling in an NF-κB-independent mechanism. Cell Signal. 2011; 23:1505–1513.
17. Zhang LF, Qi J, Zuo G, Jia P, Shen X, Shao J, Kang H, Yang H, Deng L. Osteoblast-secreted factors promote proliferation and osteogenic differentiation of bone marrow stromal cells via VEGF/heme-oxygenase-1 pathway. PloS One. 2014; 9:e99946.
18. Choi EM, Suh KS, Kim YJ, Hong SM, Park SY, Chon S. Glabridin alleviates the toxic effects of methylglyoxal on osteoblastic MC3T3-E1 cells by increasing expression of the glyoxalase system and Nrf2/HO-1 signaling and protecting mitochondrial function. J Agric Food Chem. 2016; 64:226–235.
19. Yong PH, Jeong HG. Mechanism of phytoestrogen puerarin-mediated cytoprotection following oxidative injury: estrogen receptor-dependent up-regulation of PI3K/Akt and HO-1. Toxicol Appl Pharmacol. 2008; 233:371–381.
20. Lee YJ, Jeong HY, Kim YB, Lee YJ, Won SY, Shim JH, Cho MK, Nam HS, Lee SH. Reactive oxygen species and PI3K/Akt signaling play key roles in the induction of Nrf2-driven heme oxygenase-1 expression in sulforaphane-treated human mesothelioma MSTO-211H cells. Food Chem Toxicol. 2012; 50:116–123.
21. Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008; 1784:159–185.
22. Vitaglione P, Morisco F, Caporaso N, Fogliano V. Dietary antioxidant compounds and liver health. Crit. Rev Food Sci Nutr. 2004; 44:575–586.
23. Upadhyay R, Mohan Rao LJ. An outlook on chlorogenic acids-occurrence, chemistry, technology, and biological activities. Crit Rev Food Sci Nutr. 2013; 53:968–984.
24. Tsang MS, Jiao D, Chan BC, Hon KL, Leung PC, Lau CB, Wong EC, Cheng L, Chan CK, Lam CW, Wong CK. Anti-inflammatory activities of pentaherbs formula, berberine, gallic acid and chlorogenic acid in atopic dermatitis-like skin inflammation. Molecules. 2016; 21:519.
25. Ye HY, Li ZY, Zheng Y, Chen Y, Zhou ZH, Jin J. The attenuation of chlorogenic acid on oxidative stress for renal injury in streptozotocin-induced diabetic nephropathy rats. Arch Pharm Res. 2016; 39:989–997.
26. Fu C, Xu D, Jin Y, Liu Q, Meng Q. Alpha-lipoic acid promotes osteoblastic formation in H2O2-treated MC3T3-E1 cell and prevents bone loss in ovariectomized rats. J Cell Physiol. 2015; 230:2184–2201.
27. Xu ZS, Wang XY, Xiao DM, Hu LF, Lu M, Wu ZY, Bian JS. Hydrogen sulfide protects MC3T3-E1 osteoblastic cells against H2O2-induced oxidative damage-implications for the treatment of osteoporosis. Free Radical Bio Med. 2011; 50:1314–1323.
28. Yang YH, Li B, Zheng XF, Chen JW, Chen K, Jiang SD, Jiang LS. Oxidative damage to osteoblasts can be alleviated by early autophagy througn the endoplasmic reticulum stress pathway-Implications for the treatment of osteoporosis. Free Radical Bio Med. 2014; 77:10–20.
29. Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of pathogenesis of osteoporosis. Endocr Rev. 2010; 31:266–300.
30. Gul Z, Demircan C, Bagdas D, Buyukuysal RL. Protective effects of chlorogenic acid and its metabolities on hydrogen peroxide-induced alterations in rat brain slices: a comparative study with resveratrol. Neurochem Res. 2016; 41:2075–2085.
31. Zhou Y, Zhou LL, Ruan Z, Mi S, Jiang M, Li X, Wu X, Deng Z, Yin Y. Chlorogenic acid ameliorates instestinal mitochondrial injury by increasing antioxidant effects and activity of respiratory complexes. Biosci Biotechnol Biochem. 2016; 80:962–971.
32. Zhan MJ, Hu XD. Mechanism of chlorogenic acid treatment on femoral head necrosis and its protection of osteoblasts. Biomed Rep. 2016; 5:57–62.
33. Li H, Song F, Duan LR, Sheng JJ, Xie YH, Yang Q, Chen Y, Dong QQ, Zhang BL, Wang SW. Paeonol and danshensu combination attenuates apoptosis in myocardial infarcted rats by inhibiting oxidative stress: Roles of Nrf2/HO-1 and PI3K/Akt pathway. Sci Rep. 2016; 6:1–14.
34. Wang P, Peng X, Wei ZF, Wang W, Ma WD, Yao LP, Fu YJ, Zu YG. Geraniin exerts cytoprotective effect against cellular oxidative stress by upregulation of Nrf2-mediated antioxidant enzyme expression via PI3K/AKT and ERK1/2 pathway. Biochim Biophys Acta 2015; 1850:1751–1761.
35. Mikulec C, Rundhaug JE, Simper MS, Lubet RA, Fischer SM. The chemopreventive efficacies of nonsteroidal anti-inflammatory drugs: the relationship of short-term biomarkers to long-term skin tumor outcome. Cancer Prev Res. 2013; 6:675–685.
36. Pae HO, Oh GS, Jeong SO, Lee BS, Choi BM, Lee HS, Chung HT. PGG up-regulates HO-1 expression by stimulaing Nrf2 nuclear translocation in an extracellular signal-regulated kinase dependent manner in HepG2 cells. World J Gastroentero. 2006; 12:214–221.
37. Xu X, Li H, Hou X, He S, Wan C, Yin P, Liu M, Liu F, Xu J. Punicalagin induces Nrf2/HO-1 expression via upregulation of PI3K/Akt pathway and inhibitis LPS-induced oxidative stess in RAW264.7 macrophages. Mediators Inflamm. 2015; 2015:380218.
38. Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Roles of Nrf2/HO-1 system in develpoment, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci. 2016; 73:3221–3247.
39. Guo C, Yang L, Luo J, Zhang C, Xia YZ, Ma T, Kong LY. Sophoraflavanone G from Sophora alopecuroides inhibits lipopolysaccharide-induced inflammation in RAW264.7 cells by targeting PI3K/Akt, JAK/STAT and Nrf2/HO-1 pathways. Int Immunopharmaco. 2016; 38:349–335.
40. Ardura JA, Portal-Núñez S, Castelbón-Calvo I, Marítnez de Toda I, De la Fuente M, Esbrit P. Parathyroid hormone-related protein protects osteoblastic cells from oxidative stress by activation of MKP1 phosphatase. J Cell Physiol. 2016; doi: 10.1002/jcp.25473.
41. Jang HJ, Hong EM, Kim M, Kim JH, Jang J, Park SW, Byun HW, Koh DH, Choi MH, Kae SH, Lee J. Simvastatin induces heme oxygenase-1 via NF-E2-related factor 2 (Nrf2) activation througn ERK and PI3K/Akt pathway in colon cancer. Oncotarget. 2016; 7:46219–46229. doi: 10.18632/oncotarget.10078.