
Introduction
Arsenic is one of the most notorious poisons since the ancient times. Humans are exposed to arsenic predominantly through drinking water, air, food, occupation and other environmental sources [1-3]. Especially, arsenic present in seafood is frequently as organic forms (i.e., arsenobetaine, arsenosugars, etc.), which is considered to be nontoxic or much less toxic than inorganic arsenic. [4-6] In fact, inorganic arsenic (e.g., arsenate and arsenite) is the most poisonous form which is found in the contaminated drinking water in many parts of the world [7, 8], and there are at least 40 countries in the world with arsenic concentrations in ground water are higher than 10µg/L. Importantly, it has been estimated that 200 million people in the world are at risk from health effects associated with high concentrations of arsenic in their drinking water [9-10].
As a consequence of the biggest arsenic calamity has emerged in several parts of the world, the situation of arsenic toxicity is alarming and severe health problems are reported amongst the inhabitants relying on ground water as sources of drinking water [9]. Every year, new areas are being contaminated and identified. The use of ground water for irrigation and the bioavailability of arsenic to food crops and the uptake by humans and livestock through the food chain have opened additional pathways for arsenic exposure all over the world. The magnitude of the arsenic catastrophe has projected to be the largest in history of environmental disaster that will be more serious than those at Chernobyl, Ukraine in 1986 and Bhopal, India in 1984 [11-13]. Long-term or chronic exposure to arsenic is related to severe adverse health effects including dermatitis, cardiovascular diseases, diabetes mellitus, chronic bronchitis, immune disorders, peripheral neuropathy, liver damage, renal failure, adverse reproductive outcomes, hematological effects, and other ailments [14-21]. In fact, arsenic affects almost all vital organs of human body causing the damage or dysfunction.
Along with the adverse health effects, social and economical problem also occurs for the population of the arsenic affected countries. On the other hand, arsenic trioxide (As2O3) has been used as a drug for the treatment of various diseases in ancient China and Greece for over 3000 years [22]. In addition, As2O3 (Figure 1A) has recently shown remarkable therapeutic efficacy in patients with acute promyelocytic leukemia (APL) [23]. Though As2O3 has shown excellent efficacy for the treatment of APL, however, as a single-agent, it could not provide satisfactory outcomes for the management of different solid tumors [24]. Recently, it has been reported that some organic arsenic species have strong anticancer effect against solid tumors. Thus, it is very important and need to be considered the arsenic metabolism pathway and its organic metabolites for the treatment of cancers. In addition, it would be helpful in elucidating the probable role of different arsenic species (including inorganic arsenic and its methylated metabolites) in relation to toxicities along with the uses of arsenic as a drug. Especially, the roles of the methylated metabolites in the therapeutic efficacy and toxicity are still indistinct and needs to be elucidated. Thereby, in this review, we have focused on arsenic metabolic pathways, the roles of the methylated arsenic metabolites in toxicity and in the therapeutic efficacy for the treatments of solid tumors, APL and/or non-APL malignancies. Our findings will provide a new and better understanding of arsenic toxicity and therapeutics, which will further accelerate basic understanding on the mechanism of arsenic-induced diseases and the probable role of its active trivalent intermediate metabolites in the treatment of numerous neoplastic diseases.
Influences of Arsenic Potential for Toxicity and as Anticancer Agents
Metabolism of arsenic
Epidemiological studies indicate that iAsIII methylation is incomplete and the composition of arsenic metabolites in urine varies from person to person though they were exposed to the same level of arsenic in drinking water [25]. Interindividual differences in capacity to produce methylated arsenicals are linked to the differences in susceptibility to arsenic-induced diseases [26]. The distribution of the different arsenic species depends on many factors, including the species of arsenic administered, species of the plant or animal, different types of cells within the same organism, the availability of glutathione (GSH) and other biomolecules containing thiol (-SH) groups, the principle arsenic methylating enzymes, and on the pH of the matrix [27, 28]. Generally liver is the primary target organ for the metabolism of arsenic [29, 30]. It has been demonstrated that inorganic arsenic are commonly methylated in liver in the presence of a methyl donor S-adenosylmethionine (SAM) and a co-factor glutathione (GSH) with arsenicmethyltransferase (AS3MT) to relevant monomethylated [e.g., monomethylarsonous acid (MMAIII) monomethylarsonic acid (MMAV)] and dimethylated arsenic metabolites [e.g., dimethylarsinous acid (DMAIII), dimethylarsinic acid (DMAV)], and finally excreted into urine (Figure 1B). However, in vitro study has also indicated that other tissues seem to have arsenic methylating capacity and the highest amount of arsenic methylating activity is in the cytosol of testes, followed by kidney, liver, and lungs [31, 32]. The tissue distribution and retention of arsenic depends on the bioconversion of methylated metabolites and determines its actions as a toxicant or carcinogen [33].
The chemical plausibility of different methylation schemes has been recently evaluated and questioned [33]. The pioneer work of Frederick Challenger and his colleagues first described a sequence of alternating reactions in microorganisms known as the reductive/oxidative methylation pathway, during this process, iAsV is first reduced to the more toxic iAsIII, and then methylated and oxidized to MMAV simultaneously [34, 35]. Then MMAV get reduced to MMAIII and during oxidative methylation it is transformed into DMAV, which is then reduced to DMAIII. In certain species, but not in humans, during oxidative methylation DMAV then can be converted into trimethylarsine oxide (TMAOV) [36]. Due to the lack of sufficient analytical tools, Challenger was not able to identify any of the postulated intermediate species of the pathway, later Cullen and co-workers identified methylated intermediates to provide further evidence supporting the Challenger pathway in fungal and algal cultures [33].
Obviously, Challenger and his colleagues open up and made the basement of arsenic methylation pathway, however, alternative schemes for arsenic methylation suggesting glutathione- or protein-conjugated intermediates have also been proposed. Hayakawa et. al., have postulated that iAsIII species persist during methylation reactions and that oxidation to pentavalent arsenic species occurs (somehow) after methylation. In this pathway, they proposed that the arsenic-glutathione complexes are the major substrates for AS3MT, and that the yielding of inorganic arsenic triglutathione iAsIII(GS)3 can be further methylated to form the monomethylarsonic diglutathione MMAIII(GS)2 and dimethylarsinic glutathione DMAIII(GS) in the trivalent states in the presence of methyl-group donor SAM and GSH [37]. The main limitation of this method is that, how does CH3− group leave a positive center?
William R. Cullen [33] interpreted enzymatically catalyzed methylation according to the Challenger pathway and successfully solve the problem related with Hayakawa et. al. postulated pathway. In this pathway, protein or exogenous thiol reactants would neutralize the charge on arsenic by providing one more thiolate to the arsonium center and neutralize the charge on arsenic. Resulting an opportunity for reductive removal of a disulfide and leave the protein bound trivalent methylarsenic species.
Kala and coworkers have reported that arsenic-GSH complexes [e.g., iAsIII(GS)3, MMAIII(GS)2 and DMAIII(GS)] were predominantly found in bile and excreted into the bile through the multidrug resistance protein 2/canalicular multispecific organic anion transporter (MRP2/cMOAT) [38]. Moreover, we proposed that reductive methylation is the heart of an alternative pathway (Figure 2). In this pathway, iAsIII is metabolized in the body bound to proteins in a trivalent form during the successive reductive methylation by AS3MT in the presence of GSH and SAM, then oxidized to corresponding pentavalent arsenic species [39].
Wang et al. [40] showed the effects of different reductants such as GSH, cysteine, and tris(2-carboxyethyl)phosphine (TCEP) on the kinetics of arsenic methylation catalyzed by hAS3MT. They suggested that the valence state of arsenic is unchanged during the methylation reaction occurs on hAS3MT. Reductant helps to exposing the active sites of hAS3MT for binding to iAsIII by reducing the disulfide bond between the cysteine residues of enzyme and enzyme-AdoMet-reductant. In addition, our group along with other groups found that the trivalent arsenic species are mostly in the protein-binding forms, indicating arsenic possess higher affinities to bind with cysteine rich proteins than to glutathione [39, 41, 42].
Arsenic cytotoxicity is completely dependent on its oxidation state and chemical structure. During the detoxification of iAsIII, the compound is biomethylated to mono- and dimethylated (in some cases trimethylated) arsenicals of varying toxicity. Some of these metabolites will be less while others will be more toxic than iAsIII. For instance, pentavalent methylated arsenicals are less toxic, while trivalent methylated species are highly cytotoxic and genotoxic than its precursor iAsIII [43]. Notably, methylation of arsenic is recently considered to be a bioactivation and detoxification pathways, but it seems to depend on the condition of arsenic exposure and its levels. For example, in case of chronic exposure of arsenic, it may be bioactivation process, but in others (including a single low dose and acute exposure), it may be detoxification pathway. The overall toxicity of arsenic is dependent on the rate of methylation of the iAsIII formed. Many in vitro studies have shown that the trivalent forms of MMAIII and DMAIII are much more toxic than the pentavalent forms, and trivalent methylated arsenicals are in particular more toxic than trivalent inorganic arsenic [44-46]. Moreover, it has been found that MMAIII and DMAIII can inhibit the activities of many enzymes and are more genotoxic than iAsIII [47-50]. Styblo et al. [43] reported that MMAIII was highly toxic to human cells (e.g., hepatocytes, epidermal keratocytes, and bronchial epithelial cells) compared to inorganic species.
Recent study has indicated that higher proportions of methylated arseniclas in urine of arsenic exposed population are the evidence of increased risks of skin and bladder cancer [51-53]. Depending on the improvement of analytical techniques, toxicity studies of arsenic gradually changes from oxygenated metabolites of inorganic arsenic to thiolated arsenic metabolites, and many studies found that thiolation can change arsenic toxicity. The thiolated arsenicals are readily taken up by bladder cells and quickly converted to the corresponding trivalent oxygenated form of arsenic and showed their toxicities [54, 55].
On the other hand, the anticancer effects of arsenic are not only from direct or indirect influences on the genetic levels, but also strongly related with the distinctive arsenic metabolism pathways. AS3MT catalyzes arsenic to methylated products, by utilizing S-adenosyl methionine (SAM) which is an important co-factor of DNMTs and producing SAH, resulting to bring about the DNA hypomethylation [56]. Therefore, arsenic may play a crucial role in the regulation of tumor suppressor genes by interfering with the DNA methylation patterns. More importantly, a few clinical studies have shown that different methylated arsenic species (e.g., MMAV, MMAIII, DMAV and DMAIII) were found in blood, saliva or urine of APL patients after receiving arsenic treatment [57, 58]. Drobna et al., 2009 reported that As2O3 methylates to mono-methylarsenic (MMA) and dimethylarsenic (DMA) in the liver of APL patients [59]. Arsenic methylated metabolites are also found in hepatoma (HepG2) and glioblastoma cell lines (U87MG) [60, 61]. One group reported that As2O3 is metabolized into the methylated metabolites in the NB4 cell and argue that these intracellular methylated arsenic metabolites induce DNA hypomethylation and take part in the therapeutic effects of As2O3 in APL. Thereby, we should have to consider the importance of arsenic metabolism and its metabolites for elucidating the probable role of different arsenic species (i.e., including precursor and its metabolites) in relation to toxicities as well as in therapeutic efficacies [34, 40, 62].
Arsenic uptake and transport
Arsenic as toxic agents or drugs, it needs to be accumulated in cells and exert the biological functions. Thus, arsenic toxicity is suggested to be due to its chemical species, uptake and efflux [63-65]. Among the different arsenic species, it has been identified that trivalent arsenic species are much more toxic than that of corresponding pentavalent arsenicals. Hence, to understand the role of arsenic, it seems to be essential to precisely comprehend the importance of influx and transporter pathways.
Aquaglyceroporins (AQPs) have been recognized as vital family of membrane proteins for the uptake of arsenic [66], and the most important members such as AQP3, 7 and 9 are found to be involved in the uptake of iAsIII into the cells [67]. It has been reported that AQPs mainly expressed in majority of the organs including liver, spleen, lung, adipose tissues and kidney [68]. Interestingly, AQP9 have also been identified to play an important role for uptake of organic methylated MMAIII [69]. Additionally, AQP9 was found to be highly expressed in the leukemic cell lines (e.g., NB4 cells), making the cell more sensitive towards arsenic treatment [69]. The down regulation of the AQPs in the cancer cells has shown to decrease the relative effectiveness of arsenic treatment [56]. For example, the chronic myeloid leukemia cell line (K562) is recognized less sensitive towards iAsIII treatment because of lacking adequate expression of AQP9 [69]. Moreover, when AQP3 gene was knocked down in human lung adenocarcinoma cell line (CL3), it was found that the CL3 cells became more resistant to iAsIII [70], indicating the AQPs are at least involved in cellular susceptibility towards iAsIII.
On the other hand, for the efflux of trivalent arsenicals, human multidrug resistance proteins (e.g. MRP1, 2/ABCC1, 2) have been identified as essential transporters [71]. The MRP family contains nine ATP-binding cassette transporters (ABC) and is known for the efflux of drugs from the cells [72]. MRPs are capable of facilitating the transport of numerous conjugates of drugs or metals with sulfate, glucuronide and glutathione [73-75]. Regarding the arsenic efflux, it has been reported that both MRP1 and 2 along with the phase II conjugating enzymes like glutathione transferase (GST) are the major contributors for the resistance towards specific metals and toxicants [71, 76]. Few studies have shown that GSH is required for formation of arsenic-Glutathione conjugates [i.e., iAsIII(GS)3, MMAIII(GS)2] and then transport by MRP1/2 transporters into bile [77, 78]. More importantly, the expression of MRP1 protein has been found in various tissues, nevertheless, the expression of MRP1 in tumor and cancer cells is thought to render them less sensitive towards arsenic treatment [73]. Similar results were obtained from the AR2 cells (which are derived from NB4 cells with highly expressed MRP1) or HeLa-MRP1 transfected cells, these cells showed much resistance to As2O3 when compared with parental NB4 Cells or HeLa cells, respectively [79], suggesting that MRP1 may contribute to cellular resistance to arsenicals.
Binding affinity of arsenicals to proteins
The properties of arsenic binding to proteins are one of the most important factors that have been found critical in influencing the sensitivity and resistance of a specie towards arsenic exposure. To date, it has been found that trivalent arsenic is able to inhibit more than two hundred enzymes. MMAIII has shown to display greater toxicity and/or carcinogenic potential than iAsIII. Generally speaking, trivalent arsenicals have high binding affinity to sulfhydryl groups of proteins, while pentavalent arsenicals found to be not binding with sulfhydryl groups of proteins [80]. In addition, interaction with zinc finger proteins is considered to be an important mechanism of arsenic toxicity and carcinogenesis. Recent study has shown that MMAIII acts more effectively than iAsIII to destroy the normal integrity of zinc fingers.
Among the three arsenic trivalent species (i.e., iAsIII, MMAIII and DMAIII), our previous study have shown that MMAIII have much stronger binding affinity than other two species in vivo and in vitro [39, 81] because MMAIII binds with two closely spaced cysteine residues or dithiol cofactors. Contrast to MMAIII, iAsIII needs at least three symmetrical cysteine residues in protein, indicating that MMAIII can easily bind with protein compared to iAsIII [82, 83]. On the other hand, DMAIII would have a weaker bond with cysteine residues compared to the cysteine complexes with MMAIII and iAsIII because the methyl groups change the electron density around the arsenic [84].
AS3MT, a cysteine rich enzyme that can catalyzes the biomethylation of arsenic in vivo and in vitro. Identifying the As-binding site in the AS3MT would facilitate understanding of the process by which the arsenic binds to AS3MT. In each arsenic species, for example iAsIII can bind to three, methylarsenite (MMAIII) can bind to two, and dimethylarsenite (DMAIII) can bind to one cysteine residues, respectively. It has been found that Cys156 and Cys206 in hAS3MT have been shown to be binding sites for As and recently Li et al. suggested Cys61 as the third binding site of iAsIII [85].
Interestingly, it has also been recognized that DMAIII forms a stable complex with hemoglobin of rats, rendering the animal tolerant to arsenic having minimal risk for induced toxicity with maximum arsenic accumulation [41, 86]. However, hamsters have been known to be very sensitive similar to human towards arsenicals, and found that arsenic is mostly distributed in organs of hamster rather being accumulated in red blood cells (RBCs) [86]. Due to the absence of cysteine (Cys) residue (Cys-13) in the alpha chain of hemoglobin in hamster or humans, dimethylated arsenic (DMAIII) could not binds with hemoglobin, results in redistributed and accumulated in various organs [34].
Toxicities of Arsenic
Different arsenic metabolites have different toxicities and the extent of toxicity is depends on the animal species, cell types and uptake rate of each arsenic compounds. For example, IC50 values of the same arsenic species are differ from cells to cells, in human hepatocytes is DMAIII(GS) ≈ iAsIII << MMAIII, while in human keratinocytes it is iAsIII << DMAIII(GS) < MMAIII, and in human bronchial cells it is DMAIII(GS) < iAsIII ≈ MMAIII [43]. Recently a study carried out on wild type mice stated that the majority of iAs and methylated As species were found in liver as well as in pancreas indicating the adverse effects of iAs [87]. In generally, trivalent arsenicals are much more toxic than pentavalent counterparts and the reason behind is the lower rate of accumulation of pentavalent arsenic species [43, 88, 89].
On the other hand, arsenic toxicity in vivo, it has been reported that acute arsenic poisoning is associated with nausea, vomiting, abdominal pain, and severe diarrhea. Conversely, chronic ingestion of arsenite through contaminated drinking water results in the accumulation of arsenic in vital organs and act as the incidence for diabetes, atherosclerosis, hypertension, ischemic heart diseases, hepatotoxicity, nephrotoxicity, and cancer of the skin, bladder, and lungs. However, the actual mechanism by which arsenical induced diseases has not yet been fully elucidated. The present review importantly discussed the pathogenesis of arsenic-induced toxicity and end organ damage.
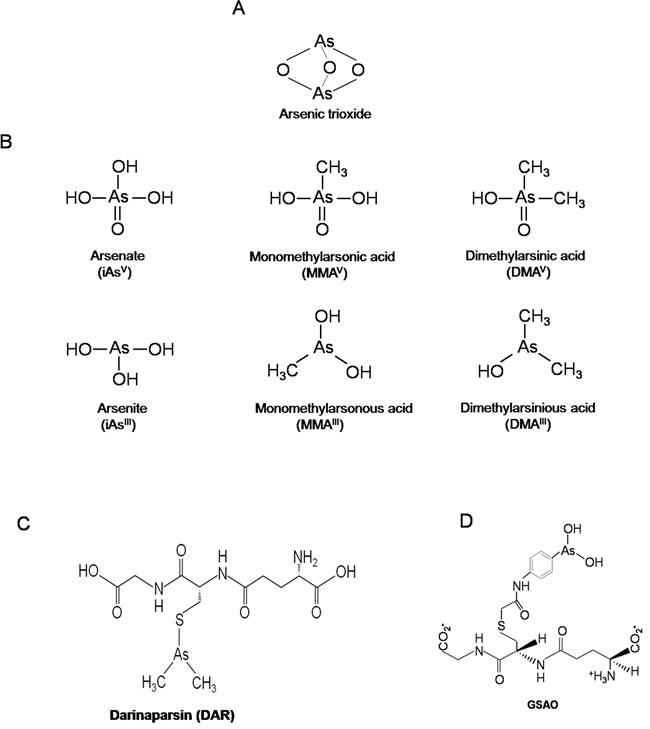
Figure 1: Structures of Different Arsenic Compounds. A. Structures of As2O3, or B. two major forms of inorganic arsenics namely; iAsV, iAsIII and major methylated metabolites including MMAIII, DMAIII, MMAV and DMAV. C. DAR and D. GSAO.
Arsenic-induced diabetes mellitus
The trivalent arsenic species (i.e., iAsIII, MMAIII and DMAIII) have found to inhibit GLUT4 recruitment to the plasma membrane of insulin stimulated adipocytes by inhibiting the activity of 3-phosphoinositide-dependent kinase-I (PDK-1) and mammalian target of rapamycin (rictor-mTOR). Activity of PDK-1 and rictor-mTOR are necessary for the phosphorylation of Akt [90].
In fact, Akt is necessary for GLUT4 translocation and glucose transport. Therefore, loss of dual phosphorylation of Akt due to the effects of arsenic and its metabolites, causes significant inhibition of insulin-dependent glucose uptake and hence results in hyperglycemia [90]. Another group suggested that the insulin producing pancreatic β-cells are among the targets for arsenic exposure, and that the inhibition of GSIS (glucose stimulated insulin secretion) by the methylated metabolites may be the key mechanism of arsenic-induced diabetes [91].
Arsenic-induced liver disease
About arsenic induced liver disease (including hepatomegaly, cirrhosis, portal tract fibrosis, etc), it was found that arsenic can mediated oxidative stress, activating c-Jun N-terminal kinases (JNK) and p38 mitogen-activated protein kinases (MAPK) pathway to induce hepatic apoptosis. Likewise, it can also induces hepatic cell death by upregulation of pro-apoptotic proteins [92-94]. Moreover, histological examination of the livers has revealed a consistent finding of portal tract fibrosis by arsenic exposure [95]. Individuals exposed more frequently to arsenic suffer from cirrhosis, which is considered to be a secondary effect of damage to the hepatic blood vessels [96]. Chronic arsenic exposure in animals can also produce liver endothelial cell damage, which subsequently damages parenchymal cells [97]. All these studies clearly revealed that exposure to arsenic are associated with hepatomegaly, hepatic fibrosis and cirrhosis. Hence, oxidative stress, apoptosis, and up regulation of apoptosis related proteins are the prospective target sites for arsenic induced hepatotoxicity [98].
Arsenic-induced cardiovascular diseases (CVD)
With respect to cardiovascular diseases (CVD) in human population, it suggested that not only arsenic but also genetic, environment and nutritional factors necessary for development of this disease. Epidemiological studies have shown that arsenic ingestion through food or water may have serious effects on the human cardiovascular system including heart damage (i.e., myocardial depolarization, hypertrophy of the ventricular wall, cardiac arrhythmias), vascular damage (Blackfoot disease), ischemic heart disease, cerebrovascular diseases, and hypertension [21, 99, 100].
It is believed that vascular endothelial cells play a pivotal role in arsenic-induced cardiovascular diseases. Increased activity of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzymes (NOX) presents in the plasma membrane of vascular endothelial cells and vascular smooth muscle cells are associated with vascular disease or hypertension [101]. Indeed, arsenic has the rapid stimulatory effects on these enzymes to generate reactive oxygen species (ROS) [102] and nitric oxide (NO) to form peroxynitrite, a strong oxidant, implicated in the up regulation of inflammatory mediators [103]. Correspondingly, arsenic can also interacting with G-protein coupled receptors (GPCR) to initiate signal amplification schemes regulating NOX-dependent redox signaling [101]. In addition, generation of ROS by arsenic increases the expression of atherosclerosis related genes such as heme oxygenase-1 (HO-1), monocyte chemo-attractant protein-1 (MCP-1), and interleukin-6 (IL-6) and thus its exposure promotes the attachment, penetration, and migration of monocytes in vascular smooth muscle cells [104]. Along with this, endothelial cell activation/dysfunction by arsenic produces several inflammatory molecules such as soluble intercellular adhesion molecule-1 (sICAM-1), soluble vascular adhesion molecule-1 (sVCAM-1), MCP-1 related to the atherosclerotic lesions [105]. On the other hand, our group has found that trivalent MMAIII has unique toxic effect on cellular differentiation of embryonic stem cells to cardiomyocytes [106]. All these observed associations suggest a potential pathway underlying the effect of arsenic exposure-related atherosclerosis leading to CVD.
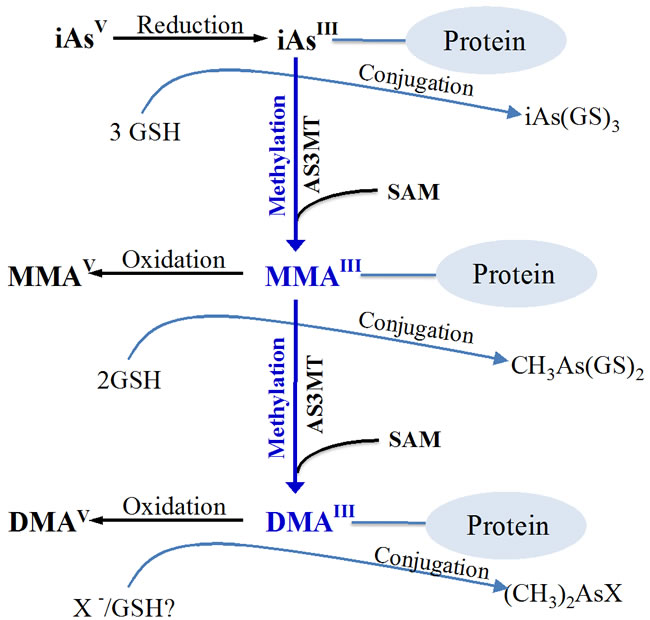
Figure 2: Proposed Arsenic Metabolism Pathway in Human. In the body, the inorganic trivalent arsenic is metabolized in a protein-bound form having successive reductive methylation by arsenicmethylatransferase (AS3MT) in the presence of glutathione and S-adenosylmethionine (SAM). In particular, monomethylarsonous acid (MMAIII), dimethylarsinous acid (DMAIII) are the trivalent intermediate metabolites which in protein-bound form are further oxidized to corresponding pentavalent monomethylarsonic acid (MMAV) and dimethylarsinic acid (DMAV), which are considered to be less toxic end products of arsenic metabolism.
Arsenic-induced nephrotoxicity and neurotoxicity
It is well known that arsenic exposure can causes the nephrotoxicity in mammals. Prevalent of chronic kidney disease (CKD) is inversely associated with the sum of inorganic and methylated arsenic concentrations in urine, and particularly with inorganic arsenic concentrations. Prospectively, inorganic plus methylated arsenic concentrations in urine, especially monomethylarsonate and dimethylarsinate concentrations were positively associated with incident of nephrotoxicity [107]. Among the three trivalent arsenicals (iAsIII, MMAIII and DMAIII), MMAIII have shown more potent cytotoxic effect on human urothelial cells in vitro [108]. Moreover, recent evidence also indicates that developmental arsenic exposure impacts renal carcinogenesis in stem cells. Tokar et al. reported that arsenic targeting the multipotent stem cells and they found that long term exposure of arsenic can causes carcinogenic transformation of stem cells, implying that the targeting of stem cells during early stages of life may be the key for the development of arsenic-induced cancers in kidney [109]. Furthermore, an interesting result has been found when rats exposed to DMAV results in development of carcinogenesis in bladder [110, 111]. However, neither the proximate carcinogen nor the mechanism of action underlying cancer development has been identified. However, the metabolites found in urine from rats those exposed to DMAV includes methylated oxoarsenicals (e.g., DMAIII and TMAOV) [112-114] and methylated thioarsenicals (e.g., dimethylmonothioarsinic acid; DMMTAV) potential proximate carcinogens [115].
In addition to above adverse toxic effect of arsenic on different organs, it has also found that low concentrations of methylated arsenic species (e.g., MMAIII) could inhibit sensory neuron and skeletal muscle formation. This may provide a mechanism for the low birth weight and changes in neurological function seen in arsenic exposed epidemiological studies [116, 117].
Effects on vital proteins/enzymes
It was found that arsenic-induced disturbance in cellular energy related nucleotides may also result in redox state alterations [118]. Moreover, increased the secretion of inflammatory factors and IL-8, TNF-α, and TGF-α expression have also been observed to be induced by arsenic exposure [119]. It is well known that arsenic species in particular trivalent methylated arsenic intermediate metabolites have high binding affinity for the cysteine residues of proteins, however, arsenic has also been recognized for inhibiting the functioning of various transcriptional co regulators, proteins and enzymes [120-123].
Arsenicals can have both genotoxic and non-genotoxic mechanisms of action, and also serve as a complete carcinogen [124]. Additionally, it has proposed that arsenic species can influence the function of the DNA repair enzymes, as in particular the trivalent arsenic compounds including iAsIII and its methylated metabolites (MMAIII and DMAIII) have been recognized for inhibiting DNA repair [83, 125-127]. Recently, it has been indicated that iAsIII can bind to the zinc finger motifs in recombinant protein which having 3 or more than 3 cysteine residues, eventually altering the function of DNA repair proteins and/or mechanisms [128, 129]. The methylated trivalent arsenic metabolites showed more efficient interacting ability with the cysteine containing repair proteins like the one involved in nucleotide excision repair (NER) as compared to their precursor; iAsIII [83]. Moreover, xeroderma pigmentosum group A protein (XPA), which is recognized for its important role in NER mechanism, has recently shown to be affected by arsenicals [130].
Notably, regarding the trivalent arsenicals interaction with Zinc finger domain of the XPA (containing Zn2+) at normal physiological condition, it has been found that only methylated metabolites; MMAIII and DMAIII (but not iAsIII) have stronger binding affinity to zinc finger proteins [119]. In fact, the studies that have shown binding of iAsIII to zinc finger peptides were mostly performed using apo-Zn finger proteins [128, 131], thus, more studies are needed to evaluate that whether iAsIII can also bind to Zn finger peptides or proteins (i.e., with Zn2+) even under normal physiological conditions.
Likewise, metallothionein (MT), a cysteine rich protein, is commonly known for protecting against toxicity of heavy metals. Thus, many researchers have also investigated the effect of iAsIII on MT, they found that iAsIII is able to induce the MT expression in vivo and in vitro [127, 132]. Additionally, it has suggested that iAsIII can probably bind to the C-terminal cysteine cluster of metal-activated transcription factor 1 (MTF1) and thereby, induce the transcription activity [133]. On investigating the binding interaction of the three trivalent arsenicals (i.e., iAsIII, MMAIII and DMAIII) with the metallothioneins, MMAIII was found to have the highest binding capacity among the three arsenic species [134], suggesting that the arsenic-induced cytotoxicity might probably have some association with arsenic-binding to proteins.
Arsenic induced skin lesions
Skin symptoms of arsenic exposure are hallmarks of the early stages of the arsenic poisoning in the population exposed to arsenic through drinking water. Nonmalignant skin lesions have a short latency period and may appear within a few years of exposure. Arsenic exposure related skin symptoms include: melanosis (diffuse and spotted), keratosis (diffuse and spotted), leucomelanosis (rain drop pigmentation), and hyperkeratosis. Melanosis a dark pigmentation on the skin and keratosis is an area of small corn like elevations (0.4-1 cm in diameter), usually appears as bilateral thickening of the palms and soles, are found at the primary stage of arsenic-induced all dermatological manifestations [135, 136], leuko-melanosis and hyperkeratosis in the second stage and ultimately may turn to skin cancer such as Bowen’s disease, basal cell and squamous cell carcinoma [11, 137, 138].
Chromosomal abnormality, oxidative stress and altered growth factors provide a basis for some of these alterations [139, 140]. Arsenic triggers ROS production in the skin and induces accumulation of unfolded proteins in ER which causes ER stress [141-143]. Under stress condition, the chaperone GRP78 dissociates from ER membrane resident sensors PERK, IRE1α and ATF6α [144], which lead to their phosphorylation or proteolysis-dependent activation. Activated IRE1α functions as a nuclease and splices XBP-1, that can initiates many signaling pathways and finally activates UPR target genes in the skin. In addition, arsenic-induced ROS can also activate p38 MAP kinase and its downstream protein MAPKAPK-2 [145]. Thereby, UPR and p38 MAP kinase are considered to be responsible for the onset of inflammation in the skin [146].
Arsenic-induced carcinogenicity
The carcinogenic potential of arsenic was recognized over 110 years ago by Hutchison, who observed an unusual number of skin cancer occurring in patients treated for various diseases with medical arsenicals [147]. The International Agency for Research on Cancer (IARC) and the US Environmental Protection Agency (EPA) has classified arsenic as a carcinogen based on epidemiological studies [7, 148]. Arsenic carcinogenesis is major concern affecting numerous organs of human. Epidemiological studies carried out in different countries have demonstrated an evident causal relationship between environmental, occupational, and medical exposure of millions of people worldwide to inorganic arsenic and increased risks of cancer of the skin, lungs, urinary bladder, kidney, prostate, liver and other sites [149-152]. Although the exact molecular mechanism of arsenic carcinogenicity is not well understood, progression of carcinogenesis by arsenic is considered to be associated with intracellular signal transduction, transcription factors activations and the abnormal genes expression.
On the other hand, promotion of oxidative stress by arsenic compounds is one of the main causes for arsenic-induced carcinogenicity [153, 154]. It has been reported that several arsenic metabolites (e.g., DMAV and TMAOV) cause an elevation of 8-hydroxydeoxyguanosine by generation of ROS, stimulates cell proliferation and induces carcinogenicity [155, 156]. Additionally, arsenic induces MAPK signal transduction pathway that alter various gene expression profile via AP-1 and NF-kB and causes cancer [157]. Arsenic causes focal adhesion kinase activation which regulates various downstream signaling pathways (e.g., EGFR, ERK, cadherins) of carcinogenesis [158]. Moreover, arsenic can damage DNA or changing methylation patterns, stimulate angiogenesis, dysregulating cell cycle, blocking physiological apoptosis and causes skin, bladder and lung cancer [159-161]. Although the precise cellular mechanism by which arsenic induces cancer is still unknown, collectively oxidative stress, genotoxic effects, growth factors expression, loss of DNA repairing mechanisms are proposed to be the possible modes of carcinogenic action of arsenic.
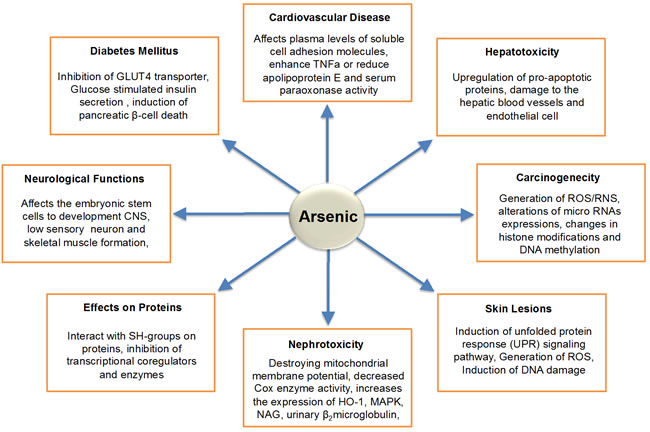
Figure 3: Schematic Diagram Represents the Toxicities of Arsenic.
Anticancer Activities of Intermediate Methylated Metabolites of Arsenic
Arsenic compounds have widely been used as traditional medicines in Mongolian, Chinese and Tibetan for the treatment of various diseases such as psoriasis, syphilis, and rheumatosis for over two thousand years [162]. Approximately, 60 different arsenic preparations have been developed and distributed during the lengthy history of this agent and many of these preparations are still in use. Although the IARC and the U.S EPA have classified arsenic as a known human carcinogen, several arsenic compounds have been recently rediscovered and formulated to treat different diseases.
Arsenic compounds such as Darinaparsin (DAR), namely DMAIII(GS), GSAO [4-(N-(S-glutathionylacetyl) amino) phenylarsonous acid] and Trisenox (As2O3) are in clinical trials of U.S. Food and Drug Administration for the treatment of cancers such as leukemia, lymphomas and solid tumors [163, 164]. In this section, we will describe about the metabolism and possible effects of these arsenic compounds in cancer treatments.
Darinaparsin
Darinaparsin (DAR) a novel organic form of arsenic that was rationally designed to provide more efficient effects to kill tumor cell through increase intracellular arsenic concentration and increased apoptosis. It is composed of dimethylated arsenic (an inorganic arsenic metabolite) and glutathione (Figure 1C). This combination of DAR in the biological environment is thought to be inert but in fact, DAR has shown to be effective with clinically acceptable doses in vitro against leukemia, myeloma, lymphoma, and various solid tumor cell lines [165, 166]. Based on promising results from the in vitro experiments, the investigation on DAR was extended to preclinical and clinical studies. In case of hematologic cancers, refractory solid tumors, multiple myeloma, and hepatocellular carcinoma the efficiency, safety and pharmacokinetics of DAR have been evaluated [167-169].
Phase II study of DAR in relapsed or refractory Hodgkin’s and non-Hodgkin’s lymphoma, including patients presenting with peripheral T-cell lymphoma (PTCL), provides preliminary evidence that DAR is active and well tolerated [170]. Over expression of the multi-drug resistant protein ABCC1 in PTCL may be the determinant factors for the decrease sensitivity to ATO, while cytotoxicity of organic arsenic species DAR is not altered in these cells because of ABCC1 is not the transporter of DAR, which resulting in increase DAR concentration in cells [171].
Additionally, the cellular uptake of DAR is different from other arsenic compounds. The presence of intracellular GSH was shown to affect the uptake of DAR. It has already been reported that DAR enter the cells in the form of DMAIII at low concentrations of exogenous glutathione or in the absence of it. Before uptake into the cells, DAR is suggested to be processed by the enzyme γ-glutamyl transpeptidase (γ-GT) on the cell surface to S-(dimethylarsenic) cysteine (DMAIII(Cys)) [172], finally the multiple cystine/cysteine importers transport DMAIII(Cys) into the cells, whereas no transporter has been yet identified that could transport intact DAR.
DAR has known to have multifarious and complex mechanistic pathways to show its effects. Figure 4 schematically represents the possible anticancer mechanistic pathways of DAR. Generally, it induces G2/M cell cycle arrest and apoptosis in tumor cells primarily through disruption of mitochondrial functions by inducing cleavage of caspase 8 and 9 (Figure 4). Additionally production of ROS can also initiate the apoptotic pathway and usually DAR initiates two major pathways for ROS generation, namely, NADPH oxidase activation and disruption of the mitochondrial transport chain. In fact, DAR activates cytosolic subunits of NADPH oxidase complex p67PHOX and p47PHOX, resulting in the translocation of NADPH oxidase to the membrane and produces ROS, which disrupt mitochondrial membrane potential and release of Cytochrome c [173]. This leads to the activation of apoptosome that ends into cellular apoptosis [165].
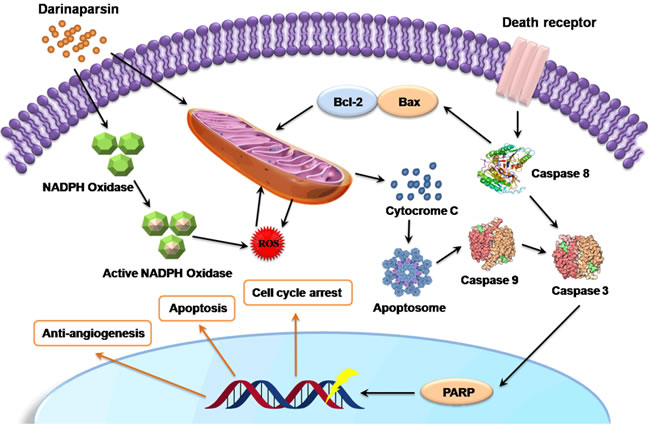
Figure 4: Effects of Darinaparsin (DAR) on Cellular Functions. Production of intracellular ROS is the key events of DAR mediated apoptosis. Generally, it occurs through either by the activation of NADPH oxidase complex or direct effect on mitochondrial membrane potential. Abnormal mitochondrial functions are associated with the release of cytochrome c resulting in the activation of apoptosome, which causes initiation of apoptosis. The ultimate result is cell cycle arrest, induction of apoptosis and anti-angiogenic activity.
GSAO
GSAO (4-(N-(S-glutathionylacetyl)amino) phenylarsonous acid) is a conjugate of glutathione and phenylarsonous acid as shown in Figure 1D. In vitro and in vivo experiments where tumor metastasis relies heavily on neovascularization GSAO exhibited potential anti-cancer effects [174-177]. More specifically, GSAO targets a key mitochondrial transporter in tumor endothelial cells and metabolism of GSAO at the cell surface is required to exert its anti-mitochondrial effect. During biotransformation, the γ-glutamyl residue of GSAO is cleaved at the cell surface by γ-glutamyl transpeptidase and 4-(N-(S-cysteinylglycylacetyl)amino) phenylarsonous acid (GCAO) forms and then enters the cell through an organic ion transporter (Figure 5). Moreover, it probably be converted to 4-(N-(S-cysteinylacetyl)amino)phenylarsonous acid (CAO) by dipeptidases, and CAO is responsible for pharmacological effects of GSAO. For instance, CAO reacts with mitochondrial adenine nucleotide translocase (ANT) and inactivates via cross-linking two of the three matrix facing cysteine thiols. Proper functioning of ANT is essential for cell viability, targeting this protein in angiogenic endothelial cells is a powerful means of blocking angiogenesis and cell proliferation, finally resulting in cell death. The possible mechanism of action of GSAO is summarized in Figure 5.
The mechanism of action of GSAO described here implies that GSAO should be more effective against γ GT-positive tumors. Notably, tumors of the breast, prostate, colon, liver, and ovary express γ GT, whereas soft tissue tumors tend not to express this enzyme [178]. Metabolism of GSAO by tumor endothelium γ GT is able to produce high concentrations of GCAO, which will block tumor angiogenesis and tumor growth. Based on the different laboratory findings, GSAO is currently being trialed in a Phase I/IIa dose escalation study in patients with solid tumors. As phase I clinical trials have not been completed, their side effects and toxicities in humans are largely unknown. Additionally, hematological cancers do not rely on new blood vessel formation for proliferation, therefore, it is only suitable for patients with solid tumors refractory (resistant) to standard therapy.
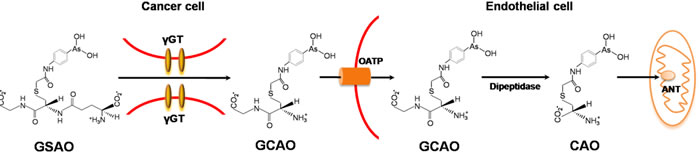
Figure 5: Molecular mechanism of GSAO. GSAO is activated by tumor γGT at the cell surface, and the following GCAO is transported across the plasma membrane by the organic anion transporting polypeptide (OATP). Additionally, GCAO is possibly further processed to CAO by dipeptidase in the cytosol and finally reacts with ANT of the inner mitochondrial membrane.
Trisenox (As2O3)
Trisenox (As2O3) one of the most effective novel anticancer agents used for the treatment of APL that received approval from the U.S. FDA in 2000 as anticancer agent. The molecular mechanism of As2O3 in front-line therapy has been largely studied among all arsenic compounds [179, 180]. Generally, in human after administration of As2O3, it is hydrolyzed to iAsIII and then metabolized into trivalent methylated intermediate metabolites; MMAIII and DMAIII by AS3MT, and the oxidized MMAV and DMAV are considered to be the end products of As2O3 [39]. However, it is still indistinct regarding the anticancer effects of its methylated metabolites (i.e., MMAIII, MMAV, DMAIII and DMAV). Therefore, it needs to be identified the precise mechanism of the effects of As2O3 and its metabolites to find out the actual benefits from this vital agent for the treatment of APL or other types of cancer. It has been found that As2O3 is able to induce differentiation of APL cells at low doses (0.25~0.5µM), while on the other hand high doses of As2O3 (1~2µM) could induce apoptosis [181]. Wang et al (2004) made an attempt to determine the arsenic species in urine of APL patients receiving As2O3 treatment by HPLC-ICP MS analysis [182]. Interestingly, they found that MMAIII, DMAIII, MMAV and DMAV were detected in the urine of these patients, particularly, methylated pentavalent arsenic species being the major arsenic species, followed by only a minor amount of trivalent methylated arsenic species found in urine samples. Similar results were also obtained from the blood sample of APL patients after As2O3 treatment, they found the order of these arsenicals concentrations (free form) in plasma to be as MMAV > DMAV > iAsIII > iAsV and no trivalent methylated arsenicals observed, indicating that As2O3 can be rapidly transformed into methylated arsenic species in body, and most of trivalent methylated arsenicals considered to be binding with proteins [58]. Interestingly, As-binding proteins have confirmed in plasma and RBCs of APL patients by HPLC column after receiving As2O3 treatment, and found that arsenicals in trivalent form bound to protein in blood [183]. Based on above findings, it was considered that arsenic metabolites (e.g., MMAIII and DMAIII) might exert vital role in APL patients after treatment with As2O3. Thus, the role of arsenic methylated metabolites in the clinical efficacy against APL or other cancer treatments should be examined.
Hikita et al. has used pentavalent organic arsenic species (e.g., MMAV, DMAV, TAOV, AsB, AsC) to determine their anticancer effects on T-lymphoblastoid leukemia cells [184]. Unexpectedly, the pentavalent organic arsenic compounds showed no inhibitory effects on the leukemia cell proliferation as compared to iAsIII. Indeed, these results clearly indicated that pentavalent organic arsenicals have no appreciable anticancer effects, . Additionally, trivalent iAsIII, MMAIII and DMAIII have also been used to determine the anticancer effects against human myeloid leukemia HL-60 cells [185], and methylated arsenic species were found to be more potent in decreasing the cell survival as compared to its precursor iAsIII. However, HL-60 cells have also exhibited to be relatively resistant to organic arsenic species than compared to other cell lines. Lower uptake and higher efflux may be the possible reason for cellular resistance to arsenicals. In order to understand the molecular mechanism of HL-60 cells resistance to arsenic, we interested to determine the expression of MRP transporters. Interestingly, MRP1 was found to be over-expressed in HL-60 cells, which confer resistance to inorganic and organic arsenic compound (i.e., iAsIII and MMAIII). Additionally, when HL-60 cells were pretreated with MK571(inhibitor of MRP1), the cells became highly sensitive to arsenic treatment, suggesting that after blocking the transporter which responsible for arsenic efflux could increase the cellular sensitivity to arsenic treatment [186].
On the other hand, MMAIII found to be specifically inhibit mitochondrial electron transport chain (ETC). Namely, MMAIII inhibit the activity of complexes II and IV, which results in electron leakage from complex I and III causing the generation of ROS in mitochondria, resulted in induction of mitochondrial dysfunction and apoptosis [187]. Differently from the MMAIII, DMAIII was found to predominantly target the endoplasmic reticulum (ER), stimulating the activation of PERK whose activation leads to the phosphorylation of eIF-2α, translation initiation of activating transcription factor 4 (ATF4) and CHOP, resulting in induction of apoptosis [188, 189], suggesting that the two methylated trivalent arsenic species use different mode of actions to showed their activity (Figure 6). Likewise, ER-stress can also activate inositol-requiring enzyme 1 (IRE1) which belongs to ER transmembrane receptors, this activation of IRE1 further recruits and forms a complex with TNF-receptor-associated factor 2 (TRAF2) recruiting apoptosis-signal-regulating kinase (ASK1) which in turn activates JNK [190], while arsenic mediated activation of JNK is recently found to be important for the anticancer effects of arsenic [191, 192].
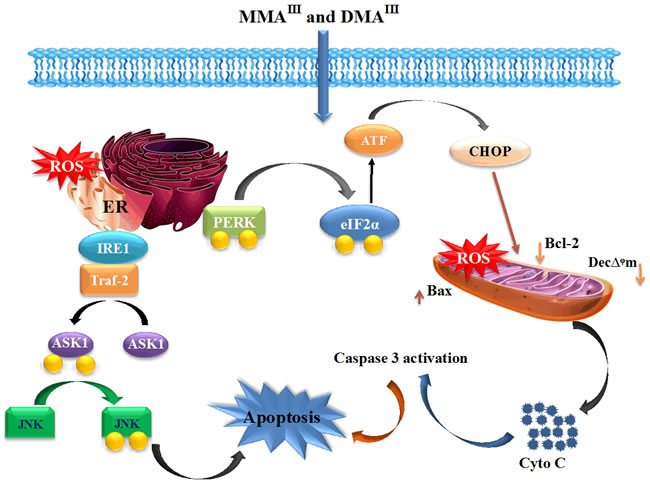
Figure 6: Toxicity and Anticancer Effects of MMAIII and DMAIII. MMAIII specifically inhibit the activity of complexes II and IV, which results in electron leakage from complex I and III causing the generation of ROS in mitochondria, resulted in induction of mitochondrial dysfunction and release Cyt c, finally inducing apoptosis through caspase-3 dependent pathway. DMAIII predominantly target ER, stimulating the activation of PERK whose activation leads to the phosphorylation of eIF-2α, translation initiation of ATF4 and CHOP, resulting in induction of apoptosis. Similarly, DMAIII-induced ER-stress also activate IRE1, this activation of IRE1 further forms a complex with TRAF2 recruiting ASK1, which in turns activates JNK to induce apoptosis.
Therefore, iAsIII and its trivalent methylated metabolites were evaluated for their potential effects. It was found that methylarsine oxide (i.e., MMAIII) followed by iododimethylarsine (i.e., DMAIII) significantly induced growth inhibition and cellular apoptosis in NB4, K562 human leukemia, lymphoma cell lines, and primary culture of chronic lymphocytic leukemia cells when compared to iAsIII [192]. However, unlike iAsIII, methylarsine oxide could not induce degradation of Promyelocytic leukemia-retinoic acid receptor alpha (PML-RARα) fusion protein in APL cells. This study suggested that though arsenic methylated metabolites could not induce cellular differentiation or PML-RARα fusion protein degradation in APL cells, but still are potent inducer of cellular apoptosis as compared to their precursor iAsIII and therefore, the arsenic metabolites might be used in leukemia and lymphoma treatments [193].
Very recently, it has been found that As2O3 induced APL cells differentiation occurs as a result of direct binding of As2O3 to cysteine residues of zinc fingers in the RING finger-B Box-Coiled Coil (RBCC) domain of PML-RARα fusion protein, which results in enhanced SUMOylation/Ubiquitination and then degradation of PML-RARα fusion protein, promoting cell differentiation leading to clinical remission. However, in in vitro, among trivalent arsenic species (i.e., iAsIII, MMAIII and DMAIII), the methylated MMAIII has shown much stronger binding affinity for various proteins including Zinc finger proteins or PML-RARα fusion proteins as compared to other two arsenic species (Figure 7) [131], but methylated arsenicals were incapable of inducing protein degradation, suggesting that arsenic induced PML-RARα fusion protein degradation is complicated. Although two methylated arsenic species could not degrade PML-RARα fusion protein in NB4 cells, these compounds have exhibited much more potent effect on induction of apoptosis, as schematically drawn in Figure 7. These are the underlying principle for the evaluation of the arsenic intermediate metabolites against solid tumors, APL and/or non-APL malignancies.
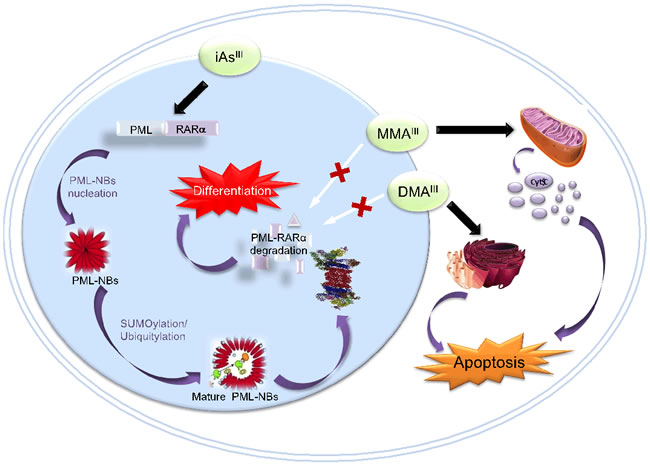
Figure 7: Molecular Mechanisms of iAsIII and its Trivalent Methylated Metabolites (i.e., MMAIII and DMAIII) Induced Cell Differentiation and Apoptosis in APL Cells. Arsenic trioxide in hydrolyzed form, i.e., iAsIII may cause formation of PML-RARα protein multimers, initiate the nucleation of PML-NBs, enhanced SUMOylation/ubiquitination, resulting degradation of PML-RARα fusion protein by ubiquitin/proteasome pathway, finally promoting cell differentiation leading to clinical remission. On the other hand, MMAIII and DMAIII have shown no effect on induction of PML-RARα protein degradation and APL cell differentiation. Conversely, they have strong effect on induction of apoptosis in APL cells through mitochondria and ER-dependent pathways.
Conclusions and Future Perspectives
The precise and appropriate metabolic pathways involved in arsenic metabolism are still a matter of question, and are of enormous value in understanding the mechanisms of arsenic induced toxicity and in determining the implication of arsenic in therapeutic advances. The actual arsenic metabolic pathway would be helpful in elucidating the probable role of different arsenic species in relation to toxicities along with the uses of arsenic as a drug.
On the other hand, arsenic as a traditional medicine, inappropriate use of arsenic may poison people, while its appropriate application cures some cancer types and saves lives. The revival of arsenic by its application in treating APL is a unique story in cancer research. The therapeutic magnitude of arsenicals deliberated over here may also be enlightening in near future with respect to the precious uses of arsenicals as medicines for the treatment of diseases. However, there are necessities to further investigate the effects of these metabolites and other arsenic species to assess whether these underlying principles can be interpreted into important clinical outcomes through carry out potential preclinical and clinical studies.
Abbreviations
Arsenic trioxide, As2O3; Acute promyelocytic leukemia, APL; Glutathione, GSH; Arsenite, iAsIII; S-adenosylmethionine, SAM; Monomethylarsonous acid, MMAIII; Monomethylarsonic acid, MMAV; Dimethylarsinous acid, DMAIII; Dimethylarsinic acid, DMAV; Arsenate, iAsV; Trimethylarsine oxide, TMAOV; Arsenic (+3 oxidation state) methyltransferase, AS3MT; Arsenic triglutathione, As(GS)3; Monomethylarsenic diglutathione, MMA(GS)2; Dimethylarsenic glutathione, DMAIII(GS); Aquaglyceroporins, AQPs; Multidrug resistance proteins, MRP; Glutathione S transferase, GST; Glutathione S-transferase π1, GSTP1; red blood cells, (RBCs); Cysteine, Cys; Glucose transporter type 4, GLUT4; 3-phosphoinositide-dependent kinase-I, PDK-1; Mammalian target of rapamycin, mTOR; Glucose stimulated insulin secretion, GSIS; Jun N-terminal kinases, JNK; mitogen-activated protein kinases, MAPK; Reactive oxygen species, ROS; Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzymes, NOX; Nitric oxide, NO; G-protein coupled receptors, GPCR; Heme oxygenase-1, HO-1; Monocyte chemo-attractant protein, MCP-1; Nucleotide excision repair, NER; xeroderma pigmentosum group A protein, XPA; metallothionein, MT; Metal-activated transcription factor 1, MTF1; Endoplasmic reticulum, ER; Activating transcription factor 4, ATF4; Inositol-requiring enzyme 1, IRE1; TNF-receptor-associated factor 2, TRAF2; Promyelocytic leukemia, PML; Retinoic acid receptor alpha, RARα; RING finger-B Box-Coiled Coil, RBCC.
Funding
The authors wish to acknowledge the National Natural Science Foundation of China (No. 81473289; 81673521; 81274138), Department of Science and Technology of Zhejiang Province (2015C33154); Key Project of Traditional Chinese Medicine Science and Technology of Zhejiang Province (2015ZZ006); Zhejiang Provincial & National Health and Family Planning Commission Project for Medical Sciences (WKJ-ZJ-1512).
Conflicts of interest
The authors declare no competing financial interest.
References
1. Mandal BK, and Suzuki KT. Arsenic round the world: a review. Talanta. 2002; 58: 201–35.
2. Roy P, and Saha A. Metabolism and Toxicity of Arsenic: A Human Carcinogen. Current Science. 2002; 82: 38-45.
3. Tchounwou PB, Wilson B, and Ishaque A. Important considerations in the development of public health advisories for arsenic and arsenic containing compounds in drinking water. Rev Environ Health. 1999; 14: 211–29.
4. Duxbury JM, Mayer AB, Lauren JG, and Hassan N. Food chain aspects of arsenic contamination in Bangladesh: effects on quality and productivity of rice. J Environ Sci Health A Tox Hazard Subst Environ Eng. 2003; 38: 61–69.
5. Meharg AA, and Rahman MM. Arsenic contamination of Bangladesh paddy field soils: implications for rice contribution to arsenic consumption. Environ Sci Technol. 2003; 37: 229–334.
6. Sun GX, Williams PN, Carey AM, Zhu YG, Deacon C, Raab A, Feldmann J, Islam RM, and Meharg AA. Inorganic arsenic in rice bran and its products are an order of magnitude higher than in bulk grain. Environ Sci Technol. 2008; 42: 7542–46.
7. IARC (International Agency for Research on Cancer). Some drinking-water disinfectants and contaminants, including arsenic. IARC Monographs on the evaluation of carcinogenic risks to humans. 2004; 84: 139–41.
8. Zheng Y, Stute M, van Geenb A, Gavrieli I, and Dhara R. Redox control of arsenic mobilization in Bangladesh groundwater. Applied Geochemistry. 2004; 19: 201–14.
9. NRC (National Research Council). Subcommittee to Update the 1999 Arsenic in Drinking Water Report. 2001. Arsenic in Drinking Water: 2001 Update. National Academy Press Washington DC. 2001.
10. Ravenscroft P, Brammer H, and Richards K. Arsenic Pollution: A Global Synthesis. pp 498-500, RGS-IBG Book Series, Wiley-Blackwell:Chichester UK. 2009.
11. Khan MM, Sakauchi F, Sonoda T, Washio M, and Mori M. Magnitude of arsenic toxicity in tube-well drinking water in Bangladesh and its adverse effects on human health including cancer: evidence from a review of the literature. Asian Pac J Cancer Prev. 2003; 4: 7–14.
12. Smith AH, Lingas EO, and Rahman M. Contamination of drinking-water by arsenic in Bangladesh: a public health emergency. Bull World Health Organ. 2000; 78: 1093–103.
13. Watanabe C, Inaoka T, Kadono T, Nagano M, Nakamura S, Ushijima K, Murayama N, Miyazaki K, and Ohtsuka R. Males in Rural Bangladeshi Communities Are More Susceptible to Chronic Arsenic Poisoning than Females: Analyses Based on Urinary Arsenic. Environ Health Perspect. 2001; 109: 1265–70.
14. Ali N, Hoque MA, Haque A, Salam KA, Karim MR, Rahman A, Islam K, Saud Z A, Khalek, MA, Akhand AA, Hossain M, Mandal A, Karim MR et al. Association between arsenic exposure and plasma cholinesterase activity: a population based study in Bangladesh. Environ Health. 2010; 9: 36.
15. Argos M, Kalra T, Rathouz PJ, Chen Y, Pierce B, Parvez F, Islam T, Ahmed A, Rakibuz-Zaman M, Hasan R, Sarwar G, Slavkovich V, van Geen A et al. Arsenic exposure from drinking water, and all-cause and chronic-disease mortalities in Bangladesh (HEALS): a prospective cohort study. Lancet. 2010; 376: 252-8.
16. Mazumder DNG, Haque R, Gosh N, De BK, Santra A, Chakraborty D, and Smith AH. Arsenic levels in drinking water and the prevalence of skin lesions in West Bengal, India. Int J Epidemiol.1998; 27: 871–7.
17. Mazumder DNG. Effect of chronic intake of arsenic-contaminated water on liver. Toxicol Appl Pharmacol. 2005; 206: 169–75.
18. Meliker JR, Wahl RL, Cameron LL, and Nriagu JO. Arsenic in drinking water and cerebrovascular disease, diabetes mellitus, and kidney disease in Michigan: a standardized mortality ratio analysis. Environ Health. 2007; 2: 4–6.
19. Tapio S, and Grosche B. Arsenic in the aetiology of cancer. Mutat Res. 2006; 612: 215–46.
20. Vahidnia A, Romijn F, van der Voet GB, and de Wolff FA. Arsenic-induced neurotoxicity in relation to toxicokinetics: Effects on sciatic nerve proteins. Chem Biol Interact. 2008; 176: 188-95.
21. Wang CH, Jeng JS, Yip PK, Chen CL, Hsu LI, Hsueh YM, Chiou HY, Wu MM, and Chen CJ. Biological gradient between long-term arsenic exposure and carotid atherosclerosis. Circulation. 2002; 105: 1804–09.
22. Swindell EP, Hankins PL, Chen H, Miodragović ÐU, and O’Halloran TV. Anticancer Activity of Small Molecule and Nanoparticulate Arsenic(III) Complexes. Inorg Chem. 2013; 52: 12292–304.
23. Mathews V, Chendamarai E, George B, Viswabandya A, and Srivastava A. Treatment of Acute Promyelocytic Leukemia with Single-Agent Arsenic Trioxide. Mediterr J Hematol Infect Dis. 2011; 3: e2011056.
24. Subbarayan PR, and Ardalan B. In the war against solid tumors arsenic trioxide needs partners. J Gastrointest Cancer. 2014; 45: 363-71.
25. Saint-Jacques N, Parker L, Brown P, and Dummer TJ. Arsenic in drinking water and urinary tract cancers: a systematic review of 30 years of epidemiological evidence. Environ Health. 2014; 13: 44.
26. Antonelli R, Shao K, Thomas DJ, Sams R, and Cowden J. AS3MT, GSTO, and PNP polymorphisms: impact on arsenic methylation and implications for disease susceptibility. Environ Research. 2014; 132: 156–67.
27. Hirano S, Kobayashi Y, Cui X, Kanno S, Hayakawa T, and Shraim A. The accumulation and toxicity of methylated arsenicals in endothelial cells: important roles of thiol compounds. Toxicol Appl Pharmacol. 2004; 198: 458–67.
28. Kobayashi Y, Hayakawa T, and Hirano S. Expression and activity of arsenic methyltransferase Cyt19 in rat tissues. Environ Toxicol Pharmacol. 2007; 23: 115-20.
29. Geubel AP, Mairlot MC, Buchet JP, and Lauwerys R. Abnormal methylation capacity in human liver cirrhosis. Int J Clin Pharmacol Res. 1988; 8: 117–22.
30. Marafante E, Vahter M, and Envall J. The role of the methylation in the detoxication of arsenate in the rabbit. Chem Biol Interact. 1985; 56: 225-38.
31. Aposhian HV. Enzymatic methylation of arsenic species and other new approaches to arsenic toxicity. Ann Rev Pharmacol Toxicol. 1997; 37: 397–419.
32. Healy SM, Casarez EA, Ayala-Fierro F, and Aposhian HV. Enzymatic methylation of arsenic compounds. V. arsenite methyltransferase activity in tissues of mice. Toxicol Appl Pharmacol. 1998; 148: 65–70.
33. Cullen WR. Chemical mechanism of arsenic biomethylation. Chem Res Toxicol. 2014; 27: 457-61.
34. Rehman K, and Naranmandura H. Arsenic metabolism and thioarsenicals. Metallomics. 2012; 4: 881-92.
35. Challenger F. Biological methylation. Chemical Reviews. 1945; 36: 315-61.
36. Pinyayev TS, Kohan MJ, Herbin-Davis K, Creed JT, and Thomas DJ. Preabsorptive metabolism of sodium arsenate by anaerobic microbiota of mouse cecum forms a variety of methylated and thiolated arsenicals. Chem Res Toxicol. 2011; 24: 475-7.
37. Hayakawa T, Kobayashi Y, Cui X, and Hirano S. A new metabolic pathway of arsenite: arsenic-glutathione complexes are substrates for human arsenic methyltransferase Cyt19. Arch Toxicol. 2005; 79: 183-91.
38. Kala SV, Neely MW, Kala G, Prater CI, Atwood DW, Rice JS, and Lieberman MW. The MRP2/cMOAT transporter and arsenic-glutathione complex formation are required for biliary excretion of arsenic. J Biol Chem. 2000; 275: 33404-8.
39. Naranmandura H, Suzuki N, and Suzuki KT. Trivalent arsenicals are bound to proteins during reductive methylation. Chem Res Toxicol. 2006; 19: 1010-8.
40. Wang S, Li X, Song X, Geng Z, Hu X, and Wang Z. Rapid equilibrium kinetic analysis of arsenite methylation catalyzed by recombinant human arsenic (+3 oxidation state) methyltransferase (hAS3MT). J Biol Chem. 2012; 287: 38790-9.
41. Suzuki KT, Katagiri A, Sakuma Y, Ogra Y, and Ohmichi M. Distributions and chemical forms of arsenic after intravenous administration of dimethylarsinic and monomethylarsonic acids to rats. Toxicol Appl Pharmacol. 2004; 198: 336-44.
42. Bogdan GM, Sampayo-Reyes A, and Aposhian HV. Arsenic binding proteins of mammalian systems: I. Isolation of three arsenite-binding proteins of rabbit liver. Toxicology. 1994; 93: 175-93.
43. Styblo M, Del Razo L, Vega L, Germolec DR, LeCluyse EL, Hamilton GA, Reed W, Wang C, Cullen WR, and Thomas DJ. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch Toxicol. 2000; 74: 289–99.
44. Mass MJ, Tennant A, Roop BC, Cullen WR, Styblo M, Thomas DJ, and Kligerman AD. Methylated trivalent arsenic species are genotoxic. Chem Res Toxicol. 2001; 14: 355–61.
45. Petrick JS, Jagadish B, Mash EA, and Aposhian HV. Monomethylarsonous acid (MMA(III)) and arsenite: LD(50) in hamsters and in vitro inhibition of pyruvate dehydrogenase. Chem Res Toxicol. 2001; 14: 651–6.
46. Cullen WR, and Reimer K. Arsenic speciation in the environment. Chem Rev. 1989; 89: 713–64.
47. Lin S, Cullen WR, and Thomas DJ. Methylarsenicals and arsinothiols are potent inhibitors of mouse liver thioredoxin reductase. Chem Res Toxicol. 1999; 12: 924–30.
48. Styblo M, Serves SV, Cullen WR, and Thomas DJ. Comparative inhibition of yeast glutathione reductase by arsenicals and arsenothiols. Chem Res Toxicol. 1997; 10: 27–33.
49. Nesnow S, Roop BC, Lambert G, Kadiiska M, Mason R, Cullen WR, and Mass MJ. DNA damage induced by methylated trivalent arsenicals is mediated by reactive oxygen species. Chem Res Toxicol. 2002; 15: 1627–34.
50. Dopp E, Recklinghausen Uvon, Diaz-Bone R, Hirner AV, and Rettenmeier AW. Cellular uptake, subcellular distribution and toxicity of arsenic compounds in methylating and non-methylating cells. Environ Res. 2010; 110: 435–42.
51. Steinmaus C, Yuan Y, Bates MN, and Smith AH. Case-control study of bladder cancer and drinking water arsenic in the western United States. Am J Epidemiol. 2003; 158: 1193–201.
52. Hirata M, Tanaka A, Hisanaga A, and Ishinishi N. Effects of glutathione depletion on the acute nephrotoxic potential of arsenite and on the arsenic metabolism in the hamster. Toxicol Appl Pharmacol. 1990; 106: 469-81.
53. Vahter M. Variation in human metabolism of arsenic, in: W.R. Chappell, C.O. Abernathy, R.L. Calderon (Eds.), Arsenic Exposure and Health Effects, Elsevier Science Ltd., Oxford, 1999, pp. 267–279.
54. Moe B, Peng H, Lu X, Chen B, Chen LWL, Stephan G, Li XF, and Le XC. Comparative cytotoxicity of fourteen trivalent and pentavalent arsenic species determined using real-time cell sensing. J Environ Sci. 2016; 49: 113–24.
55. Cohen SM, Chowdhury A, and Arnold LL. Inorganic arsenic: A non-genotoxic carcinogen. J Environ Sci. 2016; 49: 28–37.
56. Yi P, Melnyk S, Pogribna M, Pogribny IP, Hine RJ, and James SJ. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J Biol Chem. 2000; 275: 29318–23.
57. Thomas DJ, Styblo M, and Lin S. The cellular metabolism and systemic toxicity of arsenic. Toxicol Appl Pharmacol. 2001; 176: 127-44.
58. Yoshino Y, Yuan B, Miyashita SI, Iriyama N, Horikoshi A, Shikino O, Toyoda H, and Kaise T. Speciation of arsenic trioxide metabolites in blood cells and plasma of a patient with acute promyelocytic leukemia. Anal Bioanal Chem. 2009; 393: 689-97.
59. Drobna Z, Naranmandura H, Kubachka KM, Edwards BC, Herbin-Davis K, and Styblo M. Disruption of the arsenic (+3 oxidation state) methyltransferase gene in the mouse alters the phenotype for methylation of arsenic and affects distribution and retention of orally administered arsenate. Chem Res Toxicol. 2009; 22: 1713–20.
60. Chen GQ, Zhou L, Styblo M, Walton F, Jing Y, Weinberg R, Chen Z, and Waxman S. Methylated metabolites of arsenic trioxide are more potent than arsenic trioxide as apoptotic but not differentiation inducers in leukemia and lymphoma cells. Cancer Res. 2003; 63: 1853–9.
61. Falnoga I, Slejkovec Z, Pucer A, Podgornik H, and Tusek-Znidaric M. Arsenic metabolism in multiple myeloma and astrocytoma cells. Biol Trace Elem Res. 2007; 116: 5–28.
62. Rehman K, and Naranmandura H. Double-edged effects of arsenic compounds: anticancer and carcinogenic effects. Curr Drug Metab. 2013; 14: 1029-41.
63. Calatayud M, Barrios J, Velez AD, and Devesa V. In vitro study of transporters involved in intestinal absorption of inorganic arsenic. Chem Res Toxicol. 2012; 25: 446-53.
64. Calatayud M, Vazquez M, Devesa V, and Velez D. In vitro study of intestinal transport of inorganic and methylated arsenic species by Caco-2/HT29-MTX cocultures. Chem Res Toxicol. 2012; 25: 2654-62.
65. Calatayud M, Velez D, and Devesa V. Metabolism of inorganic arsenic in intestinal epithelial cell lines. Chem Res Toxicol. 2012; 25: 2402-11.
66. Csanaky I, and Gregus Z. Effect of phosphate transporter and methylation inhibitor drugs on the disposition of arsenate and arsenite in rats. Toxicol Sci. 2001; 63: 29-36.
67. Kwong YL. Arsenic trioxide in the treatment of haematological malignancies. Expert Opinion on Drug Safety. 2004; 3: 589-97.
68. Tsukaguchi H, Weremowicz Morton SCC, and Hediger MA. Functional and molecular characterization of the human neutral solute channel aquaporin-9. Am J Physiol. 1999; 277: F685-96.
69. Leung J, Pang A, Yuen WH, Kwong YL, and Tse EW. Relationship of expression of aquaglyceroporin 9 with arsenic uptake and sensitivity in leukemia cells. Blood. 2007; 109: 740-6.
70. Saito Y, Furukawa T, Obata T, and Saga T. Molecular imaging of aquaglycero-aquaporins: its potential for cancer characterization. Biol Pharm Bull. 2013; 36: 1292-8.
71. Leslie EM, Haimeur A, and Waalkes MP. Arsenic transport by the human multidrug resistance protein 1 (MRP1/ABCC1). Evidence that a tri-glutathione conjugate is required. J Biol Chem. 2004; 279: 32700-8.
72. Haimeur A, Conseil G Deeley RG, and Cole SP. The MRP-related and BCRP/ABCG2 multidrug resistance proteins: biology, substrate specificity and regulation. Curr Drug Metab. 2004; 5: 21-53.
73. Deeley RG, Westlake C, and Cole SP. Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol Rev. 2006; 86: 849-99.
74. Leslie EM, Deeley RG, and Cole SP. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol. 2005; 204: 216-37.
75. Flens MJ, Zaman GJ, van der Valk P, Izquierdo MA, Schroeijers AB, Scheffer GL, van der Groep P, de Haas M, Meijer CJ, and Scheper RJ. Tissue distribution of the multidrug resistance protein. Am J Pathol. 1996; 148: 1237-47.
76. Smitherman PK, Townsend AJ, Kute TE, and Morrow CS. Role of multidrug resistance protein 2 (MRP2, ABCC2) in alkylating agent detoxification: MRP2 potentiates glutathione S-transferase A1-1-mediated resistance to chlorambucil cytotoxicity. J Pharmacol Exp Ther. 2004; 308: 260-7.
77. Ballatori N, Hammond CL, Cunningham JB, Krance SM, and Marchan R. Molecular mechanisms of reduced glutathione transport: role of the MRP/CFTR/ABCC and OATP/SLC21A families of membrane proteins. Toxicol Appl Pharmacol. 2005; 204: 238-55.
78. Cole SP, and Deeley RG. Transport of glutathione and glutathione conjugates by MRP1. Trends in Pharmacological Sciences. 2006; 27: 438-46.
79. Carew MW, Naranmandura H, Shukalek CB, Le XC, and Leslie EM. Monomethylarsenic diglutathione transport by the human multidrug resistance protein 1 (MRP1/ABCC1). Drug Metab Dispos. 2011; 39: 2298-304.
80. Shengwen S, Li XF, Cullen WR, Weinfeld M, and Le XC. Arsenic Binding to Proteins. Chem Rev. 2013; 113: 7769−92.
81. Wang QQ, Zhou XY, Zhang YF, Bu N, Zhou J, Cao FL, and Naranmandura H. Methylated arsenic metabolites bind to PML protein but do not induce cellular differentiation and PML-RARα protein degradation. Oncotarget. 2015; 6: 25646-59. doi: 10.18632/oncotarget.4662.
82. Finnegan PM, and Chen W. Arsenic toxicity: the effects on plant metabolism. Front Physiol. 2012; 3: 182.
83. Carter DE, Aposhian HV, and Gandolfi AJ. The metabolism of inorganic arsenic oxides, gallium arsenide, and arsine: A toxicochemical review. Toxicol Appl Pharmacol. 2003; 193: 309-34.
84. Carter DE, Aposhian HV, and Gandolfi AJ. The metabolism of inorganic arsenic oxides, gallium arsenide, and arsine: a toxicochemical review. Toxicol Appl Pharmacol. 2003; 193: 309–34.
85. Li X, Geng Z, Chang J, Wang S, Song X, Hu X, and Wang Z. Identification of the Third Binding Site of Arsenic in Human Arsenic (III) Methyltransferase. PLoS One. 2013; 8: e84231.
86. Naranmandura H, Ibata K, and Suzuki KT. Toxicity of dimethylmonothioarsinic acid toward human epidermoid carcinoma A431 cells. Chem Res Toxicol. 2007; 20: 1120-25.
87. Currier JM, Douillet C, Drobná Z, and Stýblo M. Oxidation state specific analysis of arsenic species in tissues of wild-type and arsenic (+3 oxidation state) methyltransferase-knockout mice. J Environ Sci. 2016; 49: 104-12.
88. Mass MJ, Tennant A, Roop BC, Cullen WR, Styblo M, Thomas DJ, and Kligerman AD. Methylated Trivalent Arsenic Species Are Genotoxic. Chem Res Toxicol. 2001; 14: 355-61.
89. Hirano S, Cui X, Li S, Kanno S, Hayakawa T, and Shraim A. Difference in uptake and toxicity of trivalent and pentavalent inorganic arsenics in rat microvessel endothelial cells. Arch Toxicol. 2003; 77: 305-12.
90. Paul DS, Harmon AW, Devesa V, Thomas DJ, and Stýblo M. Molecular mechanisms of the diabetogenic effects of arsenic: inhibition of insulin signaling by arsenite and methylarsonous acid. Environ Health Perspect. 2007; 115: 734-42.
91. Douillet C, Currier J, Saunders J, Bodnar WM, Matoušek T, and Stýblo M. Methylated trivalent arsenicals are potent inhibitors of glucose stimulated insulin secretion by murine pancreatic islets. Toxicol Appl Pharmacol. 2013; 267: 11-15.
92. Bashir S, Sharma Y, Irshad M, Nag TC, Tiwari M, Kabra M, and Dogra TD. Arsenic induced apoptosis in rat liver following repeated 60 days exposure. Toxicology. 2006; 217: 63-70.
93 Suzuki T, and Tsukamoto I. Arsenite induces apoptosis in hepatocytes through an enhancement of the activation of Jun N-terminal kinase and p38 mitogen-activated protein kinase caused by partial hepatectomy. Toxicol Lett. 2006; 165: 257-64.
94. Jain A, Yadav A, Bozhkov AI, Padalko VI, and Flora SJ. Therapeutic efficacy of silymarin and naringenin in reducing arsenic-induced hepatic damage in young rats. Ecotoxicol Environ Saf. 2011; 74: 607-14.
95. Mazumder DNG. Effect of chronic intake of arsenic-contaminated water on liver. Toxicol Appl Pharmacol. 2005; 206: 169-75.
96. Santra A, Das Gupta J, De BK, Roy B, and Mazumder DNG. Hepatic manifestations in chronic arsenic toxicity. Indian J Gastroenterol. 1999; 18: 152-5.
97. Straub AC, Stolz DB, Ross MA, Hernández-Zavala A, Soucy NV, Klei LR, and Barchowsky A. Arsenic stimulates sinusoidal endothelial cell capillarization and vessel remodeling in mouse liver. Hepatology. 2007; 45: 205-12.
98. Li GX, Pei QL, Gao Y, Liu KM, Nie JS, Han G, Qiu YL, and Zhang WP. Protective effects of hepatocellular canalicular conjugate export pump (Mrp2) on sodium arsenite-induced hepatic dysfunction in rats. Exp Toxicol Pathol. 2007; 58: 447-53.
99. Mumford JL, Wu K, Xia Y, Kwok R, Yang Z, Foster J, and Sanders WE. Chronic Arsenic Exposure and Cardiac Repolarization Abnormalities with QT Interval Prolongation in a Population-based Study. Environ Health Perspect. 2007; 115: 690-4.
100. Hossain E, Islam K, Yeasmin F, Karim MR, Rahman M, Agarwal S, Hossain S, Aziz A, Mamun AA, Sheikh A, Haque A, Hossain M, T Hossain et al. Elevated levels of plasma Big endothelin-1 and its relation to hypertension and skin lesions in individuals exposed to arsenic. Toxicol Appl Pharmacol. 2011; 259: 187-94.
101. Lee MY, and Griendling KK. Redox signaling, vascular function, and hypertension. Antioxidants & Redox Signaling. 2008; 10: 1045–59.
102. Smith KR, Klei LR, and Barchowsky A. Arsenite stimulates plasma membrane NADPH oxidase in vascular endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2001; 280: L442–9.
103. Bunderson M, Coffin JD, and Beall HD. Arsenic induces peroxynitrite generation and cyclooxygenase-2 protein expression in aortic endothelial cells: possible role in atherosclerosis. Toxicol Appl Pharmacol. 2002; 184: 11-8.
104. Lee PC, Ho IC, and Lee TC. Oxidative stress mediates sodium arsenite-induced expression of heme oxygenase-1, monocyte chemoattractant protein-1, and interleukin-6 in vascular smooth muscle cells. Toxicol Sci. 2005; 85: 541-50.
105. Chen Y, Parvez F, Gamble M, Islam T, Ahmed A, Argos M, Graziano JH, and Ahsan H. Arsenic exposure at low-to-moderate levels and skin lesions, arsenic metabolism, neurological functions, and biomarkers for respiratory and cardiovascular diseases: review of recent findings from the Health Effects of Arsenic Longitudinal Study (HEALS) in Bangladesh. Toxicol Appl Pharmacol. 2009; 239: 184-92.
106. Dodmane PR, Arnold LL, Kakiuchi-Kiyota S, Qiu F, Liu X, Rennard SI, and Cohen SM. Cytotoxicity and gene expression changes induced by inorganic and organic trivalent arsenicals in human cells. Toxicology. 2013; 312: 18-29.
107. Zheng LY, Umans JG, Yeh F, Francesconi KA, Goessler W, Silbergeld EK, Bandeen-Roche K, Guallar E, Howard BV, Weaver VM, and Navas-Acien A. The Association of Urine Arsenic with Prevalent and Incident Chronic Kidney Disease: Evidence from the Strong Heart Study. Epidemiology. 2015; 26: 601–12.
108. Tokar EJ, Person RJ, Sun Y, Perantoni AO, and Waalkes MP. Chronic exposure of renal stem cells to inorganic arsenic induces a cancer phenotype. Chem Res Toxicol. 2013; 26: 96-105.
109. Wang QQ, Lan YF, Rehman K, Jiang YH, Maimaitiyiming YD, Zhu Y, and Naranmandura H. Effect of arsenic compounds on the in vitro differentiation of mouse embryonic stem cells into cardiomyocytes. Chem Res Toxicol. 2015; 28: 351-3.
110. Wei M, Wanibuchi H, Morimura K, Iwai S, Yoshida K, Endo G, Nakae D, and Fukushima S. Carcinogenicity of dimethylarsinic acid in male F344 rats and genetic alterations in induced urinary bladder tumors. Carcinogenesis. 2002; 23: 1387−97.
111. Cohen SM, Ohnishi T, Arnold LL, and Le XC. Arsenic-induced bladder cancer in an animal model. Toxicol Appl Pharmacol. 2007; 222: 258-63.
112. Adair BM, Moore T, Conklin SD, Creed JT, Wolf DC, and Thomas DJ. Tissue distribution and urinary excretion of dimethylated arsenic and its metabolites in dimethylarsinic acid- or arsenate-treated rats. Toxicol Appl Pharmacol. 2007; 222: 235-42.
113. Cohen SM, Arnold LL, Uzvolgyi E, Cano M, St John M, Yamamoto S, Lu X, and Le XC. Possible role of dimethylarsinous acid in dimethylarsinic acid-induced urothelial toxicity and regeneration in the rat. Chem Res Toxicol. 2002; 15: 1150-7.
114. Shen J, Wanibuchi H, Salim EI, Wei M, Kinoshita A, Yoshida K, Endo G, and Fukushima S. Liver tumorigenicity of trimethylarsine oxide in male Fischer 344 rats–association with oxidative DNA damage and enhanced cell proliferation. Carcinogenesis. 2003; 24: 1827-35.
115. Naranmandura H, Carew MW, Xu S, Lee J, Leslie EM, Weinfeld M, and Le XC. Comparative toxicity of arsenic metabolites in human bladder cancer EJ-1 cells. Chem Res Toxicol. 2011; 24: 1586-96.
116. McCoy CR, Stadelman BS, Brumaghim JL, Liu JT, and Bain LJ. Arsenic and Its Methylated Metabolites Inhibit the Differentiation of Neural Plate Border Specifier Cells. Chem Res Toxicol. 2015; 28: 1409-21.
117. Tyler CR, and Allan AM. The effects of arsenic exposure on neurological and cognitive dysfunction in human and rodent studies: a review. Curr Environ Health Rep. 2014; 1: 132-47.
118. Bornhorst J, Ebert F, Lohren H, Humpf HU, Karst U, and Schwerdtle T. Effects of manganese and arsenic species on the level of energy related nucleotides in human cells. Metallomics. 2012; 4: 297-306.
119. Liu S, Sun Q, Wang F, Zhang L, Song Y, Xi S, and Sun G. Arsenic induced overexpression of inflammatory cytokines based on the human urothelial cell model in vitro and urinary secretion of individuals chronically exposed to arsenic. Chem Res Toxicol. 2014; 27: 1934-42.
120. Pournara A, Holmlund T, Lu Y, Ceder R, Putnik M, Grafstrom R, Vahter M, and Wallberg AE. Arsenic-induced suppression of kidney cell proliferation and the, transcriptional coregulator MAML1. Metallomics. 2014; 6: 498-504.
121. Chen B, Liu Q, Popowich A, Shen S, Yan X, Zhang Q, Li XF, Weinfeld M, Cullen WR, and Le XC. Therapeutic and analytical applications of arsenic binding to proteins. Metallomics. 2014; 7: 39-55.
122. Rehman K, Chen Z, Wang WW, Wang YW, Sakamoto A, Zhang YF, Naranmandura H, and Suzuki N. Mechanisms underlying the inhibitory effects of arsenic compounds on protein tyrosine phosphatase (PTP). Toxicol Appl Pharmacol. 2012; 263: 273-80.
123. Shen S, Li XF, Cullen WR, Weinfeld M, and Le XC. Arsenic binding to proteins. Chem Rev. 2013, 113: 7769-92.
124. Erik JT, Chikara K, and Michael PW. Methylarsonous acid causes oxidative DNA damage in cells independent of the ability to biomethylate inorganic arsenic. Arch Toxicol. 2014; 88: 249-61.
125. Shen S, Wang C, Weinfeld M, and Le XC. Inhibition of nucleotide excision repair by arsenic. Chinese Science Bulletin. 2013; 58: 214-21.
126. Shen S, Lee J, Weinfeld M, and Le XC. Attenuation of DNA damage-induced p53 expression by arsenic: a possible mechanism for arsenic co-carcinogenesis. Molecular Carcinogenesis. 2008; 47: 508-18.
127. Zhou XD, Sens DA, Sens MA, Namburi VB, Singh RK, Garrett SH, and Somji S. Metallothionein-1 and -2 expression in cadmium- or arsenic-derived human malignant urothelial cells and tumor heterotransplants and as a prognostic indicator in human bladder cancer. Toxicol Sci. 2006; 91: 467-75.
128. Paul S, Banerjee N, Chatterjee A, Sau TJ, Das JK, Mishra PK, and Chakrabarti P. Bandyopadhyay A, Giri AK. Arsenic-induced promoter hypomethylation and over-expression of ERCC2 reduces DNA repair capacity in humans by non-disjunction of the ERCC2-Cdk7 complex. Metallomics. 2014; 6: 864-73.
129. Zhao L, Chen S, Jia L, Shu S, Zhu P, and Liu Y. Selectivity of arsenite interaction with zinc finger proteins. Metallomics. 2012; 4: 988-94.
130. Schwerdtle T, Walter I, and Hartwig A. Arsenite and its biomethylated metabolites interfere with the formation and repair of stable BPDE-induced DNA adducts in human cells and impair XPAzf and Fpg. DNA Repair (Amst). 2003; 2: 1449-63.
131. Zhang XW, Yan XJ, Zhou ZR, Yang FF, Wu ZY, Sun HB, Liang WX, Song AX, Lallemand-Breitenbach V, Jeanne M, Zhang QY, Yang HY, Huang QH et al. Arsenic trioxide controls the fate of the PML-RAR alpha oncoprotein by directly binding PML. Science. 2010; 328: 240-3.
132. Falnoga I, Zelenik Pevec A, Šlejkovec Z, Žnidarič MT, Zajc I, Mlakar SJ, and Marc J. Arsenic trioxide (ATO) influences the gene expression of metallothioneins in human glioblastoma cells. Biol Trace Elem Res. 2012; 149: 331-9.
133. He X; and Ma Q. Induction of metallothionein I by arsenic via metal-activated transcription factor 1: critical role of C-terminal cysteine residues in arsenic sensing. J Biol Chem. 2009; 284: 12609-21.
134. Jiang G, Gong Z, Li XF, Cullen WR, and Le XC. Interaction of trivalent arsenicals with metallothionein. Chem Res Toxicol. 2003; 16: 873-80.
135. Saha KC. Diagnosis of arsenicosis. J Environ Sci Health A Tox Hazard Subst Environ Eng. 2003; 38: 255–72.
136. Milton AH, Hasan Z, Rahman A, and Rahman M. Non-cancer effects of chronic arsenicosis in Bangladesh: preliminary results. J Environ Sci Health A Tox Hazard Subst Environ Eng. 2003; 38: 301–5.
137. Yoshida T, Yamauchi H, and Fan SG. Chronic health effects in people exposed to arsenic via the drinking water: dose-response relationships in review. Toxicol Appl Pharmacol. 2004; 198: 243–52.
138. Huang C, Ke Q, Costa M, and Shi X. Molecular mechanisms of arsenic carcinogenesis. Mol Cell Biochem. 2004; 255: 57–66.
139. Kitchin KT. Recent advances in arsenic carcinogenesis: modes of action, animal model systems, and methylated arsenic metabolites, Toxicol Appl Pharmacol. 2001; 172: 249–61.
140. Yu HS, Liao WT, and Chai CY. Arsenic carcinogenesis in the skin. J Biomed Sci. 2006; 13: 657–66.
141. Liu SX, Athar M, Lippai I, Waldren C, and Hei TK. Induction of oxyradicals by arsenic: implication for mechanism of genotoxicity. Proc Natl Acad Sci U S A. 2001; 98: 1643–8.
142. Liu SX, Davidson MM, Tang X, Walker WF, Athar M, and Ivanov V. Mitochondrial damage mediates genotoxicity of arsenic in mammalian cells. Cancer Res. 2005; 65: 3236–42.
143. Srivastava RK, Li C, Chaudhary SC, Ballestas ME, Elmets CA, Robbins DJ, Matalon S, Deshane JS, Afaq F, Bickers DR, and Athar M. Unfolded protein response (UPR) signaling regulates arsenic trioxide-mediated macrophage innate immune function disruption. Toxicol Appl Pharmacol. 2013; 272: 879-87.
144. Gardner BM, Pincus D, Gotthardt K, Gallagher CM, and Walter P. Endoplasmic Reticulum Stress Sensing in the Unfolded Protein Response. Cold Spring Harb Perspect Biol. 2013; 5: a013169.
145. Zhang K, and Kaufman RJ. Signaling the Unfolded Protein Response from the Endoplasmic Reticulum. J Biol Chem. 2004; 279: 25935–8.
146. Li C, Xu J, Li F, Chaudhary SC, Weng Z, Wen J, Elmets CA, Ahsan H, and Athar M. Unfolded protein response signaling and MAP kinase pathways underlie pathogenesis of arsenic-induced cutaneous inflammation. Cancer Prev Res. (Phila). 2011; 4: 2101–9.
147. Klassen CD. Casarette and Doull’s Toxicology: the basic science of poisons. 7th ed. USA: Mc Graw Hill. 2008; 15: 936–9.
148. IARC (International Agency for Research on Cancer). 1987. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Suppl. 7, Overall Evaluations of Carcinogenicity: An Updating of IARC Monographs IARC Press, Lyon, 1 to 42:100–106.
149. IARC (International Agency for Research on Cancer). Some drinking-water disinfectants and contaminants, including arsenic. IARC Monographs on the evaluation of carcinogenic risks to humans. 2004; 84:139–41.
150. Chen CJ, Chen CW, Wu M, and Kuo TL. Cancer potential in liver, lung, bladder and kidney due to ingested inorganic arsenic in drinking water. Br J Cancer. 1992; 66: 888–92.
151. Smith AH, Goycolea M, Haque R, and Biggs ML. Marked increase in bladder and lung cancer mortality in a region of northern Chile due to arsenic in drinking water. Am J Epidemiol. 1998; 147: 660–9.
152. Yu RC, Hsu KH, Chen CJ, and Froines JR. Arsenic methylation capacity and skin cancer. Cancer Epidemiol Biomarkers Prev. 2000; 9:1259–62.
153. Valko M, Rhodes CJ, Moncol J, Izakovic M, and Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006; 160: 1–40.
154. Cohen SM, Arnold LL, Eldan M, Lewis AS, and Beck BD. Methylated arsenicals: the implications of metabolism and carcinogenicity studies in rodents to human risk assessment. Crit Rev Toxicol. 2006; 36: 99–133.
155. Kinoshita A, Wanibuchi H, Wei M, Yunoki T, and Fukushima S. Elevation of 8-hydroxydeoxyguanosine and cell proliferation via generation of oxidative stress by organic arsenicals contributes to their carcinogenicity in the rat liver and bladder. Toxicol Appl Pharmacol. 2007; 221: 295–305.
156. Suzuki S, Arnold LL, Pennington KL, Kakiuchi-Kiyota S, and Cohen SM. Effects of co-administration of dietary sodium arsenite and an NADPH oxidase inhibitor on the rat bladder epithelium. Toxicology. 2009; 261: 41–6.
157. Yang C, and Frenkel K. Arsenic-mediated cellular signal transduction, transcription factor activation, and aberrant gene expression: implications in carcinogenesis. J Environ Pathol Toxicol Oncol. 2002; 21: 331–42.
158. Liu J, and Waalkes M. Focal adhesion kinase as a potential target in arsenic toxicity. Toxicol Sci. 2005; 84: 212–3.
159. Bates MN, Smith AH, and Cantor KP. Case-control study of bladder cancer and arsenic in drinking water. Am J Epidemiol. 1995; 141: 523–30.
160. Hays AM, Srinivasan D, Witten ML, Carter DE, and Lantz RC. Arsenic and cigarette smoke synergistically increase DNA oxidation in the lung. Toxicol Pathol. 2006; 34: 396–404.
161. Klein CB, Leszczynska J, Hickey C, and Rossman TG. Further evidence against a direct genotoxic mode of action for arsenic-induced cancer. Toxicol Appl Pharmacol. 2007; 222: 289–97.
162. Laurenzana A, Pettersson F, and Jr WHM. Role of PML/RARα in the pathogenesis of APL. Drug Discovery Today: Disease Mechanisms. 2006; 3: 499-505.
163. Lloyd NC, Morgan HW, Nicholson BK, and Ronimus RS. The Composition of Ehrlich’s Salvarsan: Resolution of a Century Old Debate. Angew Chem Int Ed Engl. 2005; 44: 941-4.
164. Mandal BK, Ogra Y, and Suzuki KT. Identification of Dimethylarsinous and Monomethylarsonous Acids in Human Urine of the Arsenic-Affected Areas in West Bengal, India. Chem Res Toxicol. 2001; 14: 371-8.
165. Mann KK, Wallner B, Lossos IS, and Miller WHJ. Darinaparsin: a novel organic arsenical with promising anticancer activity. Expert Opin Investig Drugs. 2009; 18: 1727-34.
166. Matulis SM, Morale, AA, Yehiayan L, Croutch C, Gutman D, Cai Y, Lee KP, and Boise LH. Darinaparsin induces a unique cellular response and is active in an arsenic trioxide-resistant myeloma cell line. Mol Cancer Ther. 2009; 8: 1197-206.
167. Berenson JR, Boccia RV, Hussein A, Belch A, Boise L, Schwartz B, Gale RP, and Kornblau SM. Phase-1 study of ZIO-101: A new organic arsenic active in acute myelogenous leukemia (AML) and multiple myeloma (MM). Blood. 2006; 108: 1966.
168. Tsimberidou AM, Camacho LH, Verstovsek S, Ng C, Hong DS, Uehara CK, Gutierrez C, Daring S, Stevens J, Komarnitsky PB, Schwartz B, and Kurzrock R. A phase I clinical trial of darinaparsin in patients with refractory solid tumors. Clin Cancer Res. 2009; 15: 4769-76.
169. Wu J, Henderson C, Feun L, Van VP, Gold P, Zheng H, Ryan T, Blaszkowsky LS, Chen H, Costa M, Rosenzweig B, Nierodzik M, Hochster H et al. Phase II study of darinaparsin in patients with advanced hepatocellular carcinoma. Invest New Drugs. 2010; 28: 670–6.
170. Hosein PJ, Craig MD, Tallman MS, Boccia RV, Hamilton BL, Lewis JJ, and Lossos IS. A multicenter phase II study of darinaparsin in relapsed or refractory Hodgkin’s and non-Hodgkin’s lymphoma. Am J Hematol. 2012; 87: 111-4.
171. Ohno N, Tani A, Chen ZS, Uozumi K, Hanada S, Akiba S, Ren XQ, Furukawa T, Sumizawa T, Arima T, and Akiyama SI. Prognostic significance of multidrug resistance protein in adult T-cell leukemia. Clin Cancer Res. 2001; 7: 3120-6.
172. Duzkale H, Jilani I, Orsolic N, Zingaro RA, Golemovic M, Giles FJ, Kantarjian H, Albitar M, Freireich EJ, and Verstovsek S. In vitro activity of dimethylarsinic acid against human leukemia and multiple myeloma cell lines. Cancer Chemother Pharmacol. 2003; 51: 427-32.
173. Don AS, Kisker O, Dilda P, Donoghue N, Zhao X, Decollogne S, Creighton B, Flynn E, Folkman J, and Hogg PJ. A peptide trivalent arsenical inhibits tumor angiogenesis by perturbing mitochondrial function in angiogenic endothelial cells. Cancer Cell. 2003; 3: 497-509.
174. Elliott M, Ford S, Prasad E, Dick L, Farmer H, Hogg P, and Halbert G. Pharmaceutical development of the novel arsenical based cancer therapeutic GSAO for Phase I clinical trial. Int J Pharm. 2012; 426: 67-75.
175. Cadd VA, Hogg PJ, Harris AL, and Feller SM. Molecular profiling of signalling proteins for effects induced by the anti-cancer compound GSAO with 400 antibodies. BMC Cancer. 2006; 6: 155.
176. Park D, Chiu J, Perrone GG, Dilda PJ, and Hogg PJ. The tumour metabolism inhibitors GSAO and PENAO react with cysteines 57 and 257 of mitochondrial adenine nucleotide translocase. Cancer Cell Int. 2012; 12: 1-6.
177. Dilda PJ, Decollogne S, Rossiter-Thornton M, and Hogg PJ. Para to ortho repositioning of the arsenical moiety of the angiogenesis inhibitor 4-(N-(S-glutathionylacetyl)amino)phenylarsenoxide results in a markedly increased cellular accumulation and antiproliferative activity. Cancer Res. 2005; 65: 11729–34.
178. Dilda PJ, Emma ER, Alessandro C, Alfonso P and Philip JH. Metabolism of the Tumor Angiogenesis Inhibitor 4-(N-(S-Glutathionylacetyl)amino)phenylarsonous Acid. J Biol Chem. 2008; 283: 35428–34.
179. Dilda PJ, and Hogg PJ. Arsenical-based cancer drugs. Cancer Treat Rev. 2007; 33: 542-64.
180. Lallemand-Breitenbach V, Zhu J, Chen Z, and Thé HD. Curing APL through PML/RARA degradation by As2O3. Trends Mol Med. 2012; 18: 36-42.
181. Lengfelder E, Hofmann WK, and Nowak D. Impact of arsenic trioxide in the treatment of acute promyelocytic leukemia. Leukemia. 2012; 26:433-42.
182. Wang Z, Zhou J, Lu X, Gong Z, and Le XC. Arsenic speciation in urine from acute promyelocytic leukemia patients undergoing arsenic trioxide treatment. Chem Res Toxicol. 2004; 17: 95-103.
183. Iriyama N, Yoshino Y, Yuan B, Horikoshi A, Hirabayashi Y, Hatta Y, Toyoda H, and Takeuchi J. Speciation of arsenic trioxide metabolites in peripheral blood and bone marrow from an acute promyelocytic leukemia patient. J Hematol Oncol. 2012; 5: 1.
184. Hikita E, Arai M, Tanaka S, Onda K, Utsumi H, Yuan B, Toyoda H, and Hirano T. Effects of inorganic and organic arsenic compounds on growth and apoptosis of human T-lymphoblastoid leukemia cells. Anticancer Res. 2011; 31: 4169-78.
185. Rehman K, Fu YJ, Zhang YF, Wang QQ, Wu B, Wu Y, Zhou XY, Sun WH, Sun TF, and Naranmandura H. Trivalent methylated arsenic metabolites induce apoptosis in human myeloid leukemic HL-60 cells through generation of reactive oxygen species. Metallomics. 2014; 6: 1502-12.
186. Xu S, Zhang YF, Carew MW, Hao WH, Loo JF, Naranmandura H, and Le XC. Multidrug resistance protein 1 (ABCC1) confers resistance to arsenic compounds in human myeloid leukemic HL-60 cells. Arch Toxicol. 2013; 87: 1013-23.
187. Naranmandura H, Xu S, Sawata T, Hao WH, Liu H, Bu N, Ogra Y, Lou YJ, and Suzuki N. Mitochondria are the main target organelle for trivalent monomethylarsonous acid (MMA(III))-induced cytotoxicity. Chem Res Toxicol. 2011; 24: 1094-103.
188. Naranmandura H, Xu S, Koike S, Pan LQ, Chen B, Wang YW, Rehman K, Wu B, Chen Z, and Suzuki N. The endoplasmic reticulum is a target organelle for trivalent dimethylarsinic acid (DMAIII)-induced cytotoxicity. Toxicol Appl Pharmacol. 2012; 60: 241-9.
189. Zhang YF, Zhang M, Huang XL, Fu YJ, Jiang YH, Bao LL, Maimaitiyiming Y, Zhang G J, Wang QQ, and Naranmandura H. Combination of arsenic and cryptotanshinone induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in breast cancer cells. Metallomics. 2014; 7: 165-73.
190. Szegezdi E, Logue SE, Gorman AM, and Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006; 7: 880–5.
191. Huang C, Ma WY, Li J, and Dong Z. Arsenic induces apoptosis through a c-Jun NH2-terminal kinase-dependent, p53-independent pathway. Cancer Res. 1999; 59: 3053-8.
192. Huang C, Ma WY, Li J, Goranson A, and Dong Z. Requirement of Erk, but not JNK, for arsenite-induced cell transformation. J Biol Chem. 1999; 274: 14595-601.
193. Chen GQ, Zhou L, Styblo M, Walton F, Jing Y, Weinberg R, Chen Z, and Waxman S. Methylated metabolites of arsenic trioxide are more potent than arsenic trioxide as apoptotic but not differentiation inducers in leukemia and lymphoma cells. Cancer Res. 2003; 63: 1853-9.