
INTRODUCTION
Glioblastoma multiforme (GBM) is the most malignant primary brain tumor with infiltrative growth characteristics. The survival of GBM patients is often less than 15 months from diagnosis, with the 5-year survival rate below 10% even after surgery combined with adjuvant radiotherapy and chemotherapy [1, 2]. Therefore, an advanced understanding of the molecular mechanism of GBM progression is essential to improve clinical outcome.
Valproic acid (VPA) has been widely used in seizure prophylaxis after neurosurgery including GBM, and the pharmacokinetics and toxicity of VPA have been well documented for treating epilepsy [3]. VPA was further shown to inhibit histone deacetylase (HDAC) activity causing impaired epigenetic modification and suppressed cell growth [4]. Thereafter, the antitumor effects of VPA were described in vitro and in retrospective clinical studies [5–11]. Several studies revealed that VPA sensitized GBM cells to chemotherapy and radiotherapy by increased cell apoptosis, which involved increased p21 expression and cell cycle arrest, suppression of DNA double strand break repair, and activating pro-apoptotic signaling [12–16]. Reactive oxygen species (ROS) involves tumor development. Overproduction of ROS and antioxidant system defect result in DNA repair impairment and gene expression alteration, contributing to the carcinogenesis process [17, 18].
The paraoxonase (PON) family belongs to endogenous free-radical scavenging enzyme system, which consists of PON1, PON2, and PON3 [19]. The three highly conserved genes share about 60% to 70% similarity at the amino acid and nucleotide levels, All three PON members possess antioxidant properties, but their tissue distributions and stress responses are different [19–21]. PON1 and PON3 are found mainly in the liver and are associated with high-density lipoprotein and cholesterol levels. PON2 is an intracellular protein that is expressed extensively in thorax and abdomen tissues, skeletal muscle, artery wall cells, and macrophages [22]. Previous studies have shown that people with impaired PON1 function are at increased risk of cancer development [23–25]. Overexpression of PON3 protects cancer cells from mitochondrial superoxide-mediated cell death [26].
In the present study, we observed that VPA decreased PON2 expression in GBM-derived cell lines. Impaired antioxidant genes may be associated with GBM development, and intracellular PON2 may mediate anti-apoptosis and maintain the growth of GBM. We hypothesized that VPA inhibited PON2 in GBM cells and sensitized GBM cells to oxidative damage and cell death. Our results indicate that VPA suppresses cell growth via the PON2–Bim cascade in GBM cells.
RESULTS
VPA attenuates GBM cell growth
First, we investigated whether VPA inhibits GBM cell progression. We treated the U87, GBM8401, and DBTRG-05MG GBM cell lines with 5, 10, and 20 mM VPA for 24 to 72 h. Using the MTS and Bromodeoxyuridine (BrdU) assays, the cell growth was reduced significantly by 10 to 20 mM VPA in the U87 cells, and by 5 to 20 mM VPA in the GBM8401 and DBTRG-05MG cells from 24 to 72 h (Figure 1A–1F). Thus, these GBM cells were sensitized with VPA in a time- and dose-dependent manner. Furthermore, to evaluate whether the cell cycle is influenced by VPA, the cell cycle of GBM was assessed by flow cytometry. As expected, the cell cycle was arrested at the G2/M phase at 24 and 48 h in the presence of VPA in U87, GBM8401, and DBTRG-05MG cells, indicating that numbers of GBM cells entering the S phase were significantly reduced (Figure 2A–2C). These observations suggest that VPA decreases cell growth through cell cycle arrest in the G2/M phase in GBM cells.
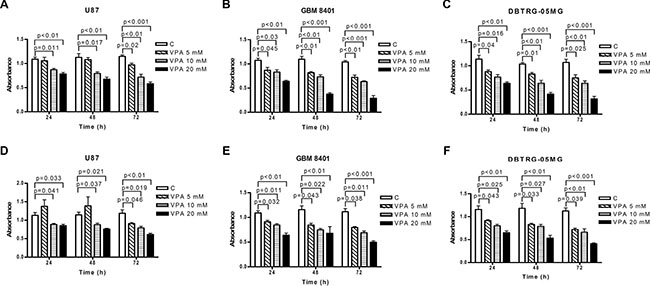
Figure 1: Valproic acid (VPA) inhibits glioblastoma cell growth. Cell proliferation was determined in U87 (A, D), GBM8401 (B, E), and DBTRG-05MG (C, F) cells after 5–20 mM VPA stimulation for 24 to 72 h using the MTS (A–C) and Bromodeoxyuridine (BrdU) (D–F) assays. The cell proliferation is significantly decreased in GBM cells using VPA in different doses. The data shown are from three independent experiments performed in triplicate. Error bars: SD. Values are shown as absorbance of VPA-treated cells relative to controls (C; cells without VPA treatment).
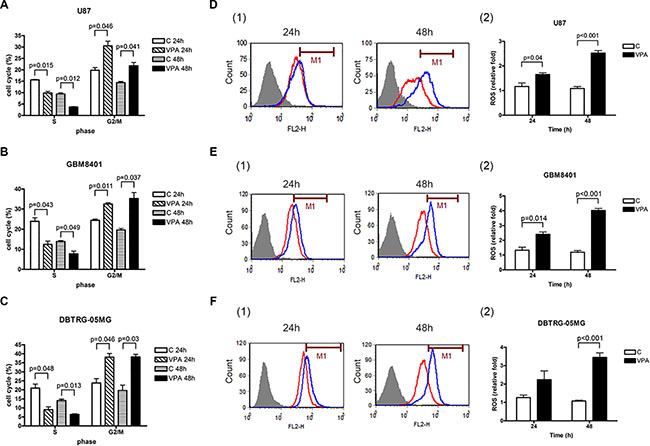
Figure 2: Valproic acid (VPA) induces cell cycle arrest at G2/M phase and increases ROS production. The cell cycle was analyzed by flow cytometry in U87 (A), GBM8401 (B), and DBTRG-05MG (C) cells treated with 5 (GBM8401 and DBTRG-05MG) or 10 mM (U87) VPA for 24 to 48 h. The levels of S and G2/M phase are quantified. A significant number of GBM cells arrested at the G2/M phase of cell cycle in GBM cells treated by VPA. The ROS pattern was analyzed by flow cytometry in U87 (D), GBM8401 (E), and DBTRG-05MG (F) cells treated with 5 (GBM8401and DBTRG-05MG) or 10 (U87) mM VPA for 24 to 48 h. The ROS levels are quantified in right panel (2) of D, E, and F, respectively. The ROS level is significantly increased by VPA treatment for 24 to 48 h in GBM cells. The data shown are from three independent experiments. Error bars: SD. Values are shown as cell cycle (%) and relative fold (ROS) of VPA-treated cells relative to controls (cells without VPA treatment). Red line: control; blue line: VPA treatment. C: control.
VPA increases ROS production
To investigate the mechanism of VPA-mediated cell growth suppression in GBM cells, the effect of VPA on the cell levels of ROS, an important factor in tumor progression [27], was tested on all three GBM-derived cell lines. The ROS level was significantly increased in U87 cells treated with 10 mM VPA for 24 to 48 h as assessed by flow cytometry (Figure 2D). Similarly, significantly increased ROS levels were observed in the GBM8401 and DBTRG-05MG cells after stimulation with 5 mM VPA for 24 to 48 h (Figure 2E, 2F). These results indicate VPA suppresses cell growth via upregulation of ROS production. Additionally, the migration and invasion ability were decreased by VPA in U87, GBM8401 and DBTRG-05MG cells (Supplementary Figure 1A–1F). However, the cell senescence was not altered with VPA by evaluating senescence associated β-galactosidase (SA-βgal) activity [28] in both U87 and GBM8401 cells (Supplementary Figure 1G). We also analyzed the apoptosis characterization. The sub-G1 phase was increased by VPA with PI staining using flow cytometry in U87, GBM8401 and DBTRG-05MG cells (Supplementary Figure 1H).
VPA influences PON2 and cell cycle–related markers
Based on the above results, we found that cell proliferation and ROS levels were altered by VPA in GBM cells. Therefore, we utilized a commercially available Proteome Profiler Array for cell stress phenotype to evaluate specific VPA-regulated molecules. Several molecules, including PON2 and p27, were identified as potential VPA targets (not shown). The regulation of these molecules was further validated by Western blotting in U87, GBM8401, and DBTRG-05MG cells. PON2 was downregulated and p27 was upregulated at 24 h by VPA (Figure 3A). Since VPA possesses an anti-cancer effect to influence tumor cell proliferation and apoptosis [29, 30], its effect on several additional proliferation and apoptotic-related molecules, such as cyclin B1, cdc2, p21, Bcl-xL, and Bim were investigated. Bim and p21 were upregulated while cyclin B1, cdc2, and Bcl-xL were downregulated at 24 h by VPA treatment in GBM cells (Figure 3A). The results suggest that VPA may regulate these molecules to influence tumor cell proliferation and apoptosis. Since PON2 has been identified as an anti-oxidative protein that decreases intracellular oxidative stress in various cell types [22, 31] and is ubiquitously expressed in a variety of tissues and organs [20], PON2 was selected for further study. Immunohistochemistry of the PON2 expression level in brain tissue of clinical glioblastoma patients revealed that PON2 expression was increased in GBM cells compared with normal brain cells (Figure 3B). We further treated GBM cells with 5 and 10 mM VPA for 24 to 72 h, the PON2 mRNA and protein levels reduced by VPA was in a dose-dependent manner using RT-PCR and Western blot (Figure 3C, 3D).
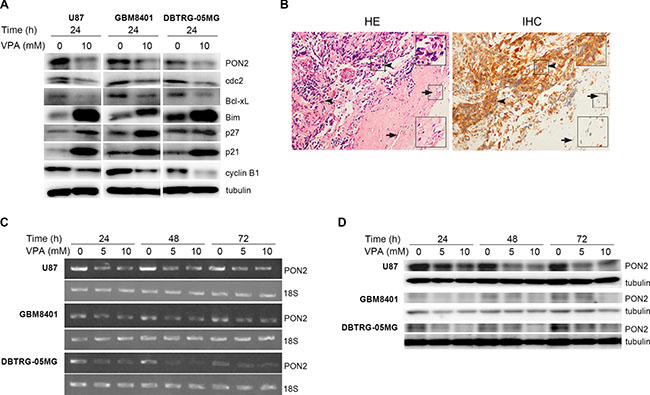
Figure 3: Oxidative stress–related molecules are analyzed in glioblastoma cells. (A) Western blot to show the expression levels of oxidative stress–related molecules such as PON2, cdc2, Bcl-xL, Bim, p27, p21 and cyclin B1, which were determined after stimulation with valproic acid (VPA) at 24 h in U87, GBM8401, and DBTRG-05MG cell lines. The expression of PON2, cdc2, Bcl-xL and cyclin B1 is decreased, and Bim, p27 and p21 is increased by VPA stimulation in GBM cells. (B) Immunohistochemistry (IHC, right panel) to show PON2 protein expression in human GBM biopsies and the left panel is hematoxylin and eosin (HE) staining. The PON2 staining is stronger in tumor cells (arrowhead) compared with normal neuron cells (arrow; magnification: 200×). Large inset: Two-fold magnification of the small inset with arrowheads or arrows. (C, D) The PON2 mRNA and protein levels were determined after stimulation with 5 and 10 mM VPA for 24 to 72 h in U87, GBM8401, and DBTRG-05MG cell lines by RT-PCR (C) and Western blot (D). The decreased PON2 induced by VPA is in a dose-dependent manner. C: control, cells without VPA treatment.
VPA inhibits PON2 at the transcriptional level
Next, we investigated the PON2 regulation mechanism by VPA. The reporter assay was performed to determine the position and clarify the regulatory effects of VPA on PON2 at the transcriptional level. The PON2 5′-flanking region encompassing nucleotides -1000/-1 (relative to the transcription initiation site) (Figure 4A) was cloned and inserted upstream of the luciferase reporter gene in pA3TK-luc (construct p1) to generate construct p2. The pA3TK-luc construct contained a minimum thymidine kinase promoter. Serial deletion fragments were additionally constructed (Figure 4A). The transcriptional activities of the PON2 promoter fragments are illustrated in Figure 4A. Among these, only the p5 construct was repressed about 2-fold by VPA in U87 and GBM8401 cells (Figure 4A). These findings suggest that VPA inhibits PON2 at the transcriptional level between position -400/-1 (p5) in glioblastoma.
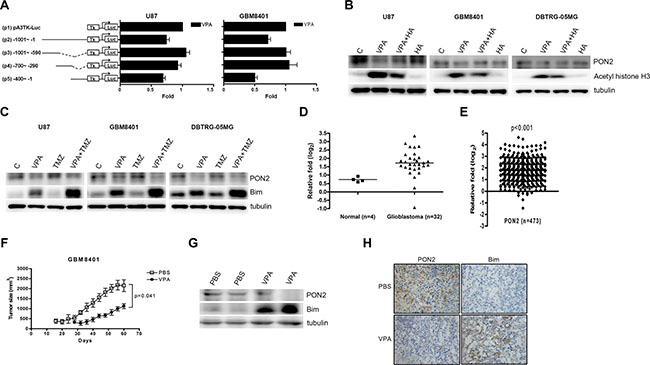
Figure 4: Valproic acid (VPA) inhibits PON2 expression in vitro and in vivo. (A) U87 and GBM8401 cells were transfected with the luciferase reporter plasmid driven by the PON2 5′-flanking region (position -1001 to -1) with or without pA3TK-luc. Promoter activities were calculated, relative to 0 nM VPA (+VPA/-VPA), and further normalized to the pA3TK-luc control as well as β-galactosidase activity (VPA-induced changes were normalized to that of β-gal). VPA decreases PON2 expression at position -400 to -1. Columns, mean values obtained from at least three independent experiments performed in triplicate; error bars, SE. (B) The glioblastoma cells were pre-treated with a HDAC activator, 1-benzoyl-3-phenyl-2-thiourea (HA, 20 μM) for 1 h followed by VPA (U87, 10 mM; GBM8401 and DBTRG-05MG, 5 mM) treatment 24 h. The acetyl histone H3 expression, an acetylation histone protein regulated by HDAC, was not increased as that by VPA. The HA did not significantly alter the PON2 expression compared with control cells. The PON2 expression was downregulated by VPA, which was abrogated by HA in U87, GBM8401 and DBTRG-05MG cells. (C) The glioblastoma cells were treated with VPA (U87, 10 mM; GBM8401 and DBTRG-05MG, 5 mM) and temozolomide (TMZ, 40 μM) for 24 hours, which has synergistic effect with VPA on Bim upregulation. PON2 downregulation was not observed in TMZ treatment. TMZ did not have synergistic effect with VPA on PON2 regulation. (D, E) PON2 mRNA expression was analyzed in 30 GBM specimens and 4 normal subjects from the Oncomine public Bredel GBM microarray dataset (GEO accession: GSE2223 [38]) (D) and 473 specimens from The Cancer Genome Atlas (TCGA) GBM microarray database (E). The PON2 level is significantly higher in GBM patients compared with normal controls. (F) The nude mice received subcutaneous xenograft with GBM8401 cells (5 × 107) were intraperitoneal injections of PBS (n = 3) or VPA (400 mg/kg; n = 3) every two days for 60 days. The tumor sizes were measured up to 60 days after inoculation of tumor cells. VPA decreased glioblastoma cells proliferation significantly in vivo. (G, H) The PON2 and Bim levels were measured in tumors of PBS and VPA conditions with GBM8401 cells inoculation by Western blot (G) and immunohistochemistry (H). The PON2 was downregulated and Bim upregulated by VPA in GBM8401 cells-injected mice. C: control, cells without treatment. p: promoter.
VPA attenuates glioblastoma growth via PON2 regulation
VPA has been shown to possess HDAC inhibitor activity [11], hence we investigated whether the VPA-regulated PON2 is affected by HDAC. HDAC decreased the acetylation of histone H3 [35, 36]. We treated GBM cells with a HDAC activator, 1-benzoyl-3-phenyl-2-thiourea [35, 36], which decreased the expression of acetyl histone H3 in GBM cells. VPA inhibited HDAC that increased the expression of acetyl histone H3 in GBM cells. This effect was attenuated by the simultaneous use of VPA and 1-benzoyl-3-phenyl-2-thiourea in these cells. The 1-benzoyl-3-phenyl-2-thiourea did not significantly alter the PON2 expression compared with control cells. The PON2 expression was downregulated by VPA, which was reversed by 1-benzoyl-3-phenyl-2-thiourea in U87, GBM8401 and DBTRG-05MG cells (Figure 4B). Additionally, VPA has been shown to be an effective sensitizing agent in combination with irradiation and chemotherapy to augmentation of therapeutic efficiency on glioblastoma [37]. We stimulated glioblastoma with VPA and temozolomide (TMZ), a DNA alkylating agent, which has synergistic effect with VPA on Bim upregulation (Figure 4C). Because TMZ induced DNA methylation and damage, PON2 downregulation was not observed in TMZ treatment. TMZ did not have synergistic effect with VPA on PON2 regulation (Figure 4C).
The finding was further confirmed by examining the PON2 expression in GBM cells from the public Bredel GBM microarray dataset (Gene Expression Omnibus [GEO] accession: GSE2223 [38]). From the dataset report, the mean PON2 mRNA level was higher in 30 GBM specimens compared with 4 normal subjects (Figure 4D). Although the public database displayed only 4 normal subjects, the average PON2 level in glioblastoma patients was about 2.3-fold that of normal subjects (3.75 vs 1.66; Figure 4D). Moreover, this phenomenon was further observed in 473 GBM specimens from The Cancer Genome Atlas (TCGA) GBM microarray database [32] (Figure 4E). To investigate whether the effects of VPA in vitro could be applied in vivo, we established a subcutaneous xenograft of GBM8401 cells in BALB/c nude mice. Subsequently, nude mice were injected with PBS or VPA (400 mg/kg) intraperitoneally every two days for 60 days [40, 41]. Tumor sizes from the two groups (PBS and VPA) of mice are shown in Figure 4F. The tumor growth of PBS group was initially detected at 14 days, but in VPA group was about 30 days. Tumors sizes of mice injected with PBS were two-fold larger than those of VPA-injected mice (Figure 4F). VPA can inhibit glioblastoma growth in vivo. In the study of mouse xenograft, the PON2 expression was downregulated and Bim expression upregulated by VPA in tumors of GBM8401 cells as shown by Western blotting and immunohistochemistry (Figure 4G, 4H).
PON2 decreases ROS production
To determine whether PON2 inhibits ROS production in GBM cells, the ROS level was assessed in PON2-overexpressed GBM cells transiently transfected with PON2 cDNA. The basal expression level of PON2 in U87, GBM8401, and DBTRG-05MG was shown in Supplemental Figure 2. The expression level of PON2 protein in U87 and GBM8401 cells was increased after PON2 overexpression (Figure 5A, 5C). The ROS level was significantly reduced in PON2-overexpressed cells stimulated with VPA compared with controls in the U87 and GBM8401 cells using flow cytometry (Figure 5B, 5D). Furthermore, we established two transient PON2-silenced GBM cells (U87 and GBM8401) in which PON2 protein expression was decreased (Figure 5E, 5G). The ROS level was significantly higher in PON2-silenced cells in the presence of VPA compared with controls (Figure 5F, 5H). These findings indicate that PON2 influences ROS production and is implicated in VPA-mediated tumor cell growth arrest in GBM cells.
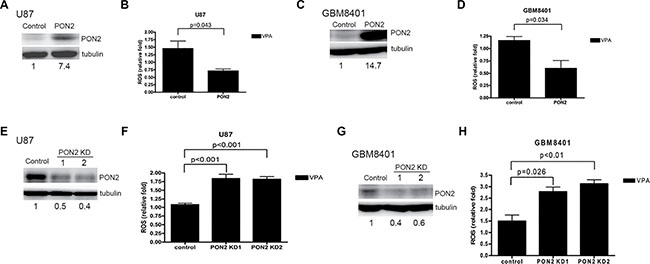
Figure 5: ROS level is inhibited by PON2. Cells overexpressing PON2 (A, C) and PON2-silenced (E, G) cells were established in U87 (A, E) and GBM8401 (C, G) as shown by Western blot. The ROS levels in U87 (B, F) and GBM8401 (D, H) cells in both PON2-overexpressed (B, D) and PON2-silenced (F, H) cells were analyzed by flow cytometry. VPA (10 mM) increases the ROS production in vector-control cells, the effect is reduced in PON2-overexpressed cells (B, D). The ROS levels in PON2-silenced cells with VPA (10 mM) treatment are higher than that of vector-control cells with VPA treatment (F, H). Control: cells transfected with empty vector only; KD: knockdown.
VPA increases tumor cell ROS production through PON2–Bim signaling
To investigate the downstream signaling pathway involved in PON2-mediated glioblastoma cell growth arrest, we established two transient PON2-transfectants overexpressing PON2 in U87 and GBM8401 cells. The expression level of PON2 protein in these transfected U87 and GBM8401 cells was increased compared to levels present in the vector-control cells (Figure 6A, 6B, lanes 1 vs 3). PON2 protein was, however, decreased after stimulation with VPA in the U87 and GBM8401 cells transfected by vector, but not in the PON2 -overexpressed transfectants (Figure 6A, 6B, lanes 1 vs 2, lanes 3 vs 4). We speculate that the expression of PON2 was saturated in PON2-overexpressed cells, hence the PON2 level was not significantly altered after VPA treatment. However, the level of Bim was elevated after VPA treatment (Figure 6A, 6B, lanes 1 vs 2, lanes 3 vs 4), and the expression level was attenuated in the PON2-overexpressed cells with VPA treatment compared with controls (Figure 6A, 6B, lanes 2 vs 4). Similar effects were observed in PON2-silenced U87 and GBM8401 cells. The expression level of PON2 protein in U87 and GBM8401 cells was silenced (Figure 6C, 6D, lanes 1 vs 3). Furthermore, the expression of PON2 was decreased after VPA stimulation (Figure 6C, 6D, lanes 1 vs 2, lanes 3 vs 4). Bim expression was activated by VPA in both controls and PON2-silenced cells (Figure 6C, 6D, lanes 1 vs 2, lanes 3 vs 4), and the levels were increased in PON2-silenced cells compared with controls in U87 and GBM8401 cells (Figure 6C, 6D, lanes 2 vs 4). To investigate whether the Bim regulation is specific in VPA-mediated reducing cell proliferation, we further silenced Bim expression in GBM cells. The Bim expression was efficiently suppressed. The decrease in cell proliferation was abrogated in the Bim-silenced (Bim KD) condition compared with the vector-control cells in the VPA stimulation in U87 and GBM8401 cells (Figure 6E, 6F). Hence we suggest the Bim is specific in the PON2-Bim pathway of VPA-mediated reducing GBM cell proliferation.
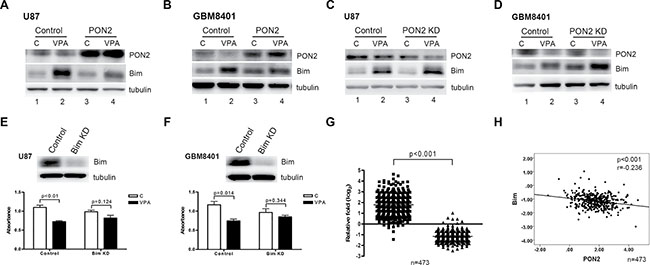
Figure 6: PON2-Bim cascade is involved in VPA-increased ROS. The expression levels of PON2 and Bim were determined by Western blot after VPA stimulation in U87 (A, C) and GBM8401 (B, D) cells in both PON2-overexpressed (PON2) (A, B) and PON2-silenced (PON2 KD) (C, D) conditions. The expression of Bim in VPA treated PON2-overexpressed cells is lower than that of VPA treated vector-control cells (A, B; lanes 2 vs 4). The levels of Bim in VPA-treated PON2-silenced cells is higher than that in VPA-treated vector-control cells (C, D; lanes 2 vs 4). (E, F) The Bim expression and cell proliferation were determined by Western blot and MTS assay after VPA stimulation in U87 and GBM8401 cells with Bim-silenced (Bim KD) condition. The decreasing cell proliferation was abrogated in the Bim-silenced (Bim KD) condition compared with the vector-control cells in the VPA stimulation cells (G, H) The mRNA levels of PON2 and Bim were analyzed in 473 specimens from The Cancer Genome Atlas (TCGA) GBM microarray database [32]. The mRNA expression levels of PON2 and Bim in the individuals were significantly different in 473 GBM specimens. The PON2 level is inversely correlated with Bim (Pearson r = –0.236, p < 0.001). Control: cells transfected with empty vector only; C; cells without VPA treatment; KD: knockdown.
To confirm the clinical significance of the PON2–Bim axis, we determined whether the observed expression values between the PON2 and Bim molecules could be applied in human GBM. The expression levels of the PON2 and Bim genes were retrieved from the published TCGA GBM microarray dataset. The mRNA expression levels of PON2 and Bim in the individuals were significantly different in 473 GBM specimens (Figure 6G), which further revealed PON2 and Bim were significantly inversely correlated (Figure 6H). Taking the data together, we suggest that VPA attenuation of GBM cell proliferation may involve significant upregulation of ROS production via the PON2–Bim cascade.
DISCUSSION
Here we have shown that cell proliferation is reduced by VPA in U87, GBM8401, and DBTRG-05MG cells. VPA suppresses GBM cell growth via the upregulation of ROS production. PON2 overexpression and silencing influence ROS production in GBM cells. VPA appears to increase ROS production via the PON2–Bim cascade, leading to cancer cell death.
Our study demonstrated that VPA suppressed the growth of GBM-derived cell lines and induced cell cycle arrest in a time- and dose-dependent manner. ROS level was increased in GBM cells after VPA treatment. Results from protein array analysis revealed the possible mechanisms of inhibitory effects of VPA on GBM cells. These included elevated p27, resulting in cell proliferation suppression and reduced cyclin B1/cdc2, which in turn results in cell cycle arrest at G2/M phase [42], decreased levels of pro-survival Bcl2 and increased pro-apoptotic Bim proteins, which could then result in cell death [43]. Hence, VPA influence may inhibit GBM progression through changes in proliferation and apoptosis.
VPA has been identified as an HDAC inhibitor influencing tumor cell apoptosis, differentiation, and proliferation [29, 30]. Moreover, ROS is an important factor implicated in tumor progression [27]. We also showed that VPA augmented ROS production in GBM cells thus supporting a potential role of VPA in cell growth suppression through the upregulation of ROS production.
A decrease of the anti-oxidant PON2 protein was also detected after using VPA. This deceased PON2 expression could potentiate the cytotoxic effects of ROS and enhance VPA-induced cell cycle arrest. The use of the model of transfectants overexpressing PON2 provided further support for the VPA-induced GBM cell growth suppression being mediated by increased ROS production and that the effect was augmented by decreased PON2.
In a study of non-Hispanic whites, an increased risk of glioma was associated with the single nucleotide polymorphism of SOD3 [45]. Single nucleotide polymorphisms in SOD2, SOD3, GPX1, and NOS1 were found to significantly increase the risk of glioma development in a Chinese population. These data suggest that oxidative stress gene variation might contribute to the etiology of glioma [46]. PON2 was identified in subcellular mitochondrial fractions [31]. PON2 is able to inhibit the generation of mitochondrial superoxide and apoptosis [47]. We observed that GBM cells expressed a higher PON2 protein level compared with normal brain tissue, providing additional support for the importance of PON2 for cancer cell survival.
In agreement with our findings, overexpression of PON2 is observed in solid cancers derived from prostate, liver, pancreas, kidney, lung and thymus [48–50], and T-cell leukemia virus-infected lymphocytes [51]. Kang et al. reported that upregulation of PON2 can be used as a predictor for acute lymphoblastic leukemia with poor prognosis [52]. PON2 accelerates chemoresistance in leukemia cells, and silencing PON2 has resulted in spontaneous apoptosis in various human cancer cells [50]. In keeping with the above observations, our results support PON2 as a tumor promoter via the downregulation of ROS production.
VPA is a relatively weak HDAC inhibitor with activity at millimolar concentrations [53]. The docking and molecular dynamic simulations showed that HDAC inhibited by VPA was through the carboxyl group of VPA coordinating with Zn atom and other local residues (H141-142 and Y360) located at the catalytic site of HDAC. VPA bound with highest affinity at a site located at the acetyl-releasing channel [54]. VPA, either in in vitro studies or phase I/II clinical trials, also demonstrated cell growth inhibition effects on both benign cells, such as vascular pericytes, and cancers, such as acute myeloid leukemia and solid malignancies [39, 56]. As the clinical kinetics and bioavailability of VPA are well established, it has promise as an anticancer agent indicated for a reduction of glioma growth either as mono-therapy or as part of a combined treatment. Previous reports have shown that VPA synergistically interacts with chemotherapy or radiotherapy to enhance cytotoxicity in GBM cells by promoting HDAC-dependent transcriptional repression and histone hyperacetylation in vitro and in vivo, which improves survival in GBM patients [12, 56–62]. Tsai et al. displayed that VPA may be efficacious for GBM with increased histone acetylation, and early administration of VPA for patients within 2 weeks of diagnosis as an adjunct to temozolomide chemotherapy may benefit survival [63]. GBM patients treated with VPA had a significantly longer survival than those who had received other anti-epileptic drugs [11], and VPA may be preferred in patients with glioblastoma who require an anti-epileptic drug during temozolomide-based chemoradiotherapy [37, 64, 65]. For example, patients treated with VPA in combination with temozolomide had a median survival for 69 weeks, longer than those without VPA treatment (61 weeks, p = 0.016) [61]. Patients taking VPA had a more favorable median overall survival in (16.9 months), compared to those taking other anti-epileptic drugs (13.6 months, p = 0.016) [66].
Our data demonstrate that VPA may suppress GBM cell growth in GBM-derived cell lines through the upregulation of ROS production via the PON2–Bim signaling pathway. In the present study, the selected 5 to 10 mM concentration of VPA (molecular weight 144.21) used in in vitro is equivalent to 10-times therapeutic serum level of VPA in human (50–125 μg/mL; the toxic level is greater than 150 μg/mL) [67]. Such a concentration for in vitro study could be much lower than true therapeutic level in a culture dish, because of the protein binding proportion was neglected. So, effective dose of valproic acid for in vitro study may require higher dose to exhibit cytotoxic effects. The higher dose of VPA for in vivo study refers to the previous reports [40, 41]. However, humans cannot tolerate high doses of VPA. High-dose VPA experimental data contribute to the clinical efficacy of the drug, which may direct treatments using combination treatment with radiotherapy or chemotherapy, or design other drug formulations in patients. VPA may, thus, represent a potential important therapeutic agent for the treatment for malignant glioma. Further work is needed to study the clinical prognosis of GBM patients treated with VPA to reveal if the PON2 can also be used as a diagnostic marker for predicting therapeutic effect of VPA on GBM patients.
MATERIALS AND METHODS
Cell culture
The human glioblastoma cell lines U87, Denver Brain Tumor Research Group 05MG (DBTRG-05MG; ATCC, Manassas, VA) , and GBM8401 (Bioresource Collection Research Center, Hsinchu, Taiwan) were routinely cultured at 37°C in a humidified atmosphere of 95% air and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Grand Island, NY) and Roswell Park Memorial Institute 1640 medium (RPMI 1640; Invitrogen) supplemented with 10% fetal bovine serum (Hyclone, Road Logan, UT).
Cell proliferation assay
Cell proliferation rates were examined using the (3-[4,5-dimethylthiazol-2-yl]-5-[3-carboxymethoxyphenyl]-2-[4-sulfophenyl]-2H-tetrazolium) (MTS) assay (Promega, Madison, WI). Cells (2 × 103) were seeded on 96-well plates overnight. After 6 h starvation, cells were treated with various doses of VPA (2–20 mM) for 48–72 h, and 20 μL MTS solution (10× dilution of 5 mg/mL MTS in DMEM without serum) was added to each well for 3 h at 37°C. Finally, absorbance at 490 nm was measured using a SpectraMax microplate reader.
BrdU assay
A BrdU incorporation assay was performed using a Cell Proliferation ELISA, BrdU Kit (Roche, Mannheim, Germany). Briefly, the U87, GBM8401 and DBTRG-05MG cells were cultured in 96-well plates at a density of 5000 cells/100 μL/well in a complete growth medium. After stimulating with VPA for 24 to 72 h, the cells were labelled using 100 μL BrdU (1:1000) per well and incubated for 18 h that followed by the procedures as manufacturer’s instructions. The reaction was quantified by measuring the absorbance using a scanning multi-well spectrophotometer (Tecan, Mannedorf, Switzerland) at 450 nm, with a reference wavelength of 540 nm.
Flow cytometry studies on cell cycle and apoptosis
U87, GBM8401, and DBTRG-05MG cells were fixed in ethanol/PBS (7:3, v/v) for 1 h at–20°C. Cells were centrifuged at 3000 rpm for 3 min and pellets were stained with propidium iodide (PI; Sigma-Aldrich, St. Louis, MO) in 250 μL PBS (0.5% Triton X-100) containing 0.125 mg DNase-free RNase A (Roche, South San Francisco, CA) and 6.25 μg PI (Sigma-Aldrich) for 15 min at 4°C in the dark. The cell cycle was examined by flow cytometry. Data were collected and analyzed using FACScan (Becton Dickinson, San Jose, CA) running CellQuest software.
ROS detection
Cells were trypsinized and resuspended in PBS with RedoxSensor Red CC-1 (Molecular probes, Eugene, OR) for 15 min at room temperature. The ROS level was then determined by flow cytometry using RedoxSensor™ Red CC-1 kit (Molecular Probes) as manufacturer’s instructions.
In vitro migration and invasion assays
The influence of VPA on glioblastoma motility was determined as described previously [34]. Briefly, 100 μL cell suspension (1 × 105 cells/ml) was seeded on upper chambers of either non-Matrigel-coated (migration) or Matrigel-coated (invasion; BD Biosciences, San Diego, CA) Transwell with 8-μm-pore size (Falcon BD, Franklin Lakes, New Jersey). The medium in the upper chamber was serum-free medium, whereas the lower chamber medium contained 10% FBS. After incubation for 24 h at 37°C, the cells traversing the filter from the upper to lower chamber were examined via cell counting. Experiments were performed at least three times.
Cell senescence assay
The senescence assay is to analyze the SA-βgal activity [28]. The U87 and GBM8401 cells (2 × 105 cells) were seeded on 6-well plate. After stimulation with 10 mM VPA for 24 h, cells were washed with PBS twice and fixed with 3.7 % formaldehyde for 10 min at room temperature, followed by staining with SA-βgal staining solution (5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 150 mM NaCl, 2 mM MgCl2 and 1 mg/mL X-gal) overnight at 37°C. The blue SA-βgal positive cells were defined as senescent cells.
Proteome ProfilerTM Array (human cell stress array kit)
U87 cells were either treated with 10 μM VPA for 24 h or left untreated. The cell lysates of U87 were incubated with a Proteome Profiler Array to study cell stress phenotype overnight at 4°C. The array included carbonic anhydrase IX, cited-2, cytochrome C, dickkopf Wnt signaling pathway inhibitor 4, fatty acid binding protein 1, hypoxia-inducible factor 1alpha, heat shock protein (HSP) 60, HSP70, phosphorylated c-Jun N-terminal kinase, nuclear factor–kappaB, p21, p27, PON1, PON2, thioredoxin-1, silent mating type information regulation 2 homolog, and superoxide dismutase 2. The array membrane was washed, followed by incubation with streptavidin-horseradish peroxidase buffer for 30 min at room temperature. Subsequently, the intensity of molecules on the membrane was examined by the chemiluminescence method using a chemiluminescence detection kit (Amersham Biosciences, Piscataway, NJ).
Western blot analysis
Cells were treated with VPA (5–10 mM), 1-benzoyl-3-phenyl-2-thiourea (HDAC activator, 20 μM, Sigma-Aldrich) and temozolomide (TMZ, 40 μM, Sigma- Aldrich) as the indicated times. Total cell lysate 20-μg proteins were fractionated on 12% sodium dodecyl sulfate-polyacrylamide gel. Separated proteins were transferred onto a nitrocellulose membrane (pH 7.9, Amersham Biosciences), blocked with 5% nonfat powdered milk in PBS, incubated with the specific primary antibodies PON2 (LifeSpan BioSciences, Seattle, WA), Bcl-xL (Santa Cruz Biotechnology, Dallas, TX), Bim, p21, p27, acetyl histone H3 (Cell Signaling, Danvers, MA), cyclin B1, cdc2, and tubulin (Millipore, Billerica, MA) at 4°C overnight, and subsequently hybridized with the appropriate horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Finally, immune complexes were visualized using the chemiluminescence method with a chemiluminescence detection kit (Amersham Biosciences).
Immunohistochemistry
Usage of archived formalin-fixed, paraffin-embedded tissue block was approved by the Institutional Review Board of MacKay Memorial Hospital. Tissue slides from glioblastoma patients were evaluated by immunohistochemistry and hematoxylin/eosin staining using polyclonal antibody against PON2 (LifeSpan BioSciences) through the avidin-biotin complex method, as described previously [33]. Immunoreactivity for PON2 was visualized using DAB/nickel substrate (Vector Laboratories, Burlingame, CA).
Reverse transcription polymerase chain reaction (RT-PCR)
Total RNA of glioblastoma cells was extracted using the TRIzol reagent (Life Technologies, Carlsbad, CA) according to the manufacturer’s protocol, and cDNA was synthesized using oligodeoxythymidine (Promega, Madison, WI) and a Superscript II reverse transcriptase (Invitrogen). The cDNA was amplified via PCR for 30 cycles at 95°C for 1 min, 58°C for 1 min, and 72°C for 1 min. 18s rRNA was used as an internal control. PCR products were checked by 1% agarose gel (Amresco, Solon, OH) electrophoresis. The primers were displayed in Table 1.
Table 1: The primer sequences used for RT-PCR. cloning. siRNA and shRNA
RT-PCR |
||
|---|---|---|
Gene |
Forward (5′–3′) |
Reverse (5′–3′) |
PON2 |
ATGGGGCGGCTGGTGGCT |
TTAGAGTTCACAATACAAGGCTCTG |
18s rRNA |
TAGAGCTAATACATGCCGACGG |
GGGCCTCGAAAGAGTCCTGTATT |
Gene |
Cloning |
|
|---|---|---|
PON2 ORF |
ATGGGGCGGCTGGTGGCT |
TTAGAGTTCACAATACAAGGCTCTG |
PON2 P1 |
TGTTGAAAATTGTCAAGTGTGACCA |
CTGCGTCGGGGCCGCGG |
PON2 P2 |
TGTTGAAAATTGTCAAGTGTGACCA |
GTGTGTGGAGACCGAGTCTT |
PON2 P3 |
GGCTCATTCCTGTAATTCTAGC |
GGAAAGTAACGGGATGCGTC |
PON2 P4 |
AAAAAAAGTATTACGAGGCACAGG |
CTGCGTCGGGGCCGCGG |
Gene |
siRNA SMART pool (5′–3′) |
|
|---|---|---|
Bim |
UGAGUGUGACCGAGAAGGUAGACAA |
|
UUGUCUACCUUCUCGGUCACACUCA |
||
CAUGAGUUGUGACAAAUCAACACAA |
Gene |
siRNA (5′–3′) |
|---|---|
TRCN0000051643 |
CCGGGCACATTTCTATGCCACAAATCTCGAGATTTGTGGCATAGAAATGTGCTTTTTG |
TRCN0000051644 |
CCGGGCACTCAGAAATCGACTTAAACTCGAGTTTAAGTCGATTTCTGAGTGCTTTTTG |
Abbreviations: ORF: open reading frame; P: promoter.
Cloning and activities of PON2 promoter fragments
The PON2 promoter (positions -1001 to -1, relative to the transcription initiation site) were ligated into the pA3TK vector, based on the published sequence. Several serial deletion fragments of the PON2 promoter were amplified via PCR and cloned into the pA3TK vector. The constructed promoter sequences were confirmed using automatic DNA sequencing. U87 and GBM8401 cells were cotransfected with 0.3 μg DNA/well of pA3TK vector containing the PON2 promoter sequence and 0.3 μg of SVβ plasmid, a β-galactosidase expression vector (Clontech, Palo Alto, CA), followed by treated with 10 and 5 mM VPA for 24 h in 24-well plates using Lipofectamine 2000 reagent following manufacturer’s instructions (Invitrogen) to determine the transcriptional activities of the PON2 promoter. At the end of the treatment period, cells were lysed, luciferase and β-galactosidase activities were measured. Luciferase activity was normalized against that of β-galactosidase. The primers were displayed in Table 1.
Oncomine® database
Oncomine is a cancer microarray database and integrated data platform available at www.oncomine.org. The Bredel GBM microarray dataset in Oncomine was searched using the criteria of the PON2 gene and glioblastoma cell.
TCGA database
TCGA is public and comprehensive databases that consists of various cancer types [32]. We searched the glioblastoma microarray database containing 473 biospecimens from the Data Coordinating Center of TCGA to analyze the PON2 and Bim expression levels.
Animal study
The BALB/c nude mice (Jackson Immuno Research Laboratories, West Grove, PA) were inoculated subcutaneously with GBM8401 cells (5 × 107 cells) mixed with 100 μl Matrigel TM (5 mg/mL, BD Biosciences), to enhance tumor growth ability in nude mice [55]. After 1 day of the implantation, VPA (400 mg/kg) [40, 41] or PBS was injected intraperitoneally every two days for 60 days, and PBS group were used as control. Once palpable tumors were established after the inoculation, the tumor xenografts were measured with two dimensions by caliper every two days. These nude mice were sacrificed at 60 days after tumor inoculation. Tumor volume was calculated by the following equation: length × height × width (mm3). The Institutional Animal Care and Use Committee of the Mackay Memorial Hospital approved the use of animals in this study.
Cloning of PON2
Total RNA (1 μg) was reverse-transcribed using Superscript II reverse transcriptase (Invitrogen) and oligodeoxythymidine to synthesize template cDNA. The PON2 cDNA was amplified via a polymerase chain reaction for 30 cycles at 95°C for 1 min, 58°C for 1 min, and 72°C for 1 min. The PON2 open reading frame was ligated into a pcDNA 3.0 expression vector (Promega) and the resulting construct was sequenced to confirm the presence of the gene. The PON2 open reading frame primers were displayed in Table1.
Establishment of U87 and GBM8401 cell lines transiently overexpressing PON2
The U87 and GBM8401 cells were transiently transfected with the PON2 cDNA construct in 6-cm cell culture dishes using serum free Opti-MEMi medium and Lipofectamine 2000 reagent following manufacturer’s instructions (Invitrogen). After 24 h, the medium of the transfected cells was replaced with regular medium. Subsequently, the expression of PON2 protein in U87 and GBM8401 cells was examined using Western blot analysis. PON2 cDNA was constructed into pcDNA 3.0 expression vector (Promega), hence, pcDNA 3.0 (Promega) was used as vector control.
Effect of knockdown Bim and PON2 expression
The siRNA silencing Bim was purchased from Invitrogen Corporation. The clone TRCN0000051643 and TRCN0000051644 of short hairpin (sh)RNA targeting PON2 were purchased from the National RNAi Core Facility (Institute of Molecular Biology, Academia Sinica, Taiwan). The sequence of targeting PON2 was constructed into pLKO.1 vector (Academia Sinica), which was used as vector control. Transfection of siRNA or shRNA against the endogenous Bim and PON2 genes in U87 and GBM8401 cells were transiently performed using Lipofectamine 2000 reagent (Invitrogen). The expression of Bim and PON2 was confirmed by Western blot analysis. The Bim siRNA and PON2 shRNA sequences were displayed in Table 1.
Statistical analysis
All values are reported as mean ± SD. Differences were evaluated by Student’s t-test or Wilcoxon signed rank test, when appropriate. In search for bivariate correlations, this investigation used Pearson’s correlation analysis and scatter plot for two continuous variables. P < 0.05 was considered significant.
Abbreviations
Bcl2, B-cell lymphoma 2; BrdU, Bromodeoxyuridine; DMEM, Dulbecco’s modified Eagle’s medium; DBTRG-05MG, Denver Brain Tumor Research Group 05MG; GBM, glioblastoma multiforme; GEO, Gene Expression Omnibus; HDAC, histone deacetylase; HSP, heat shock protein; MTS, (3-[4,5-dimethylthiazol-2-yl]-5-[3-carboxymethoxyphenyl]-2-[4-sulfophenyl]-2H-tetrazolium); PON2, paraoxonase 2; PI, propidium iodide; ROS, reactive oxygen species; sh, short hairpin; TCGA, The Cancer Genome Atlas; VPA, valproic acid;
ACKNOWLEDGMENTS
This work was supported by a grant from Mackay Memorial Hospital (MMH-E 103001 to C.-P Chen).
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest.
Authors’ contributions
CPC conceived, designed the experiments. JHT, CYC and CPC wrote the paper. JHT, CYC, PCC, CCF performed the experiments. SHH, YCL, JHT, CYC and CPC analyzed the data. CPC contributed reagents/materials/analysis tools.
REFERENCES
1. McLendon RE, Halperin EC. Is the long-term survival of patients with intracranial glioblastoma multiforme overstated? Cancer. 2003; 98:1745–1748.
2. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009; 10:459–466.
3. Yeow WS, Ziauddin MF, Maxhimer JB, Shamimi-Noori S, Baras A, Chua A, Schrump DS, Nguyen DM. Potentiation of the anticancer effect of valproic acid, an antiepileptic agent with histone deacetylase inhibitory activity, by the kinase inhibitor Staurosporine or its clinically relevant analogue UCN-01. Br J Cancer. 2006; 94:1436–1445.
4. Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001; 276:36734–36741.
5. Masoudi A, Elopre M, Amini E, Nagel ME, Ater JL, Gopalakrishnan V, Wolff JE. Influence of valproic acid on outcome of high-grade gliomas in children. Anticancer Res. 2008; 28:2437–2442.
6. Driever PH, Knupfer MM, Cinatl J, Wolff JE. Valproic acid for the treatment of pediatric malignant glioma. Klin Padiatr. 1999; 211:323–328.
7. Kim MS, Blake M, Baek JH, Kohlhagen G, Pommier Y, Carrier F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 2003; 63:7291–7300.
8. Witt O, Schweigerer L, Driever PH, Wolff J, Pekrun A. Valproic acid treatment of glioblastoma multiforme in a child. Pediatr Blood Cancer. 2004; 43:181.
9. Driever PH, Wagner S, Hofstadter F, Wolff JE. Valproic acid induces differentiation of a supratentorial primitive neuroectodermal tumor. Pediatr Hematol Oncol. 2004; 21:743–751.
10. Oberndorfer S, Piribauer M, Marosi C, Lahrmann H, Hitzenberger P, Grisold W. P450 enzyme inducing and non-enzyme inducing antiepileptics in glioblastoma patients treated with standard chemotherapy. J Neurooncol. 2005; 72:255–260.
11. Guthrie GD, Eljamel S. Impact of particular antiepileptic drugs on the survival of patients with glioblastoma multiforme. J Neurosurg. 2013; 118:859–865.
12. Das CM, Aguilera D, Vasquez H, Prasad P, Zhang M, Wolff JE, Gopalakrishnan V. Valproic acid induces p21 and topoisomerase-II (alpha/beta) expression and synergistically enhances etoposide cytotoxicity in human glioblastoma cell lines. J Neurooncol. 2007; 85:159–170.
13. Bangert A, Cristofanon S, Eckhardt I, Abhari BA, Kolodziej S, Hacker S, Vellanki SH, Lausen J, Debatin KM, Fulda S. Histone deacetylase inhibitors sensitize glioblastoma cells to TRAIL-induced apoptosis by c-myc-mediated downregulation of cFLIP. Oncogene. 2012; 31:4677–4688.
14. Camphausen K, Cerna D, Scott T, Sproull M, Burgan WE, Cerra MA, Fine H, Tofilon PJ. Enhancement of in vitro and in vivo tumor cell radiosensitivity by valproic acid. Int J Cancer. 2005; 114:380–386.
15. Chinnaiyan P, Cerna D, Burgan WE, Beam K, Williams ES, Camphausen K, Tofilon PJ. Postradiation sensitization of the histone deacetylase inhibitor valproic acid. Clin Cancer Res. 2008; 14:5410–5415.
16. Shabason JE, Tofilon PJ, Camphausen K. Grand rounds at the National Institutes of Health: HDAC inhibitors as radiation modifiers, from bench to clinic. J Cell Mol Med. 2011; 15:2735–2744.
17. Pourahmad J, Eskandari MR, Kaghazi A, Shaki F, Shahraki J, Fard JK. A new approach on valproic acid induced hepatotoxicity: involvement of lysosomal membrane leakiness and cellular proteolysis. Toxicol In Vitro. 2012; 26:545–551.
18. Sha K, Winn LM. Characterization of valproic acid-initiated homologous recombination. Birth Defects Res B Dev Reprod Toxicol. 2010; 89:124–132.
19. Hassett C, Richter RJ, Humbert R, Chapline C, Crabb JW, Omiecinski CJ, Furlong CE. Characterization of cDNA clones encoding rabbit and human serum paraoxonase: the mature protein retains its signal sequence. Biochemistry. 1991; 30:10141–10149.
20. Shih DM, Lusis AJ. The roles of PON1 and PON2 in cardiovascular disease and innate immunity. Curr Opin Lipidol. 2009; 20:288–292.
21. Blatter MC, James RW, Messmer S, Barja F, Pometta D. Identification of a distinct human high-density lipoprotein subspecies defined by a lipoprotein-associated protein, K-45. Identity of K-45 with paraoxonase. Eur J Biochem. 1993; 211:871–879.
22. Ng CJ, Wadleigh DJ, Gangopadhyay A, Hama S, Grijalva VR, Navab M, Fogelman AM, Reddy ST. Paraoxonase-2 is a ubiquitously expressed protein with antioxidant properties and is capable of preventing cell-mediated oxidative modification of low density lipoprotein. J Biol Chem. 2001; 276:44444–44449.
23. Elkiran ET, Mar N, Aygen B, Gursu F, Karaoglu A, Koca S. Serum paraoxonase and arylesterase activities in patients with lung cancer in a Turkish population. BMC Cancer. 2007; 7:48–55.
24. Aksoy-Sagirli P, Cakmakoglu B, Isbir T, Kaytan-Saglam E, Kizir A, Topuz E, Berkkan H. Paraoxonase-1 192/55 polymorphisms and the risk of lung cancer in a Turkish population. Anticancer Res. 2011; 31:2225–2229.
25. Vecka M, Jachymova M, Vavrova L, Kodydkova J, Macasek J, Urbanek M, Krechler T, Slaby A, Duskova J, Muravska A, Zak A. Paraoxonase-1 (PON1) status in pancreatic cancer: relation to clinical parameters. Folia Biol (Praha). 2012; 58:231–237.
26. Schweikert EM, Devarajan A, Witte I, Wilgenbus P, Amort J, Forstermann U, Shabazian A, Grijalva V, Shih DM, Farias-Eisner R, Teiber JF, Reddy ST, Horke S. PON3 is upregulated in cancer tissues and protects against mitochondrial superoxide-mediated cell death. Cell Death Differ. 2012; 19:1549–1560.
27. Salcher S, Hagenbuchner J, Geiger K, Seiter MA, Rainer J, Kofler R, Hermann M, Kiechl-Kohlendorfer U, Ausserlechner MJ, Obexer P. C10ORF10/DEPP, a transcriptional target of FOXO3, regulates ROS-sensitivity in human neuroblastoma. Mol Cancer. 2014; 13:224–240.
28. Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009; 4:1798–1806.
29. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006; 5:769–784.
30. Chavez-Blanco A, Perez-Plasencia C, Perez-Cardenas E, Carrasco-Legleu C, Rangel-Lopez E, Segura-Pacheco B, Taja-Chayeb L, Trejo-Becerril C, Gonzalez-Fierro A, Candelaria M, Cabrera G, Duenas-Gonzalez A. Antineoplastic effects of the DNA methylation inhibitor hydralazine and the histone deacetylase inhibitor valproic acid in cancer cell lines. Cancer Cell Int. 2006; 6:2–10.
31. Horke S, Witte I, Wilgenbus P, Kruger M, Strand D, Forstermann U. Paraoxonase-2 reduces oxidative stress in vascular cells and decreases endoplasmic reticulum stress-induced caspase activation. Circulation. 2007; 115:2055–2064.
32. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455:1061–1068.
33. Chen CY, Chung IH, Tsai MM, Tseng YH, Chi HC, Tsai CY, Lin YH, Wang YC, Chen CP, Wu TI, Yeh CT, Tai DI, Lin KH. Thyroid hormone enhanced human hepatoma cell motility involves brain-specific serine protease 4 activation via ERK signaling. Mol Cancer. 2014; 13:162–178.
34. Chen CY, Chi LM, Chi HC, Tsai MM, Tsai CY, Tseng YH, Lin YH, Chen WJ, Huang YH, Lin KH. Stable isotope labeling with amino acids in cell culture (SILAC)-based quantitative proteomics study of a thyroid hormone-regulated secretome in human hepatoma cells. Mol Cell Proteomics. 2012; 11:M111 011270–19.
35. Singh RK, Cho K, Padi SK, Yu J, Haldar M, Mandal T, Yan C, Cook G, Guo B, Mallik S, Srivastava DK. Mechanism of N-Acylthiourea-mediated activation of human histone deacetylase 8 (HDAC8) at molecular and cellular levels. J Biol Chem. 2015; 290:6607–6619.
36. Singh RK, Mandal T, Balsubramanian N, Viaene T, Leedahl T, Sule N, Cook G, Srivastava DK. Histone deacetylase activators: N-acetylthioureas serve as highly potent and isozyme selective activators for human histone deacetylase-8 on a fluorescent substrate. Bioorg Med Chem Lett. 2011; 21:5920–5923.
37. Weller M, Gorlia T, Cairncross JG, van den Bent MJ, Mason W, Belanger K, Brandes AA, Bogdahn U, Macdonald DR, Forsyth P, Rossetti AO, Lacombe D, Mirimanoff RO, et al. Prolonged survival with valproic acid use in the EORTC/NCIC temozolomide trial for glioblastoma. Neurology. 2011; 77:1156–1164.
38. Bredel M, Bredel C, Juric D, Harsh GR, Vogel H, Recht LD, Sikic BI. Functional network analysis reveals extended gliomagenesis pathway maps and three novel MYC-interacting genes in human gliomas. Cancer Res. 2005; 65:8679–8689.
39. Ververis K, Hiong A, Karagiannis TC, Licciardi PV. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Biologics. 2013; 7:47–60.
40. Li XN, Shu Q, Su JM, Perlaky L, Blaney SM, Lau CC. Valproic acid induces growth arrest, apoptosis, and senescence in medulloblastomas by increasing histone hyperacetylation and regulating expression of p21Cip1, CDK4, and CMYC. Mol Cancer Ther. 2005; 4:1912–1922.
41. Fu J, Shao CJ, Chen FR, Ng HK, Chen ZP. Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro Oncol. 2010; 12:328–340.
42. Kawamoto H, Koizumi H, Uchikoshi T. Expression of the G2-M checkpoint regulators cyclin B1 and cdc2 in nonmalignant and malignant human breast lesions: immunocytochemical and quantitative image analyses. Am J Pathol. 1997; 150:15–23.
43. Campone M, Noel B, Couriaud C, Grau M, Guillemin Y, Gautier F, Gouraud W, Charbonnel C, Campion L, Jezequel P, Braun F, Barre B, Coqueret O, et al. c-Myc dependent expression of pro-apoptotic Bim renders HER2-overexpressing breast cancer cells dependent on anti-apoptotic Mcl-1. Mol Cancer. 2011; 10:110–123.
44. Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001; 20:6969–6978.
45. Rajaraman P, Hutchinson A, Rothman N, Black PM, Fine HA, Loeffler JS, Selker RG, Shapiro WR, Linet MS, Inskip PD. Oxidative response gene polymorphisms and risk of adult brain tumors. Neuro Oncol. 2008; 10:709–715.
46. Zhao P, Zhao L, Zou P, Lu A, Liu N, Yan W, Kang C, Fu Z, You Y, Jiang T. Genetic oxidative stress variants and glioma risk in a Chinese population: a hospital-based case-control study. BMC Cancer. 2012; 12:617–626.
47. Altenhofer S, Witte I, Teiber JF, Wilgenbus P, Pautz A, Li H, Daiber A, Witan H, Clement AM, Forstermann U, Horke S. One enzyme, two functions: PON2 prevents mitochondrial superoxide formation and apoptosis independent from its lactonase activity. J Biol Chem. 2010; 285:24398–24403.
48. Li Y, Tang R, Xu H, Qiu M, Chen Q, Chen J, Fu Z, Ying K, Xie Y, Mao Y. Discovery and analysis of hepatocellular carcinoma genes using cDNA microarrays. J Cancer Res Clin Oncol. 2002; 128:369–379.
49. Ribarska T, Ingenwerth M, Goering W, Engers R, Schulz WA. Epigenetic inactivation of the placentally imprinted tumor suppressor gene TFPI2 in prostate carcinoma. Cancer Genomics Proteomics. 2010; 7:51–60.
50. Witte I, Altenhofer S, Wilgenbus P, Amort J, Clement AM, Pautz A, Li H, Forstermann U, Horke S. Beyond reduction of atherosclerosis: PON2 provides apoptosis resistance and stabilizes tumor cells. Cell Death Dis. 2011; 2:e112.
51. Pise-Masison CA, Radonovich M, Mahieux R, Chatterjee P, Whiteford C, Duvall J, Guillerm C, Gessain A, Brady JN. Transcription profile of cells infected with human T-cell leukemia virus type I compared with activated lymphocytes. Cancer Res. 2002; 62:3562–3571.
52. Kang H, Chen IM, Wilson CS, Bedrick EJ, Harvey RC, Atlas SR, Devidas M, Mullighan CG, Wang X, Murphy M, Ar K, Wharton W, Borowitz MJ, et al. Gene expression classifiers for relapse-free survival and minimal residual disease improve risk classification and outcome prediction in pediatric B-precursor acute lymphoblastic leukemia. Blood. 2010; 115:1394–1405.
53. Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res. 2011; 3:166–179.
54. Bermudez-Lugo JA, Perez-Gonzalez O, Rosales-Hernandez MC, Ilizaliturri-Flores I, Trujillo-Ferrara J, Correa-Basurto J. Exploration of the valproic acid binding site on histone deacetylase 8 using docking and molecular dynamic simulations. J Mol Model. 2012; 18:2301–2310.
55. Akbasak A, Toevs CC, Laske DW. Reconstituted basement membrane (matrigel) enhances the growth of human glioma cell lines in nude mice. J Neurooncol. 1996; 27:23–30.
56. Karen J, Rodriguez A, Friman T, Dencker L, Sundberg C, Scholz B. Effects of the histone deacetylase inhibitor valproic acid on human pericytes in vitro. PLoS One. 2011; 6:e24954.
57. Jung M, Velena A, Chen B, Petukhov PA, Kozikowski AP, Dritschilo A. Novel HDAC inhibitors with radiosensitizing properties. Radiat Res. 2005; 163:488–493.
58. Van Nifterik KA, Van den Berg J, Slotman BJ, Lafleur MV, Sminia P, Stalpers LJ. Valproic acid sensitizes human glioma cells for temozolomide and gamma-radiation. J Neurooncol. 2012; 107:61–67.
59. Chen Y, Tsai YH, Tseng SH. Valproic acid affected the survival and invasiveness of human glioma cells through diverse mechanisms. J Neurooncol. 2012; 109:23–33.
60. Zhou Y, Xu Y, Wang H, Niu J, Hou H, Jiang Y. Histone deacetylase inhibitor, valproic acid, radiosensitizes the C6 glioma cell line. Oncol Lett. 2014; 7:203–208.
61. Kerkhof M, Dielemans JC, van Breemen MS, Zwinkels H, Walchenbach R, Taphoorn MJ, Vecht CJ. Effect of valproic acid on seizure control and on survival in patients with glioblastoma multiforme. Neuro Oncol. 2013; 15:961–967.
62. Zhao GX, Xu LH, Pan H, Lin QR, Huang MY, Cai JY, Ouyang DY, He XH. The BH3-mimetic gossypol and noncytotoxic doses of valproic acid induce apoptosis by suppressing cyclin-A2/Akt/FOXO3a signaling. Oncotarget. 2015; 6:38952–38966. doi: 10.18632/oncotarget.5731.
63. Tsai HC, Wei KC, Tsai CN, Huang YC, Chen PY, Chen SM, Lu YJ, Lee ST. Effect of valproic acid on the outcome of glioblastoma multiforme. Br J Neurosurg. 2012; 26:347–354.
64. Thotala D, Karvas RM, Engelbach JA, Garbow JR, Hallahan AN, DeWees TA, Laszlo A, Hallahan DE. Valproic acid enhances the efficacy of radiation therapy by protecting normal hippocampal neurons and sensitizing malignant glioblastoma cells. Oncotarget. 2015; 6:35004–35022. doi: 10.18632/oncotarget.5253.
65. Chiou HY, Lai WK, Huang LC, Huang SM, Chueh SH, Ma HI, Hueng DY. Valproic acid promotes radiosensitization in meningioma stem-like cells. Oncotarget. 2015; 6:9959–9969. doi: 10.18632/oncotarget.3692.
66. Barker CA, Bishop AJ, Chang M, Beal K, Chan TA. Valproic acid use during radiation therapy for glioblastoma associated with improved survival. Int J Radiat Oncol Biol Phys. 2013; 86:504–509.
67. Brunton LL, Chabner BA, Knollmann BC. Goodman & Gilman’s the Pharmacological Basis of Therapeutics, 12e. Pharmacotherapy of the Epilepsies, Valproic Acid; 2011.