
INTRODUCTION
Paragangliomas (PGLs) are neuroendocrine tumors derived from cells of the parasympathetic or sympathetic ganglia. Parasympathetic PGLs occur most commonly in the head and neck region (carotid body, glomus jugulare, and glomus typanicum), are typically benign, and are rarely associated with catecholamine secretion [1, 2]. PGLs arising from the sympathetic ganglia occur in the abdomen and thorax, often secrete catecholamines, and are associated with a higher risk of malignancy. Pheochromocytomas (PCCs) are generally benign paragangliomas that arise in the chromaffin cells of the adrenal medulla, but are frequently associated with hypertension due to excessive catecholamine secretion [3].
Germline mutations in genes encoding subunits of succinate dehydrogenase (SDH, complex II of the mitochondrial respiratory chain) are the most common genetic cause of PGL/PCC, occurring in up to 25% of all cases [4, 5]. SDH is a heterotetramer consisting of two catalytic subunits, SDHA and SDHB, and two membrane-spanning subunits, SDHC and SDHD. SDHAF2 encodes an accessory factor required for the flavination of SDHA. SDH is an essential component of the tricarboxylic acid (TCA) cycle and the mitochondrial respiratory chain. A puzzling aspect of SDHx-related disease is that despite the close functional relationship of the SDH proteins, mutations lead to marked differences in both tumor location and clinical phenotype.
Another striking difference is that only mutations in SDHD and SDHAF2, both located on chromosome 11, show a parent-of-origin inheritance effect in which carriers develop tumors almost exclusively following paternal transmission of the mutation [6, 7]. An important role in causing this inheritance pattern has been ascribed to the loss of the entire maternal copy of chromosome 11 in SDHD-linked tumors. A cluster of maternally expressed imprinted genes is located on chromosome 11p15, which formed the basis for a hypothesis now known as the ‘Hensen model’. The model proposes that maternal chromosomal 11 loss results in the simultaneous deletion of the SDHD wild type gene and an as yet unidentified exclusively maternally expressed gene (or genes), resulting in tumor formation [7]. This hypothesis predicts that loss of the maternal copy of chromosome 11 might be similarly important for SDHAF2-linked tumors, but has yet to be demonstrated.
To further clarify the role of loss of the maternal copy of 11p in relation to loss of the long arm of chromosome 11 in paragangliomas, we used several genetic approaches to determine the allelic status of chromosome 11 in SDHAF2, SDHD, SDHB, and VHL mutant PGLs/PCCs. SDHB and VHL-related tumors were included because these genes map to other chromosomes than 11. The results show that tumorigenesis in SDHAF2-related tumors are fully compatible with the Hensen model, and that 11p loss is less important in SDHB-related tumors than for the other three tumor subgroups.
RESULTS
Loss of heterozygosity in SDHAF2-related PGL
Since SDHAF2-related PGLs show a parent-of-origin effect, similar to SDHD mutations [7], we hypothesized that their tumorigenesis might also critically depend on loss of the maternal copy of chromosome 11. We first established SDHAF2 mutation status in germline DNA from 9 patients from SDHAF2-related families. All patients showed a missense mutation of SDHAF2, c.232G>A (p.Gly78Arg), in a conserved region of the gene [8]. We then sequenced DNA isolated from tumors of all nine patients, and compared this to matched DNA from blood samples. This comparison showed that the wild type allele (guanine (G) nucleotide – arrow, Figure 1A) is underrepresented in tumor DNA (Figure 1B), and the mutant allele is (adenine (A) nucleotide) overrepresented, indicating loss of the wild type allele (loss of heterozygosity - LOH) in the tumor. Partial retention of the wild type allele is characteristic of LOH in PGLs, and is due to admixture with normal cells that proliferate together with tumor cells [9].
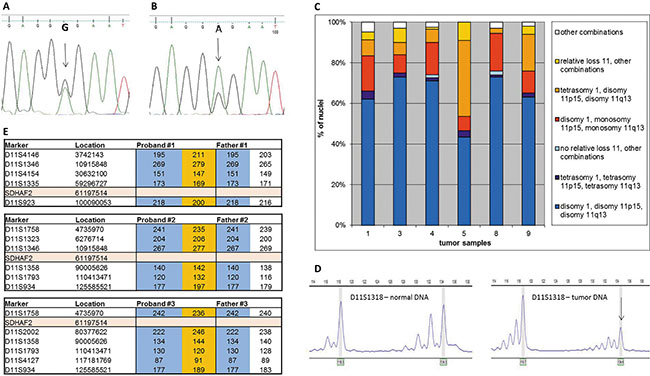
Figure 1: Sanger sequencing of SDHAF2 in normal (A) and tumor (B) DNA. Arrows indicate the relevant nucleotides in the heterozygous patient. (C) Interphase FISH results from isolated whole nuclei isolated from paraffin-embedded material of 6 SDHAF2-related patients. Frequency distribution of signals obtained with probes for centromere 1 (PUC1.77), telomere 11p (371C18) and telomere 11q (469N6). (D) A typical profile of microsatellite marker alleles showing loss of heterozygosity. Arrows indicate the allele lost. (E) Chromosome 11 haplotypes of family members. Microsatellite markers are shown with genomic location and the position of SDHAF2 is indicated. Alleles in orange blocks represent the probable disease haplotype, present in the proband and absent in the father. Alleles in blue blocks represent alleles from the father.
Loss of chromosome 11 in SDHAF2-related PGL
To investigate whether the entire copy of chromosome 11 is lost in SDHAF2-related tumors, we performed fluorescence in situ hybridization (FISH) studies on 6 SDHAF2-linked tumors using a probe for the centromere of chromosome 1 as a ploidy reference as described earlier [7], and BAC probes for the subtelomeric regions of 11p and 11q. Simultaneous loss of both probes located on chromosome 11 relative to centromere 1 was found in all SDHAF2-related PGLs, in 15–44% of nuclei (Figure 1C, red and orange). Loss of one of the two probes located on chromosome 11 relative to the other was observed in only a very small minority of nuclei (< 0.5%), demonstrating that the observed relative loss involves the entire copy of chromosome 11.
Parental origin of chromosomal loss in SDHAF2-related PGL
To further evaluate LOH in all SDHAF2-related tumors and study the parental origin of chromosomal loss, tumor DNA was analyzed for LOH using 24 highly polymorphic microsatellite markers selected from a custom microsatellite database. In the 9 SDHAF2-related PGLs investigated, 8 (89%) showed chromosome 11-wide LOH, with allelic imbalance ratios of < 0.7 or >1.3 (Figure 1D, Supplementary Table 2). Parental blood DNA samples were available for 3 SDHAF2-related patients. Microsatellite analysis of parental DNA confirmed that the copy of chromosome 11 lost in all 3 SDHAF2-mutated tumors was maternal (Figure 1E).
As parental DNA was not available for the remaining cases, we investigated the methylation status of two 11p15.5 DMRs, KvDMR (maternal allele methylated) and H19-DMR (paternal allele methylated). In the presence of two chromosomes with normal methylation levels, each of these DMRs should show an average 50% methylation rate (one chromosome methylated, opposite chromosome unmethylated). This analysis showed hypermethylation of the H19-DMR and hypomethylation in the KvDMR in 8 (89%) SDHAF2 mutant PGLs (Table 1). These findings are consistent with loss of the maternal allele [10]. The mean methylation rates (± sd) of 7 SDHAF2-related tumors differed significantly from the normal methylation rates in matched blood DNA (H19-DMR 0.83 ± 0.13 versus 0.52 ± 0.03 (p = 0.008) and KvDMR 0.06 ± 0.07 versus 0.50 ± 0.10 (p = 0.006)). In each of the tumors with chromosome 11 loss, the ratio of the methylation rate of H19-DMR/KvDMR was > 3, while the ratio of H19-DMR/KvDMR in blood DNA was ~1. The one SDHAF2-related tumor without LOH for chromosome 11 by microsatellite marker analysis, demonstrated normal methylation of both H19-DMR and KvDMR, comparable to blood DNA.
Table 1: Methylation status of 11p15 imprinted regions KvDMR and H19-DMR in SDHAF2 mutant PGLs with and without chromosome 11 LOH
Gene mutation and sample ID |
11p15 status |
11q12 status |
Methylation rate of tumor DNA |
H19-DMR/KvDMR methylation rate Ratio of tumor DNA |
Methylation rate of matched blood DNA |
|---|---|---|---|---|---|
SDHAF2 (1) |
LOH |
LOH (maternal) |
KvDMR: 0.07 |
8.1 |
|
SDHAF2 (2) |
LOH |
LOH |
KvDMR: 0.01 |
> 10 |
KvDMR: 0.59 |
SDHAF2 (3) |
LOH |
LOH |
KvDMR: 0.001 |
> 10 |
|
SDHAF2 (4) |
LOH |
n.i |
KvDMR: 0.03 |
>10 |
|
SDHAF2 (5) |
LOH |
LOH (maternal) |
KvDMR: 0.08 |
9.8 |
KvDMR: 0.47 |
SDHAF2 (6) |
No LOH |
No LOH |
KvDMR: 0.63 |
0.9 |
KvDMR: 0.57 |
SDHAF2 (7) |
LOH |
LOH (maternal) |
KvDMR: 0.22 |
3.7 |
KvDMR: 0.37 |
SDHAF2 (8) |
LOH |
LOH |
KvDMR: 0.03 |
> 10 |
|
SDHAF2 (9) |
LOH |
LOH |
KvDMR: 0.0001 |
- |
LOH: loss of heterozygosity. n.i : not informative.
Frequent loss of maternal chromosome 11 in SDHD and VHL-related tumors
To investigate the extent and nature of chromosome 11 loss across the various paraganglioma subgroups, we assembled a panel of 26 SDHD, 13 SDHB, and 8 VHL-related PGLs/PCCs. Of the 26 SDHD-related tumors investigated using polymorphic microsatellite marker analysis, LOH at all informative markers of chromosome 11 was observed in 22 (85%) tumors (Table 2). In four SDHD-related tumors almost all markers showed allele ratios between 0.8 and 1, indicating retention of heterozygosity (Supplementary Table 3 – tumor 23, 26, 27 and 36). Methylation analysis of H19-DMR and KvDMR demonstrated hypermethylation of H19-DMR and hypomethylation of KvDMR in all SDHD mutant PGLs with LOH for chromosome 11, consistent with loss of the maternal allele and significantly different from methylation rates of H19-DMR (p = 0.004) and KvDMR (p = 0.002) in blood DNA (Table 2). In the four SDHD-related tumors without chromosomal loss, the ratio of H19-DMR to KvDMR was 1, comparable to blood DNA.
Table 2: Methylation status of KvDMR and H19-DMR in SDHD mutant PGLs with and without chromosome 11 LOH
Gene mutation and sample ID |
11p15 status |
11q23 status |
KvDMR methylation rate |
H19-DMR methylation rate |
H19-DMR/KvDMR methylation rate. Ratio |
|---|---|---|---|---|---|
SDHD (10) |
LOH |
LOH |
0.01 |
0.62 |
> 10 |
SDHD (11) |
LOH |
LOH |
0.0 |
1.0 |
> 10 |
SDHD (12) |
LOH |
LOH |
0.01 |
0.96 |
>10 |
SDHD (13) |
LOH |
LOH |
0.10 |
0.86 |
8.6 |
SDHD (14) |
LOH |
n.i |
0.04 |
0.97 |
> 10 |
SDHD (15) |
LOH |
LOH |
0.07 |
1 |
> 10 |
SDHD (16) |
LOH |
n.i |
0.02 |
0.75 |
>10 |
SDHD (17) |
n.i |
LOH |
0.03 |
0.45 |
> 10 |
SDHD (18) |
LOH |
LOH |
0.02 |
0.96 |
> 10 |
SDHD (19) |
LOH |
n.i |
0.0 |
0.91 |
> 10 |
SDHD (20) |
LOH |
LOH |
0.0 |
0.87 |
> 10 |
SDHD (21) |
LOH |
LOH |
0.08 |
0.76 |
> 10 |
SDHD (22) |
LOH |
n.i |
0.14 |
0.90 |
6.4 |
SDHD (23) |
No LOH |
No LOH |
0.51 |
0.56 |
1 |
SDHD (24) |
LOH |
LOH |
0.01 |
0.7 |
> 10 |
SDHD (25) |
LOH |
LOH |
0.04 |
0.86 |
> 10 |
SDHD (26) |
No LOH |
No LOH |
0.57 |
0.59 |
1 |
SDHD (27) |
No LOH |
No LOH |
0.53 |
0.57 |
1 |
SDHD (28) |
LOH |
LOH |
0.17 |
0.66 |
3.9 |
SDHD (29) |
LOH |
LOH |
0.15 |
0.68 |
4.4 |
SDHD (30) |
LOH |
LOH |
0.21 |
0.70 |
3.3 |
SDHD (31) |
LOH |
LOH |
0.16 |
0.68 |
4.4 |
SDHD (32) |
LOH |
LOH |
0.12 |
0.71 |
5.9 |
SDHD (33) |
LOH |
LOH |
0.13 |
0.65 |
5.2 |
SDHD (34) |
LOH |
LOH |
0.11 |
0.70 |
6.4 |
SDHD (35) |
LOH |
LOH |
0.13 |
0.69 |
5.4 |
SDHD (36) |
No LOH |
No LOH |
0.48 |
0.61 |
1.2 |
LOH: loss of heterozygosity. n.i : not informative.
All 8 VHL-associated PCCs demonstrated loss of chromosome 11 (Table 3), although in 3 tumors, microsatellite markers were uninformative at 11p15 or at 11q23, while other markers on chromosome 11 showed allelic imbalance ratios of < 0.7 or > 1.3 (Supplementary Table 4).
Table 3: Methylation status of KvDMR and H19-DMR in VHL mutant PCCs with and without chromosome 11 LOH
Gene mutation and sample ID |
11p15 status |
11q23 status |
KvDMR methylation rate |
H19-DMR methylation rate |
H19-DMR/KvDMR Methylation rate. Ratio |
|---|---|---|---|---|---|
VHL (37) |
LOH |
n.i |
0 |
0.65 |
> 10 |
VHL (38) |
LOH |
LOH |
0.01 |
0.89 |
> 10 |
VHL (39) |
LOH |
LOH |
0.03 |
0.75 |
> 10 |
VHL (40) |
LOH |
LOH |
0.01 |
1 |
> 10 |
VHL (41) |
LOH |
LOH |
0.01 |
1 |
> 10 |
VHL (42) |
LOH |
LOH |
0.01 |
0.83 |
> 10 |
VHL (43) |
n.i |
LOH |
0.01 |
failed |
- |
VHL (44) |
n.i |
LOH |
0 |
failed |
- |
LOH: loss of heterozygosity. n.i : not informative
Methylation status of H19-DMR and KvDMR revealed loss of the maternal copy of chromosome 11 in 6 of 8 (75%) VHL-associated PCCs (Table 3), while the methylation status of the H19-DMR could not be determined in 2 (25%) tumors. However, both these tumors showed hypomethylated KvDMR, suggestive of maternal allele loss.
Low frequency maternal chromosome 11 loss in SDHB-related tumors
Almost all (92%) SDHB-mutated tumors retained heterozygosity in the 11q region, while 4 (31%) tumors showed LOH exclusively in the 11p15 region, in 1 tumor microsatellite markers were uninformative at 11p15 (Table 4). Moreover, in all cases with LOH, this LOH affected multiple small regions of chromosome 11 alternated with regions of retention of heterozygosity (Supplementary Table 5). Methylation analysis of H19-DMR and KvDMR in 8 (62%) SDHB mutant tumors showed the ratio of H19-DMR to KvDMR was ~1, comparable to blood DNA. Of the 4 tumors with indications for LOH of 11p15, 1 (tumor 51) showed hypermethylation of H19-DMR and hypomethylation of KvDMR. Tumors 48 and 50 showed hypermethylation of H19-DMR but normal methylation of KvDMR, resulting in a ratio of H19-DMR/KvDMR < 3 (Table 4). In the remaining SDHB mutant tumor (tumor 49), no methylation was detected at KvDMR, whereas the methylation rate of H19-DMR was normal. These results are in stark contrast to the unequivocal findings in SDHAF2, SDHD and VHL-related PGLs/PCCs. To investigate whether SDHB-mutated tumors show a scattered pattern of LOH on other chromosomes, we used microsatellite markers from chromosomes known to be affected in PGL/PCC [11], including chromosomes 1, 3, 5, 14 and 17. All SDHB mutant tumors showed LOH for chromosome 1p, presumably affecting the SDHB wild type allele (Figure 2). In addition, LOH of other chromosomes, defined as allelic imbalance ratios of < 0.7 or > 1.3, was observed in most SDHB-related tumors, in contrast to SDHD mutant tumors (Figure 2, Supplementary Table 6).
Table 4: Methylation status of KvDMR and H19-DMR in SDHB mutant tumors with and without chromosome 11 LOH
Gene mutation and sample ID |
11p15 status |
11q23 status |
KvDMR methylation rate |
H19-DMR methylation rate |
H19-DMR/KvDMR methylation rate Ratio |
Methylation rate of matched blood DNA |
|---|---|---|---|---|---|---|
SDHB (45) |
No LOH |
No LOH |
0.39 |
0.45 |
1.1 |
KvDMR: 0.39 |
SDHB (46) |
No LOH |
No LOH |
0.68 |
0.60 |
0.9 |
|
SDHB (47) |
No LOH |
No LOH |
0.48 |
0.31 |
0.6 |
|
SDHB (48) |
LOH |
No LOH |
0.57 |
0.81 |
1.4 |
KvDMR: 0.30 |
SDHB (49) |
LOH |
No LOH |
0.09 |
0.43 |
4.8 |
KvDMR: 0.37 |
SDHB (50) |
LOH |
No LOH |
0.33 |
0.91 |
2.8 |
|
SDHB (51) |
LOH |
LOH |
0.04 |
0.86 |
> 10 |
|
SDHB (52) |
n.i |
No LOH |
0.004 |
0.47 |
> 10 |
|
SDHB (53) |
No LOH |
No LOH |
0.41 |
0.69 |
1.7 |
|
SDHB (54) |
No LOH |
No LOH |
0.89 |
0.97 |
1.1 |
KvDMR: 0.50 |
SDHB (55) |
No LOH |
No LOH |
0.91 |
0.92 |
1.0 |
|
SDHB (56) |
No LOH |
No LOH |
0.46 |
0.52 |
1.1 |
|
SDHB (57) |
No LOH |
No LOH |
0.23 |
0.33 |
1.4 |
LOH: loss of heterozygosity. n.i : not informative
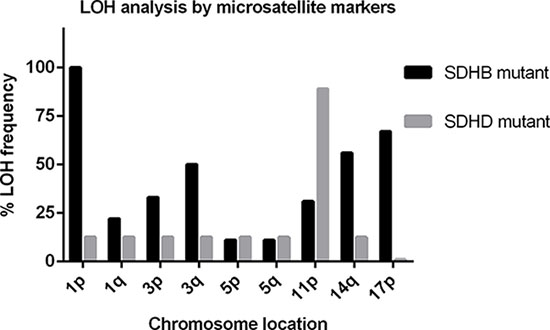
Figure 2: Frequency (%) plot of loss of heterozygosity (LOH) of different chromosomes in SDHB (black bars) and SDHD (grey bars) mutant tumors, determined by microsatellite marker analysis. A higher frequency of LOH of chromosomes 1, 3, 14, and 17 is observed in SDHB-related tumors compared to SDHD-related tumors. LOH of chromosome 11p is the most frequent event in SDHD mutant tumors.
Greater genomic instability in SDHB tumors compared to SDHD and SDHAF2-related tumors
To further explore genomic instability in these tumors, we analyzed genome-wide copy number changes and LOH in a total of 28 tumors (12 SDHD, 4 SDHAF2, 9 SDHB and 3 VHL-related PGLs/PCCs) using SNP array analysis. In agreement with our microsatellite marker results and with other studies [11–15], the most frequent copy number alterations in these tumors were deletions of 1p (48%), 3p/q (28%/32%), and 11p/q (88%/68%) (Figure 3). Although the SDHB, VHL, SDHD, and SDHAF2 genes are located in these chromosomal regions, losses occurred independently of the presence of germline mutations in these genes (Supplementary Figure 2). SNP array analysis revealed patterns of chromosomal gains and losses that were more heterogeneous in SDHB mutant tumors compared to SDHD and SDHAF2-mutated tumors. We evaluated the level of chromosomal instability in each tumor by calculating the ‘Fraction of Aberrant Arms’ (i.e. the proportion of chromosome arms altered over more than 40% of their length [16]). This analysis confirmed the greater degree of genome instability in SDHB mutant tumors (mean 12%) compared to SDHD mutant tumors (mean 4%) or to SDHAF2 mutant tumors (mean 4.5%). One SDHD and two SDHB-linked tumors appeared to be tetraploid as determined by the 2Log (test/reference) ratios (Supplementary Figure 3). The most commonly affected chromosomal regions in SDHB-related tumors were gain of 1q (57%), chromosome 7 (28%) and 17q (28%), and loss of 1p (100%) (SDHB locus) and 17p (57%). These regions have also been shown to be affected in RET, NF1 and sporadic paragangliomas/pheochromocytomas [11], indicating the potential presence of driver genes on these autosomes.
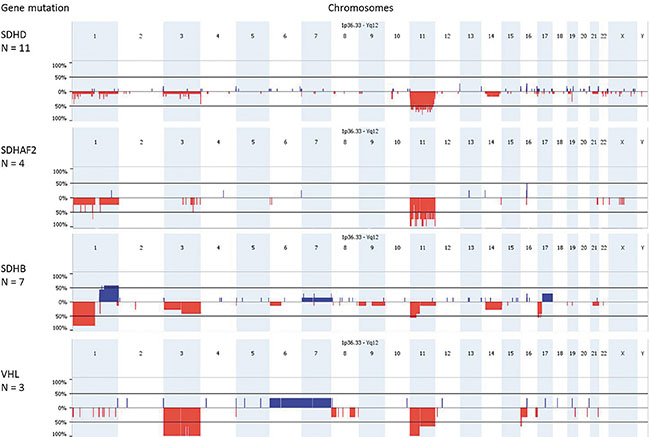
Figure 3: SNP array results of SDHx and VHL-mutated paragangliomas/pheochromocytomas. Genomic frequency plots of gains (in blue) and deletions (in red) among 11 SDHD, 4 SDHAF2, 7 SDHB and 3 VHL-mutated tumors, obtained with Nexus Express. SDHD, SDHAF2 and VHL mutant paragangliomas/pheochromocytomas have the highest frequency of 11p loss, while the loss of 1p is a frequent event in SDHB mutant tumors. The X-axis shows the genomic position along the chromosomes and the Y-axis shows the frequency (%) of copy number gains and losses.
DISCUSSION
In this study, we showed that loss of maternal chromosome 11 is also a cardinal feature of SDHAF2-linked paragangliomas. The selective loss of maternal chromosome 11 conforms to the Hensen model and explains why SDHAF2-linked tumors arise in principal upon paternal transmission of the mutation, comparable to most SDHD-linked tumors [7, 12, 17]. The presence of a paternal parent-of-origin effect in both SDHAF2 and SDHD-related PGLs, a phenomenon absent in other SDH-related PGLs, argues that their location on chromosome 11 reveals the fundamental role of another chromosome 11 gene in tumorigenesis.
The specific loss of maternal chromosome 11 in VHL-related PCCs suggests that a maternally-expressed gene on chromosome 11 also plays a crucial role in these tumors. In this case, no parent-of-origin effect for the VHL mutation is predicted because loss of the wild type VHL allele on chromosome 3 can occur independently of maternal chromosome 11 loss. These findings agree with earlier reports [10, 11, 14, 18] showing a high frequency of chromosome 11p loss in VHL mutant tumors. It is likely that loss of chromosome 11p confers further growth advantage to the tumors besides the inactivation of the VHL gene. Interestingly, chromosome 11 loss in VHL-related tumors is specific to PGL/PCC and has not been shown in VHL-related renal cell carcinomas [19, 20].
The 11p15 region contains several imprinted genes that are exclusively maternally expressed and paternally silenced. LOH of maternal chromosome 11 will result in complete loss of expression of these genes. Our results lend further support to the notion that loss of an as yet unidentified locus (or loci) in 11p15 could contribute to tumor formation in SDHD, SDHAF2 and VHL-related PGLs/PCCs.
Of the potential candidate genes, the paternally expressed growth promoter insulin like growth factor 2 (IGF2) and the maternally expressed candidate tumor suppressor genes CDKN1C and H19, have been most consistently implicated in imprinting disorders, such as Beckwith-Wiedemann Syndrome and Silver-Russell Syndrome [21]. Interestingly, we recently found loss of CDKN1C and SLC22A18 expression in SDHD-related PGLs compared with normal carotid body tissue and established that knockdown of SDHD together with SLC22A18 or with CDKN1C led to small increases in cell proliferation and resistance to apoptosis in neuronal cells [22]. Results from the cell line-based functional assays were further supported by the finding that SDHD mutant tumors with either retention or loss of chromosome 11 showed equally low levels of SLC22A18 and CDKN1C protein expression [22]. SDHx-related tumors are associated with hypermethylation and histone methylation, suggesting a possible mechanism underlying the lowered expression of SLC22A18 and/or CDKN1C in SDHD mutant tumors with retention of chromosome 11. Further studies are needed to clarify the role of imprinted genes located on 11p15 in tumor development of SDHx and VHL mutant PGL/PCC.
While partial or entire loss of chromosome 11 is a signature event in a proportion of SDHB-related tumors, many SDHB tumors exhibit gains and losses confined to other chromosomes. Compared to SDHD and SDHAF2, SDHB tumors show a more complex pattern of chromosome 11 loss and characteristic changes affecting other chromosomes. Closer analysis to the allele ratios of various microsatellite markers in all tumors revealed a scattered segmental LOH pattern (Supplementary Table 5). While loss might be partly masked by tumor heterogeneity and copy neutral LOH, either of which could impair the detection of genomic alterations [23], the complex pattern of chromosome 11 loss we observed was specific to SDHB tumors. Nevertheless, 4 (31%) SDHB-related tumors showed loss of maternal chromosome 11p, perhaps signifying a role for the same modifier genes that play such a prominent role in SDHD and SDHAF2 tumors. A low frequency of chromosome 11 loss in SDHB-related tumors is in agreement with previous reports [11, 15]. While a proportion of the greater heterogeneity of chromosomal gains and losses we observed in SDHB mutant tumors might simply be a byproduct of genomic instability, many changes are recurrent and thus apparently under the influence of selection, especially losses on chromosomes 1p, 3q, 11p, and 17p and somatic gain of chromosome 1q. One or more modifier genes on these autosomes may work in specific synergistic combinations to initiate or promote tumor growth. These recurring and often non-overlapping chromosomal changes also point to a potential redundancy in modifiers, and as such, altered expression of different groups of modifier genes might be involved in SDHB tumorigenesis. Analysis of a much larger number of SDHB tumors will be required to resolve this question.
Interestingly, recent work [24, 25] showed that engineering in vitro the loss of chromosome 8p in cells alters fatty acid and ceramide metabolism. The shift in lipid metabolism triggered tumorigenic potential under stress conditions. Such a complex metabolic shift is difficult to ascribe to a dosage effect of a single gene, and is more likely the result of multiple genes on 8p coordinately undergoing a dosage change. This mechanism might also be at work in SDHx-related tumors, with chromosome 11p loss necessary and sufficient to trigger SDHD and SDHAF2 tumorigenesis, whereas SDHB tumors require amplification or deletion of multiple driver genes located on different chromosomes.
This speculation is supported by the striking difference in penetrance. A characteristic feature of SDHD and SDHAF2-related mutations is very high penetrance (90–100%) [8, 26], in contrast to SDHB mutations that have an estimated penetrance of only 20–30% [27–29]. This striking difference cannot be readily explained by functional differences between the respective proteins and therefore suggests a role for genetic effects, such as chromosomal location. In this scenario, tumorigenesis in SDHD and SDHAF2 mutation carriers requires only a single somatic genetic event (chromosome 11 loss), as opposed to the two or more independent somatic events required in SDHB mutation carriers (loss of the respective wild type allele, together with loss or gain of other chromosomal regions). In conclusion, our results clearly show that SDHB tumors follow a more complex and possibly different path to tumorigenesis compared to SDHD and SDHAF2-related PGLs, involving loss or gain of a greater proportion of the genome.
Despite the apparently integrated function of the SDH subunits, mutations in individual subunit genes result in a number of striking genetic, phenotypic and clinical differences.
Our data now highlight further differences between SDHB-related PGL/PCC compared to SDHD, SDHAF2 or VHL mutant PGL/PCC in terms of maternal chromosome 11 loss and additional genomic instability. Loss of maternal chromosome 11 is a highly specific and statistically significant event in the latter tumors, suggesting an important role for a still unidentified chromosome 11 factor in the genesis of paraganglioma.
MATERIALS AND METHODS
Patients and samples
A total of 44 formalin-fixed, paraffin-embedded (FFPE) tissue samples of PGL/PCC from 41 different patients were used for DNA extraction, including 12 SDHB, 16 SDHD, 9 SDHAF2 and 8 VHL-related tumors. In addition, we included 12 fresh frozen tumor samples for DNA extraction; 11 SDHD and 1 SDHB-mutated tumors. The histology of all tumors was reviewed (JVMGB, JPB, ASH) and the mutation detection was confirmed by routine SDHA and SDHB immunohistochemical staining, as described previously [30] (Supplementary Figure 1). For 7 SDHAF2 mutant tumors and 4 SDHB mutant tumors, paired blood lymphocyte DNA samples were available. In addition, parental blood lymphocyte DNA was available for 3 SDHAF2-linked patients. Following the original identification of the SDHAF2 mutation, c.232G>A, p.Gly78Arg, all patients were analyzed by sequencing for the presence of the mutation [8]. The following primers were used for the amplification of exon 2 of the SDHAF2 gene: 5′-GTTGACCTTCCCAGGCTC-3′ (forward) and 5′-GAGGTTCAGCTGCTTTTCTG-3′ (reverse). Thirty nanograms of genomic DNA from each patient was amplified, and primer annealing was performed at 58°C. PCR fragments were purified using the Nucleospin gel and PCR clean-up kit (Macherey-Nagel). Sequencing was performed using standard protocols. Sequences were analyzed using the Mutation Surveyor software package (Softgenetics).
The SDHB-related samples were obtained from Radboud UMC, Nijmegen, The Netherlands, from UMCG, Groningen, The Netherlands and from University Hospital Southampton, UK. SDHD-related samples were obtained from the LUMC, Leiden, The Netherlands. SDHAF2-related samples were obtained from Radboud UMC and LUMC. VHL-related samples were obtained from the Erasmus MC, Rotterdam, The Netherlands. Written informed consent was obtained for DNA testing, further analyses and publication of all results, according to protocols approved by the Ethics Committees of the Erasmus MC, Radboud UMC, and University Hospital Southampton. Tissues from UMCG were used anonymously in accordance with the code for adequate secondary use of tissue, code of conduct: “Proper Secondary Use of Human Tissue” established by the Dutch Federation of Medical Scientific Societies (http://www.federa.org). Oral informed consent was obtained from patients according to protocols approved by the Ethics Committees of the LUMC, Protocol P12.082. Patients’ clinical and genetic data of the tumors included in our study is provided in Supplementary Table 1.
Triple colour interphase FISH on nuclei isolated from paraffin-embedded tissue
The PUC1.77 probe for the centromeric alphoid repeat DNA of chromosomes 1 was kindly provided by Dr J Wiegant (Department of Molecular Cell Biology, LUMC, Leiden, The Netherlands) [31, 32]. The BAC probes 371C18 (telomere 11p) and 469N6 (telomere 11q) were obtained from the Children’s Hospital Oakland Research Institute (Peter de Jong BAC library RP11). All probes were labelled by standard nick translation with biotin-16-aUTP, digoxigenin-11-dUTP or fluorescein-12-dUTP (Roche, Basel, Switzerland).
Isolation of intact nuclei, hybridization and immunodetection were performed as previously described [33], with some modifications. The hybridization mix contained 50% formamide, 3 ng/μl of each of the three probes (either PUC1.77, pLC11A and 3F7 or PUC1.77, 371C18 and 469N6) and a 50-fold excess of human Cot-1 DNA (Invitrogen Life tech., Paisley, UK). A volume of 5 μl of the mix was applied directly onto the slides and covered with an 18 × 18 mm2 coverslip. After a denaturation step of 8 min at 80°C, the slides were incubated overnight at 37°C in a moisture chamber. A total of 200 nuclei were analysed for each sample and probe combination by two independent investigators (EFH and ESJ).
LOH analysis by microsatellite genotyping
Representative tumor areas from FFPE samples were selected to punch 3 cores of 0.6 mm in diameter for DNA isolation. Microdissection was performed on 8 SDHB-related tumors, using a total of two 10 μm thick sections for each case. A tumor percentage of greater than 80% was achieved for all tumors. FFPE and fresh frozen tumor samples were incubated overnight with proteinase K at 60°C and DNA was isolated using the Qiagen FFPE DNA kit or QIAamp DNA Mini Kit (Qiagen Benelux B.V., Venlo, The Netherlands), respectively, according to the manufacturer’s instructions. Tumor and blood samples were genotyped for microsatellite markers located on chromosome 11, as described in the results section. Primer sequences of the microsatellite markers are described in Supplementary Table 7. For each marker, 40 ng of DNA was amplified over 40 cycles using FastStar Taq DNA Polymerase (Roche). Forward primers were labeled with 6-FAM, HEX or NED fluorophore (Sigma-Aldrich, St. Louis, MO, USA). Amplicons of microsatellite markers were run on an ABI 3730 genetic analyzer and data were analyzed using Gene Marker software (Soft Genetics, State College, PA 16803, USA), using ABI GeneScan Rox 400 as the internal size standards. LOH was calculated using the allelic imbalance ratio: AIR = (Tumor1/Tumor2)/(Normal1/Normal2). Tumors were regarded as positive for LOH when the mean allele ratio between tumor and blood was < 0.7 for all informative markers, as described earlier [34]. In cases where no matching blood lymphocyte DNA sample was available, allele peak ratios were compared to DNA samples with the same or very similar allele combinations. Some markers were either not informative in the patient or did not perform well enough on tumor DNA samples to give a reliable result and were therefore excluded.
Methylation analysis of H19-DMR and KvDMR
Bisulfite conversion of 250 ng of tumor DNA was performed with the EZ DNA Methylation Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s instructions. Bisulfite treated DNA was amplified by PCR with primers specific for modified DNA, designed using Methprimer [35]. Primer sequences for the H19-DMR were 5′-GGTTT TAGTGTGAAATTTTTTT-3′ (forward) and 5′-CCATAAATATCCTATTCCCAAATAAC-3′ (reverse) and for the KvDMR 5′-TTGAGGAGTTTTTTGGAGGTT-3′ (forward) and 5′-ACCC AACCAATACCTCATAC-3′ (reverse). The PCR program consisted of an initial denaturation step at 94°C for 15 minutes followed by 44 cycles of 20 seconds at 94°C, 30 seconds at 55°C for the KvDMR and 52.5°C for the H19-DMR, followed by 5 minutes at 72°C. PCR fragments were purified using the Nucleospin gel and PCR clean-up kit (Macherey-Nagel, Düren, Germany). Sanger sequencing was performed using standard protocols and methylation rates were evaluated using ESME software [36].
Oncoscan analysis
Twelve SDHD, four SDHAF2, nine SDHB, and three VHL mutant tumors (Supplementary Table 1) were further investigated for whole genome copy number by OncoScan analysis (molecular inversion probe technology), as described in [37]. This array consists of ~335.000 probes of which the majority (~283.000) are SNP-based. DNA was processed by the Affymetrix Research Services Laboratory (Santa Clara, California, USA) using the OncoScan™ FFPE Assay. The normalized OncoScan data (2Log (test/reference)-ratios and B-allele frequency plots) were analyzed with the Nexus Express software version 3.1 (Biodiscovery, Inc, El Segundo, California, USA) for copy number calling.
Statistical analysis
IBM SPSS Statistics 20.0 for Windows software package (SPSS, Armonk, NY: IBM Corp) was used to analyze the results. Statistical significance between two groups was determined by Mann-Whitney U test. P < 0.05 was considered statistically significant.
ACKNOWLEDGMENTS AND FUNDING
We thank the clinicians and patients involved for their cooperation. This work was supported by the Dutch Cancer Society (Grant 2011-5025 to JPB/PD).
CONFLICTS OF INTEREST
The authors have nothing to disclose.
REFERENCES
1. Jansen JC, van den BR, Kuiper A, Van Der Mey AG, Zwinderman AH, Cornelisse CJ. Estimation of growth rate in patients with head and neck paragangliomas influences the treatment proposal. Cancer. 2000; 8812:2811–2816.
2. Lack EE, Cubilla AL, Woodruff JM. Paragangliomas of the head and neck region. A pathologic study of tumors from 71 patients. Hum Pathol. 1979; 102:191–218.
3. Galan SR, Kann PH. Genetics and molecular pathogenesis of pheochromocytoma and paraganglioma. Clin Endocrinol.(Oxf). 2013; 782:165–175.
4. Baysal BE, Willett-Brozick JE, Lawrence EC, Drovdlic CM, Savul SA, McLeod DR, Yee HA, Brackmann DE, Slattery WH, III, Myers EN, Ferrell RE, Rubinstein WS. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J Med Genet. 2002; 393:178–183.
5. Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002; 34619:1459–1466.
6. Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CW, Schiffman JD, Bentz BG, Gygi SP, Winge DR, Kremer H, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009; 3255944:1139–1142.
7. Hensen EF, Jordanova ES, van Minderhout IJHM, Hogendoorn PCW, Taschner PEM, van der Mey AGL, Devilee P, Cornelisse CJ. Somatic loss of maternal chromosome 11 causes parent-of-origin-dependent inheritance in SDHD-linked paraganglioma and phaeochromocytoma families. Oncogene. 2004; 2323:4076–4083.
8. Kunst HP, Rutten MH, De Monnink JP, Hoefsloot LH, Timmers HJ, Marres HA, Jansen JC, Kremer H, Bayley JP, Cremers CW. SDHAF2 (PGL2-SDH5) and Hereditary Head and Neck Paraganglioma. Clin Cancer Res. 2011; 172:247–254.
9. Douwes Dekker PB, Corver WE, Hogendoorn PC, Van Der Mey AG, Cornelisse CJ. Multiparameter DNA flow-sorting demonstrates diploidy and SDHD wild-type gene retention in the sustentacular cell compartment of head and neck paragangliomas: chief cells are the only neoplastic component. J Pathol. 2004; 2024:456–462.
10. Margetts CDE, Astuti D, Gentle DC, Cooper WN, Cascon A, Catchpoole D, Robledo M, Neumann HPH, Latif F, Maher ER. Epigenetic analysis of HIC1, CASP8, FLIP, TSP1, DCR1, DCR2, DR4, DR5, KvDMR1, H19 and preferential 11p15.5 maternal-allele loss in von Hippel-Lindau and sporadic phaeochromocytomas. Endocrine-Related Cancer. 2005; 121:161–172.
11. Castro-Vega LJ, Letouze E, Burnichon N, Buffet A, Disderot PH, Khalifa E, Loriot C, Elarouci N, Morin A, Menara M, Lepoutre-Lussey C, Badoual C, Sibony M, et al. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat Commun. 2015; 6:6044.
12. Dannenberg H, de Krijger RR, Zhao J, Speel EJ, Saremaslani P, Dinjens WN, Mooi WJ, Roth J, Heitz PU, Komminoth P. Differential loss of chromosome 11q in familial and sporadic parasympathetic paragangliomas detected by comparative genomic hybridization. Am J Pathol. 2001; 1586:1937–1942.
13. Edstrom E, Mahlamaki E, Nord B, Kjellman M, Karhu R, Hoog A, Goncharov N, Teh BT, Backdahl M, Larsson C. Comparative genomic hybridization reveals frequent losses of chromosomes 1p and 3q in pheochromocytomas and abdominal paragangliomas, suggesting a common genetic etiology. Am J Pathol. 2000; 1562:651–659.
14. Lui WO, Chen J, Glasker S, Bender BU, Madura C, Khoo SK, Kort E, Larsson C, Neumann HP, Teh BT. Selective loss of chromosome 11 in pheochromocytomas associated with the VHL syndrome. Oncogene. 2002; 217:1117–1122.
15. Sandgren J, Diaz de ST, Andersson R, Menzel U, Piotrowski A, Nord H, Backdahl M, Kiss NB, Brauckhoff M, Komorowski J, Dralle H, Hessman O, Larsson C, et al. Recurrent genomic alterations in benign and malignant pheochromocytomas and paragangliomas revealed by whole-genome array comparative genomic hybridization analysis. Endocr.Relat Cancer. 2010; 173:561–579.
16. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, Clement B, Balabaud C, Chevet E, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012; 446:694–698.
17. Riemann K, Sotlar K, Kupka S, Braun S, Zenner HP, Preyer S, Pfister M, Pusch CM, Blin N. Chromosome 11 monosomy in conjunction with a mutated SDHD initiation codon in nonfamilial paraganglioma cases. Cancer Genet.Cytogenet. 2004; 1502:128–135.
18. Flynn A, Benn D, Clifton-Bligh R, Robinson B, Trainer AH, James P, Hogg A, Waldeck K, George J, Li J, Fox SB, Gill AJ, McArthur G, et al. The genomic landscape of phaeochromocytoma. J Pathol. 2015; 2361:78–89.
19. Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, Shimamura T, Sato-Otsubo A, Nagae G, Suzuki H, Nagata Y, Yoshida K, Kon A, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013; 45:860–867.
20. The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013; 499:43–49.
21. Azzi S, Abi HW, Netchine I. Beckwith-Wiedemann and Russell-Silver Syndromes: from new molecular insights to the comprehension of imprinting regulation. Curr Opin Endocrinol Diabetes Obes. 2014; 211:30–38.
22. Hoekstra AS, Addie RD, Ras C, Seifar RM, Ruivenkamp CA, Briaire-de Bruijn IH, Hes FJ, Jansen JC, Corssmit EP, Corver WE, Morreau H, Bovee JV, Bayley JP, et al. Parent-of-origin tumorigenesis is mediated by an essential imprinted modifier in SDHD-linked paragangliomas: SLC22A18 and CDKN1C are candidate tumor modifiers. Hum Mol Genet. 2016.
23. Corver WE, Middeldorp A, ter Haar NT, Jordanova ES, van PM, van ER, Cornelisse CJ, Fleuren GJ, Morreau H, Oosting J, van WT. Genome-wide allelic state analysis on flow-sorted tumor fractions provides an accurate measure of chromosomal aberrations. Cancer Res. 2008; 6824:10333–10340.
24. Cai Y, Crowther J, Pastor T, Abbasi AL, Baietti MF, De TM, Vazquez I, Talebi A, Renzi F, Dehairs J, Swinnen JV, Sablina AA. Loss of Chromosome 8p Governs Tumor Progression and Drug Response by Altering Lipid Metabolism. Cancer Cell. 2016; 295:751–766.
25. Tschaharganeh DF, Bosbach B, Lowe SW. Coordinated Tumor Suppression by Chromosome 8p. Cancer Cell. 2016; 295:617–619.
26. Hensen EF, Jansen JC, Siemers MD, Oosterwijk JC, Vriends AH, Corssmit EP, Bayley JP, Van Der Mey AG, Cornelisse CJ, Devilee P. The Dutch founder mutation SDHD.D92Y shows a reduced penetrance for the development of paragangliomas in a large multigenerational family. Eur J Hum Genet. 2010; 181:62–66.
27. Hes FJ, Weiss MM, Woortman SA, de Miranda NF, van Bunderen PA, Bonsing BA, Stokkel MP, Morreau H, Romijn JA, Jansen JC, Vriends AH, Bayley JP, Corssmit EP. Low penetrance of a SDHB mutation in a large Dutch paraganglioma family. BMC Med Genet. 2010; 11:92.
28. Schiavi F, Milne RL, Anda E, Blay P, Castellano M, Opocher G, Robledo M, Cascon A. Are we overestimating the penetrance of mutations in SDHB? Hum Mutat. 2010; 316:761–762.
29. Solis DC, Burnichon N, Timmers HJ, Raygada MJ, Kozupa A, Merino MJ, Makey D, Adams KT, Venisse A, Gimenez-Roqueplo AP, Pacak K. Penetrance and clinical consequences of a gross SDHB deletion in a large family. Clin Genet. 2009; 754:354–363.
30. Hoekstra AS, de Graaff MA, Briaire-de Bruijn IH, Ras C, Seifar RM, van Minderhout I, Cornelisse CJ, Hogendoorn PC, Breuning MH, Suijker J, Korpershoek E, Kunst HP, Frizzell N, et al. Inactivation of SDH and FH cause loss of 5hmC and increased H3K9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget. 2015; 636:38777–38788. doi: 10.18632/oncotarget.6091.
31. Cooke HJ, Hindley J. Cloning of human satellite III DNA: different components are on different chromosomes. Nucleic Acids Res. 1979; 610:3177–3197.
32. Waye JS, Creeper LA, Willard HF. Organization and evolution of alpha satellite DNA from human chromosome 11. Chromosoma. 1987; 953:182–188.
33. Jordanova ES, Riemersma SA, Philippo K, Giphart-Gassler M, Schuuring E, Kluin PM. Hemizygous deletions in the HLA region account for loss of heterozygosity in the majority of diffuse large B-cell lymphomas of the testis and the central nervous system. Genes Chromosomes. Cancer. 2002; 351:38–48.
34. Bayley JP, Oldenburg RA, Nuk J, Hoekstra AS, van der Meer CA, Korpershoek E, McGillivray B, Corssmit EP, Dinjens WN, de Krijger RR, Devilee P, Jansen JC, Hes FJ. Paraganglioma and pheochromocytoma upon maternal transmission of SDHD mutations. BMC Med Genet. 2014; 15:111.
35. Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002; 1811:1427–1431.
36. Lewin J, Schmitt AO, Adorjan P, Hildmann T, Piepenbrock C. Quantitative DNA methylation analysis based on four-dye trace data from direct sequencing of PCR amplificates. Bioinformatics. 2004; 2017:3005–3012.
37. Wang Y, Carlton VE, Karlin-Neumann G, Sapolsky R, Zhang L, Moorhead M, Wang ZC, Richardson AL, Warren R, Walther A, Bondy M, Sahin A, Krahe R, et al. High quality copy number and genotype data from FFPE samples using Molecular Inversion Probe (MIP) microarrays. BMC Med Genomics. 2009; 2:8.