
INTRODUCTION
Esophageal carcinoma is a common malignant tumor of digestive system with the mortality rate ranking the sixth in the world [1]. Surgical resection is preferred for patients with early esophageal cancer. However, patients in clinical mainly suffer from advanced esophageal cancer [2–3] which is often accompanied by lymph node metastasis or other complications. Radiation is one of most important and effective therapeutic methods for esophageal cancer. However, esophageal cancer belongs to the moderately sensitive tumors to radiotherapy. The 3-year overall survival rate has been reported to be only about 20% after radiotherapy alone while the recurrence rate is as high as 60%–80% [4]. Although increasing the irradiation intensity could improve the therapeutic effect, the normal tissues might be also damaged and radioresistance might be another major challenge resulting in locoregional recurrence and metastasis [5]. Thus, it is essential to develop radiation sensitizer with high efficiency and low toxicity for the treatment of esophageal cancer [6].
It has been extensively reported that targeting telomerase is effective for cancer treatment [7] and the most direct way is to inhibit the activity of telomerase is targeting telomerase RNA (hTR) or reverse transcriptase protein subunit (hTERT). Imetelstat, a 13-mer oligonucleotide which is covalently modified with lipid extensions, competitively suppresses enzymatic activity of telomerase [8]. It has been found to target the template region of hTR and serve as an oligonucletide template antagonist [9]. Covalently bonded liposomes are prone to be uptaken by cell, which ensures a better and reliable bioavailability. Meanwhile, thio-phosphoramidate internucleoside linkages in imetelstat provide a longer half-life for the inhibitor in vivo [10–11]. Furthermore, it has been reported that telomerase activity was remarkably elevated and inhibition of telomerase blocks proliferation of esophageal adenocarcinoma cells both in vitro and in vivo [12], while there was no report focusing on esophageal squamous cell carcinoma.
Radiation produce large amounts of free radicals which further leads to DNA breakage. DNA double strand breaks (DSBs) is not only the most serious injury caused by radiation, but also the basis of radiation to kill tumor cells [13]. Once DSBs is induced, cells themselves react quickly and activate DNA damage responses which recruit large amounts of protein such as ATM, γ-H2AX, p53 to sense, amplify and transduce DNA damage signal rapidly [14]. Eventually, cells respond to these signals to protect themselves, including cell cycle checkpoints, regulation of gene expression and cell apoptosis.
In the present study, we investigated the increased radiosensitization effect of imetelstat on esophageal squamous cell carcinoma in vitro and in vivo. The effect of imetelstat on proliferation and radiosensitization was correlated with its ability to increase cell apoptosis.
RESULTS
Imetelstat enhanced Kyse410 and Kyse520 sensitivity to radiotherapy
Prior investigations have revealed that inhibitor imetelstat treated esophageal squamous cells is able to increase the number of double-strand breaks induced by high energy X-Ray [9]. To further study the radiosensitization of imetelstat on esophageal squamous carcinoma cells, the present study was carried out on the basis of previous researches. As shown in Figure 1A, exposure to imetelstat alone resulted in 69.7% cell survival for Kyse410 and 75.9% cell survival for Kyse520 compare to sense treatment. Kyse410 and Kyse520 cells were exposed to radiation of 0, 2, 4, 6 and 8 Gy X-ray in the presence or absence of imetelstat to study cell survival. Cell survival after 2, 4, 6 and 8 Gy irradiation of Kyse410 was 64.7%, 39.1%, 3.9% and 3.3%, whereas the expected additive survival should be 45.0%, 27.2%, 2.7% and 2.3%. However, the achieved cell survival after combination was less, 30.1%, 20.3%, 3.3% and 0.9%. For Kyse520, the expected additive cell survival is approximately 61.4%, 38.3%, 17.4% and 6.5% theoretically. Whereas, the achieved cell survival of Kyse520 exposed to 2, 4, 6 and 8 Gy irradiation and imetelstat combination was 47.6%, 30.2%, 11.0% and 5.5%. Figure 1B showed the survival curves of Kyse410 and Kyse520 after exposure to X-ray combined with the treatment of 5 μM imetelstat, which reflected the radiosensitizing effect. As indicated, imetelstat treated cells were normalized to sense treated controls, the figure only reflected the raidosensitizing effect. We observed that imetelstat could radiosensitize both Kyse410 and Kyse520 cells, the obtained SERs were 1.9 and 1.6, respectively.
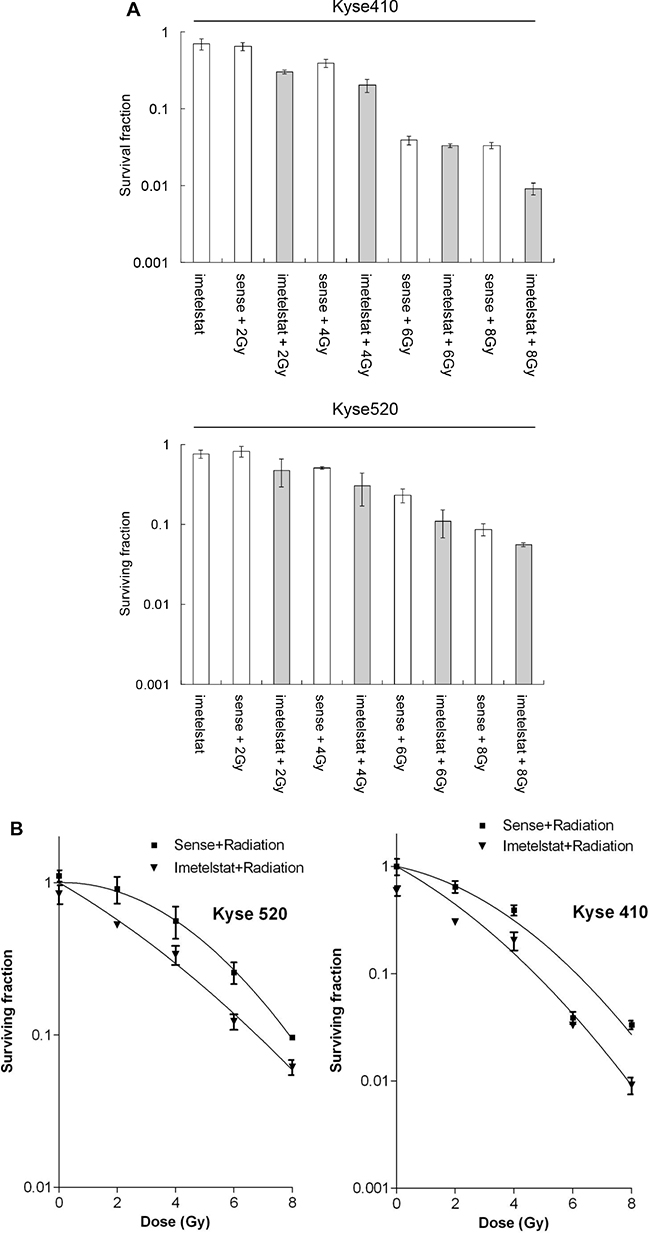
Figure 1: Imetelstat radiosensitizes Kyse410 and Kyse520 cells by using clonogenic cell survival following imetelstat and/or X-ray irradiation (2, 4, 6 and 8 Gy). (A) shows survival fraction as a function of imetelstat, radiation dose and their combination. (B) shows survival plots as a function of radiation dose with or without imetelstat. Survival data were fitted to S = exp (-αD-βD2) where D is the dose in Gy and α and β are fitting parameters, n ≥ 6, Error bars represent standard deviation. All imetelstat treatment had P-values less than 0.05 except imetelstat + 6 Gy treatment for Kyse410.
Imetelstat increases cell apoptosis of Kyse410 and Kyse520 cells
Apoptosis, a mode of cell death in response to radiation, is an important method for cancer therapy. To test whether Kyse410 and Kyse520 cell apoptosis was enhanced by imetelstat, annexin V - FITC staining were applied. Temozolomide (TMZ) can form DNA double strand breaks and induce cell apoptosis. In later studies, TMZ was selected to mimic the function of radiation in vitro. As shown in Figure 2, TMZ significantly increased apoptosis percentage for both cell lines compared with the control group (p < 0.05). Apoptosis rate for Kyse410 and Kyse520 cells was approximately 24% and 14% with the exposure to TMZ/sense. Imetelstat administration increased the apoptosis percentage to 38% for Kyse410 and 18% for Kyse520, significantly higher than those treated with sense (p < 0.05).
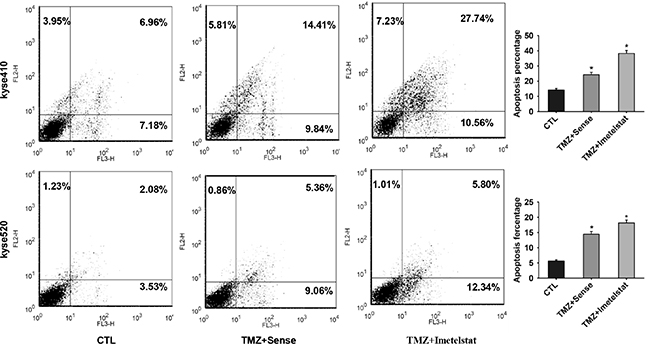
Figure 2: Imetelstat increases cell apoptosis of Kyse410 and Kyse520 cell. Kyse410 and Kyse520 cells were incubated and treated with 5 μM sense or imetelstat for 72 h and at the same time exposed to 10 μM TMZ. Results of early and late apoptosis were added together to calculate the total amount of apoptosis. Values represent the mean±SD and are representative of three independent experiments. *p < 0.05, statistical significance was established by t test.
Cells react to DSBs by triggering the DNA damage checkpoint response, which arrests cell-cycle progression until the DNA damage has been removed. CHK1 is a multifunctional protein kinase that plays essential roles in cell survival and cell cycle checkpoints. As shown in Figure 3A, Kyse cells were treated with 10 μM TMZ and showed increased phosphorylation of CHK1. Furthermore, imetelstat treatment could increase CHK1 phosphorylation compare with sense treatment. The magnitude and rate of DNA double-strand breaks repair were assessed by the analysis of γ-H2AX using western blot. Cell treated with TMZ over a dose range of 5, 10 and 20 μM showed increased γ-H2AX in the imetelstat-treated samples for both Kyse410 and Kyse520 cells (Figure 3B and 3C), while the relative protein levels in Kyse410 cell was higher than that in Kyse520 cells. Human tumor suppressor gene p53 is known to be implicated in DNA repair and induce cell apoptosis. As illustrated in Figure 3B, the expression of p53 in Kyse410 cells was up-regulated with the treatment of imetelstat. Besides, caspase family plays a critical important role in mediating the process of apoptosis, wherein caspase3 is key execution molecule. It was also found that the expression of caspase3 in Kyse410 and Kyse520 cells were up-regulated with the treatment of 5 μM imetelstat (Figure 3B and 3C).
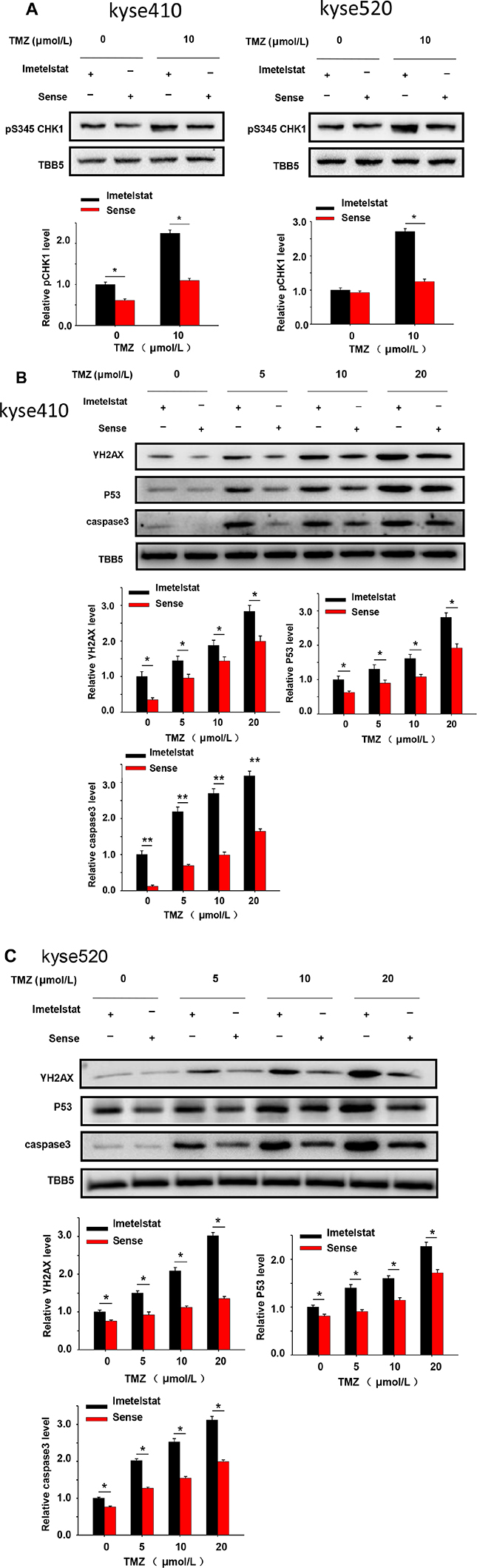
Figure 3: DNA repair and apoptosis signaling protein were upregulated by the treatment of imetelstat in Kyse410 and Kyse520 cells. (A) Kyse410 and Kyse520 cells were treated with 5 μM imetelstat or 5 μM sense for 72 h and meanwhile exposed to 10 μM TMZ. The expressions of phosphorylation of CHK1 at S345 were detected, taking the expression of TBB5 as inner control. (B and C) Kyse410 and Kyse520 cells were treated with 5 μM imetelstat or 5 μM sense for 72 h together with TMZ (0, 5, 10, 20 μM). Expression levels of γ-H2AX, p53 and caspase 3 were detected by immunoblotting. The intensity of each protein band was quantified using the BandScan software (Glyko) and normalized against tublin. *represents statistical significant (p < 0.05).
Imetelstat sensitizes esophageal cancer cells to radiation in vivo
To study whether telomerase inhibition and telomere dysfunction was associated with a progressive impairment in the cell growth in vivo as well as in vitro, the tumor growth curve was pictured. Kyse cells were pretreated with imetelstat and tumors in nude mice were subjected to 2 Gy of irradiation for 5 consecutive days. As seen in Figure 4, the volume of tumor in nude mice treated with X-ray exhibited a decrease in contrast with the untreated and sense group. As a telomerase inhibitor, imetelstat itself could significantly inhibit the growth of tumor compared with those in the sense group. More importantly, Kyse520 in imetelstat/radiation group showed a lag in tumor growth compared with mice receiving 2 Gy of irradiation alone or irradiation with sense, which confirmed that imetelstat made tumor more sensitive to radiotherapy.
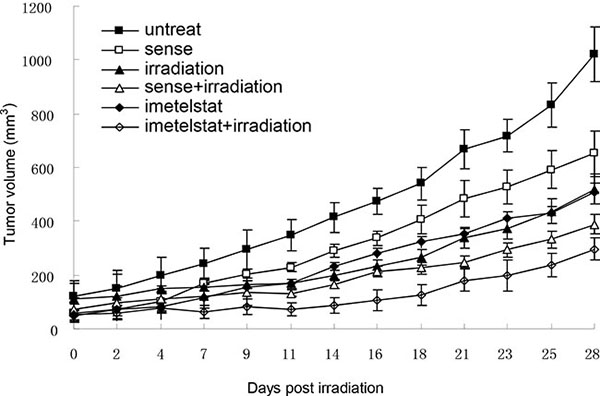
Figure 4: Imetelstat sensitizes esophageal cancer cell Kyse520 to radiation in vivo. Kyse520 cells were pretreated with imetelstat or sense for 7 weeks before injected into nude mice to form tumors. When the size of tumor reached approximately 50 mm3, mice were randomly divided into 6 different groups, including untreated group, radiation group, sense group, imetelstat group, sense + radiation group and imetelstat + radiation group. Tumors in nude mice were subjected to 2 Gy of irradiation for 5 consecutive days to mimic a fractionated weekly scheme. The sizes of tumor were measured 3 times per week to obtain cell growth curve. At days 13, 31 and 45 postinjection, imetelstat were injected intraperitoneally to ensure inhibition of telomerase activity. Data are presented as mean tumor volume ± SE. Imetelstat with irradiation significantly decreased tumor growth compared with mice receiving irradiation alone or irradiation with sense (p < 0.05).
Imetelstat increases cell apoptosis and decreases cell proliferation in vivo
Pathological changes of esophageal squamous carcinoma were evaluated by H&E staining. As seen in Figure 5, tumor samples from the untreated group were tightly aligned with a large blue-hued nucleus. Besides, samples from mice subjected to imetelstat alone displayed more vacuoles in contrast with those in the sense group. Tumor isolated from mice in the imetelstat/radiation group showed condensed cytoplasm and desmosome complexes, fragmented nuclear, separated cells in comparison with those in the sense/radiation group, suggesting the potential sensitizing effect.
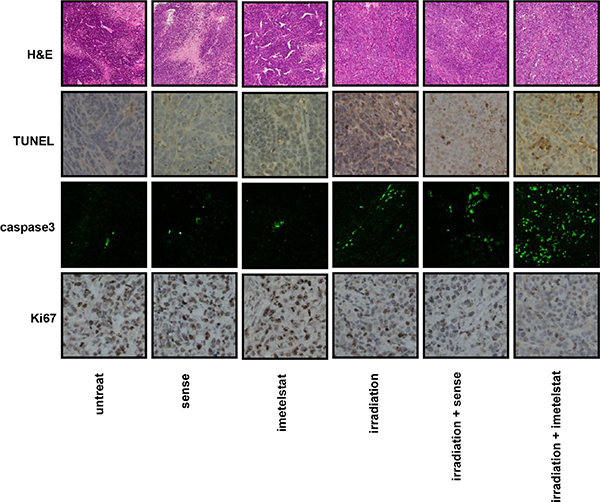
Figure 5: Imetelstat increases cell apoptosis and decreases cell proliferation in vivo. Pathological changes of esophageal squamous carcinoma were evaluated by H&E staining. Apoptotic cells were labeled by TUNEL assay and further evaluated by immunofluorescence staining of caspase 3. Imetelstat promoted radiation-induced cell apoptosis compared with radiation-treated tumor. The cell proliferation was evaluated by Ki67 staining in tumor tissue. Imetelstat combined with radiation showed less Ki67 expression compared to other treatment groups.
Apoptotic cells were labeled by TUNEL assay as shown in Figure 5. Although TUNEL staining for tumor treated with sense or imetelstat showed relatively small number of apoptotic cells, the enhanced staining intensity in imetelstat group was still stronger than that in the sense group. Moreover, tumor exposed to the combination of radiation and sense also showed less apoptosis staining than those subjected to imetelstat/radiation. The apoptosis induced by imetelsat and radiation was further analyzed by using caspase3 immunofluorescence staining. The expression of caspase3 in radiation-treated tumor in vivo was up-regulated, while imetelstat promoted radiation-induced cell apoptosis as reflected by the further up-regulated level of caspase3 (Figure 5). More importantly, the expression of caspase3 in imetelstat/radiation group was much higher than that in the sense/radiation group, which implies more apoptosis. Tumor subjected to sense or imetelstat only showed almost no expression of caspase3, which confirmed that imetelstat was not toxic to mice in vivo and the principal function of it was enhancing tumor sensitivity to radiation therapy.
The expression of Ki67 is indispensable in cell proliferation. Radiation-treated mice showed the down-regulation of Ki67 and imetelstat enables tumor more sensitive to radiation treatment, which was revealed by weaker expression of Ki67 (Figure 5). As observed in Figure 5, the staining of Ki67 in sense or imetelstat treated group was stronger compared with that exposed to radiation. More importantly, tumor pretreated with sense combined irradiation displayed higher level of Ki67 compared with that in the imetelstat/radiation group, which indicated radiosensitization effect of imetelstat. These findings suggested that imetelstat inhibited tumor growth in mice subjected to radiation was associated with the enhanced apoptosis and suppressed cell proliferation in cancer cells.
DISCUSSION
Telomerase activation is considered as a key step in cell immortalization and tumorigenesis [15]. It has been reported that positive rate of telomerase activity is 80% to 90% in squamous cell carcinoma, which is much higher than those in the surrounding normal tissues [16]. This discrepancy between cancer and normal cells provides a considerable therapeutic window for telomerase suppression-based therapy.
Our prior work has verified that imetelstat blocks telomerase activity in esophageal cancer cells, decreases cell proliferation and colony formation ability. More importantly, our research group has reported imetelstat increases radiation induced DNA breaks, which provided theoretical foundation for further researches [9]. In this study, we found that long-term imetelstat treatment could radiosensitize Kyse cells in both vitro and vivo. This is consistent with one study on breast cancer cells, in which long-term treatment of imetelstat showed radiosensitization activity in vitro colony formation assay and in vivo tumor growth assay [17]. Further analysis showed that SER was larger for Kyse410 than Kyse520, 1.9 and 1.6 respectively. These results indicated that Kyse520 is more radioresistant than Kyse410 cells, consisting with our previous results that radiation-induced DSB foci in Kyse410 are more in number and larger in size compared with the DSB foci in Kyse520 cells [9].
Multiple DNA damage repair pathways are implicated in maintaining the genetic integrity of a cell after its exposure to radiation. Wong et al. reported that telomerase deficient mouse developed a radio-sensitivity syndrome because of the delayed DNA break repair [18]. Our previous study reported that treatment of imetelstat and radiation showed synergistic increase and prolonged higher expression of DSB foci compared to cells treated with sense/radiation [9]. In this study, western blot results showed that increased expression of phospho-CHK1 and γ-H2AX in imetelstat treatment group which reflect the magnitude and rate of DSB repair. Our results are consistent with one report in glioblastoma cells that increased levels of γ-H2AX after imetelsat treatment [19]. Cells react to DSBs by triggering the DNA damage signaling, which will further activate intracellular pathways including apoptotic signaling [20–22] One our previous research reported that 17-AAG sensitize esophageal squamous cells to radiation by inhibiting cell proliferation and promote cell apoptosis [23]. Ding et al. reported that sunitinib increased radiation induced DNA double-strand breaks and promoted the apoptosis of human esophageal squamous cell carcinoma cells [24]. Our study showed that imetelstat combined TMZ which mimic the function of radiation could significantly increase cell apoptosis compared with sense/TMZ treatment (Figure 2). Western blot demonstrated that imetelstat treatment could increase expressions of caspase3 and p53, which promoted cell apoptosis and reflected increased DNA repair signaling (Figure 3B and 3C). Apoptotic cells labeled by TUNEL assay in tumor tissue were increased in imetelstat treatment group which is further proved by caspase3 immunofluorescence staining (Figure 5).
Although in vitro data provide an important preliminary background to advance the potential use of imetelstat, these results must be confirmed and validated in vivo. The clinical application of therapeutic irradiation relies on fractionated does of radiation that are given daily for several weeks. In our study, we extended in vivo results to a fractionated weekly scheme consistent with clinical treatment protocols. Our experiments demonstrated that imetelstat combine with radiation induce tumor growth delay compared to imetelstat treatment or sense/radiation treatment. The results comfirmed that imetelstat can radiosensitize esophageal cancer in vivo. As a marker of cell proliferation, antigen Ki67 was also detected to confirm the tumor growth delay induced by imetelstat treatment.
In conclusion, imetelstat increases radiation sensitivity in esophageal cancer by inducing cell apoptosis and decreasing cell proliferation. In vitro and vivo results support imetelstat as a promising adjuvant cancer treatment in combination with radiation therapy.
MATERIALS AND METHODS
Main reagents and kits
The human ESCC (esophageal squamous cell carcinoma) cell Kyse410 and Kyse520 (Deutsche Sammlung von Mikroorganismenund Zellkulturen GmbH, Braunschweig, Germany) were cultivated in PRMI-1640 medium at 37°C in a humidified 5% CO2 incubator, supplemented with 10% fetal bovine serum, 100 U penicillin and streptomycin. Primary antibodies against γ-H2AX, p53, caspase3, and pS345-CHK1 were purchased from Cell Signaling Technology (Beverly, MA). Anti-Ki-67 and tublin antibodies was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). PRMI-1640 medium was from GIBCO and fetal bovine serum (FBS) was purchased from Hyclone (Logan, UT, USA). Penicillin and streptomycin were purchased from Amresco Inc (Solon, OH).
Radiosensitization experiment
Kyse410 and Kyse520 were pretreated with imetelstat and sense at a concentration of 1 μM for 40 days. During this time, cells were passaged every three days and fresh antagonist was added. After that, cells were exposed to radiation of 0, 2, 4, 6 and 8 Gy of 6 MeV X-ray at room temperature, respectively. To assay clonogenic cell survival, cells were trypsinized and plated onto 10-cm tissue culture dishes. Cells were incubated for 10–12 days, fixed with ethanol and stained with haematoxylin. Cells able to form a colony of at least 50 cells were considered as clonogenic cells. Survival data were fitted to S = exp (-αD-βD2) where D is the dose in Gy and α and β are fitting parameters. Radiosensitivity was quantified as area under curve (AUC) and the effect of imetelstat on radiosensitivity is presented as sensitizer enhancement ratio (SER), defined as AUC control/AUC treated.
Apoptosis assay
Kyse410 and Kyse520 cells were incubated and treated with 5 μM imetelstat or 5 μM sense for 72 h and together with 10 μM temozolomide (TMZ). Cells were harvested, washed, resuspended in PBS and then stained with the Annexin V/PI Cell Apoptosis Detection Kit (Franklin Lakes, NJ) according to the manufacturer’s instructions. Data acquisition and analysis were conduct with a Becton–Dickinson FACS Calibur flow cytometer using Cell-Quest software (BD Biosciences, Franklin Lakes, NJ).
Western blot
Kyse410 and Kyse520 cells were incubated and treated with 5 μM imetelstat or 5 μM sense for 72 h and meanwhile exposed to increased concentration of TMZ (0, 5, 10, 20 μM). Subsequently, proteins in Kyse410 and Kyse520 cells were extracted with RIPA lysis buffer containing a protease inhibitor cocktail, PMSF for 30 min on ice respectively and then centrifuged at 12000 rpm for 5 min at 4°C. Total protein concentration in supernatant of cell lysates was determined by Pierce BCA protein assay kit (Thermo, Massachusetts, USA). Then we used γ-H2AX, p53, caspase3 and pS345-CHK1 Abs in the following SDS-PAGE and Western blot detection. Antigens were detected after incubation with an HRP-conjugated secondary antibody followed by visualization using an ECL detection system. The intensity of each protein band was quantified using the BandScan software (Glyko) and normalized against tublin in the same samples blotted by anti-tublin antibody.
Animals care and experimental protocol
All animal maintenance and experimental procedures were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male nude mice were purchased from Beijing HFK bioscience Company Limited. Mice were maintained in an air-conditioned pathogen-free room under conditions of controlled lighting of 12 h light/day. Five million Kyse520 cells cultured were cultured for 7 weeks adding 1 μM imetelstat or sense. Esophageal squamous Kyse520 cell suspension was injected subcutaneously into right hind limb groin of nude mice to induce the formation of tumor. When the size of tumor reached approximately 50 mm3, mice were randomly divided into 6 different groups, including untreated group, radiation group, sense group, imetelstat group, sense + radiation group and imetelstat + radiation group. After that, tumors in nude mice were subjected to 2 Gy of irradiation for 5 consecutive days, while other parts were protected by lead. The sizes of tumor were measured 3 times per week to obtain cell growth curve. Mice were sacrificed 50 days later and tumors were dissected subsequently for further analysis. At days 13, 31 and 45 postinjection, inhibitor imetelstat (30 mg/kg) and control sense (30 mg/kg) were injected intraperitoneally to ensure inhibition of telomerase activity.
TUNEL assay
DNA fragments in tissue sections were detected using TUNEL assay kit (BD Pharmingen, CA) according to the manufacturer’s instructions. Briefly, the enzyme terminal deoxynucleotidyl transferase (TdT) was used to incorporate digoxigenin-conjugated dUTP at the ends of DNA fragments. The signal of TdT-mediated dUTP nick-end labeling was then detected using an anti-digoxigenin antibody conjugated with peroxidase. TUNEL slides were photographed with a confocal laser scanning microscope (Fluoview FV1000, Olympus, Tokyo, Japan). Positive staining of cells within each slide were measured and expressed as a ratio of apoptotic cells to total number of tumor cells.
Immunohistochemistry and immunofluorescence assay
Tumor tissues were fixed in 10% (V/V) neutral buffered formalin solution for at least 24 h at room temperature, dehydrated in graded ethanol, cleared in xylene and embedded in paraffin. Perpendicular tumor sections (4 μm thickness) were mounted on glass slides and de-waxed. Then, tissue sections were prepared for Hematoxylin-Eosin (HE) staining. Pathological changes were observed by light microscopy. The expression of caspase3 in tumor was detected by immunofluorescence assay. Tumor tissues was processed and mounted on glass slides. Briefly, sections were pre-incubated with 1% Triton X-100 and 10% bovine serum albumin (BSA) in phosphate buffered saline (PBS) for 1 h at room temperature. After that, samples were incubated with specific antibodies against caspase3 for 2 h at room temperature. Fluorescent secondary antibodies were performed for another 1 h at room temperature according to the manufacturer’s instructions. The expression of Ki67, proliferation cell-associated nuclear antigen in tumor tissues, was assessed by immunohistochemistry. The paraffin sections were heat fixed, deparaffinized in xylene, rehydrated by graded ethanol to distilled water, boiled in sodium citrate buffer and incubated in 3% hydrogen peroxide after cooling. Each slide was blocked with 3% BSA at room temperature prior to the incubation with primary antibody overnight at 4°C, secondary antibody for 20 min at 37°C and streptavidin-HRP 20 min at 37°C, respectively. Samples were then stained with DAB and counterstained with hematoxylin. After dehydrating and drying, the sections were mounted with neutral gum.
Statistical analysis
All data were presented as means ± standards deviation (SDs). One-way analysis of variance (ANOVA) followed by Tukey multiple comparison test was performed to evaluate differences between groups. The values were considered statistically significantly different at p < 0.05.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by National Nature Science Foundation of China (No. 81301938, 81670747), Medical Science and technology development Foundation, Nanjing Department of Health (JQX14007). The work is also supported by Ministry of Science and Technology of China (2013CB911600), Jiangsu Provincial Natural Science Foundation (BK20130044, BK20130061), CJ20160051, The Research Fund for the Doctoral Program of Higher Education of China (RFDP) (20133207110005), The Program for New Century Excellent Talents in University of Ministry of Education of China (NCET-13- 0868), the Priority Academic Program Development Award for Jiangsu Higher Education Institutions. We thank Sergei Gryaznov for providing imetelstat and sense.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Tirumani H, Rosenthal MH, Tirumani SH, Shinagare AB, Krajewski KM, Ramaiya NH. Esophageal Carcinoma: Current Concepts in the Role of Imaging in Staging and Management. Can Assoc Radiol J. 2015; 66:130–9. doi: 10.1016/j.carj.2014.08.006.
2. Reeh M, Nentwich MF, Asani S, Uzunoglu FG, Bockhorn M, Sauter G, Rösch T, Izbicki JR, Bogoevski D. Locally advanced esophageal carcinoma: is there still a role of surgery alone without neoadjuvant treatment? J Gastrointest Surg. 2015; 19:587–93. doi: 10.1007/s11605-015-2762-y.
3. Sjoquist KM, Burmeister BH, Smithers BM, Zalcberg JR, Simes RJ, Barbour A, Gebski V. Survival after neoadjuvant chemotherapy or chemoradiotherapy for resectable oesophageal carcinoma: an updated meta-analysis. Lancet Oncol. 2011; 12:681–92. doi: 10.1016/S1470-2045(11)70142-5.
4. Song YP, Ma JB, Hu LK, Zhou W, Chen EC, Zhang W. Phase I/II study of hypofractioned radiation with three-dimensional conformal radiotherapy for clinical T3-4N0-1M0 stage esophageal carcinoma. Technol Cancer Res Treat. 2011; 10:25–30. doi: 10.7785/tcrt.2012.500176.
5. Chang L, Graham P, Hao J, Bucci J, Malouf D, Gillatt D, Li Y. Proteomics discovery of radioresistant cancer biomarkers for radiotherapy. Cancer Lett. 2015; 369:289–97. doi: 10.1016/j.canlet.2015.09.013.
6. Ghorab MM, Ragab FA, Heiba HI, El-Hazek RM. Anticancer and radio-sensitizing evaluation of some new thiazolopyrane and thiazolopyranopyrimidine derivatives bearing a sulfonamide moiety. Eur J Med Chem. 2011; 46:5120–6. doi: 10.1016/j.ejmech.2011.08.026.
7. Harley CB. Telomerase and cancer therapeutics. Nat Rev Cancer. 2008; 8:167–79. doi: 10.1038/nrc2275.
8. Baerlocher GM, Oppliger Leibundgut E, Ottmann OG, Spitzer G, Odenike O, McDevitt MA, Röth A, Daskalakis M, Burington B, Stuart M, Snyder DS. Telomerase Inhibitor Imetelstat in Patients with Essential Thrombocythemia. N Engl J Med. 2015; 373:920–8. doi: 10.1056/NEJMoa1503479.
9. Wu X, Smavadati S, Nordfjäll K, Karlsson K, Qvarnström F, Simonsson M, Bergqvist M, Gryaznov S, Ekman S, Paulsson-Karlsson Y. Telomerase antagonist imetelstat inhibits esophageal cancer cell growth and increases radiation-induced DNA breaks. Biochim Biophys Acta. 2012; 1823:2130–5. doi: 10.1016/j.bbamcr.2012.08.003.
10. Herbert BS, Gellert GC, Hochreiter A, Pongracz K, Wright WE, Zielinska D, Chin AC, Harley CB, Shay JW, Gryaznov SM. Lipid modification of GRN163, an N3′→P5′ thio-phosphoramidate oligonucleotide, enhances the potency of telomerase inhibition. Oncogene. 2005; 24: 5262–8. doi: 10.1038/sj.onc.1208760
11. Dikmen ZG, Ozgurtas T, Gryaznov SM, Herbert BS. Targeting critical steps of cancer metastasis and recurrence using telomerase template antagonists, Biochim Biophys Acta-Mol Basis Dis. 2009; 240–7. doi: 10.1016/j.bbadis.2009.01.018.
12. Shammas MA, Qazi A, Batchu RB, Bertheau RC, Wong JY, Rao MY, Prasad M, Chanda D, Ponnazhagan S, Anderson KC, Steffes CP, Munshi NC, De Vivo I et al. Telomere maintenance in laser capture microdissection-purified Barrett’s adenocarcinoma cells and effect of telomerase inhibition in vivo. Clin Cancer Res. 2008; 14: 4971–80. doi: 10.1158/1078-0432.CCR-08-0473.
13. Raymond AA, Benhamouche S, Neaud V, Di Martino J, Javary J, Rosenbaum J. Reptin regulates DNA double strand breaks repair in human hepatocellular carcinoma. PLoS One. 2015; 10:e0123333. doi: 10.1371/journal.pone.0123333.
14. Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint. Science. 2002; 298:1435–8. doi: 10.1126/science.1076182.
15. Takubo K, Nakamura K, Izumiyama N, Mafune K, Tanaka Y, Miyashita M, Sasajima K, Kato M, Oshimura M. Telomerase activity in esophageal carcinoma. J Surg Oncol. 1997; 66:88–92. doi:10.1002/(SICI)1096-9098(199710)66:2<88: AID-JSO3>3.0.CO;2-H.
16. Shay JW, Bacchetti S. A survey of telomerase activity in human cancer, Eur J Cancer. 1997; 33:787–91. doi: 10.1016/S0959-8049(97)00062-2.
17. Gomez-Millan J, Goldblatt M, Gryaznov M, Mendonca S, Herbert S. Specific telomere dysfunction induced by GRN163L increases radiation sensitivity in breast cancer cells. Int J Radiat Oncol Biol Phys. 2007; 67:897–905. doi: 10.1016/j.ijrobp.2006.09.038.
18. Wong KK, Chang S, Weiler SR, Ganesan S, Chaudhuri J, Zhu C, Artandi SE, Rudolph KL, Gottlieb GJ, Chin L, Alt FW, DePinho RA. Telomere dysfunction impairs DNA repair and enhances sensitivity to ionizing radiation. Nat Genet. 2000; 26:85–88. doi: 10.1038/79232.
19. Marian CO, Cho SK, McEllin BM, Maher EA, Hatanpaa KJ, Madden CJ, Mickey BE, Wright WE, Shay JW, Bachoo RM. The telomerase antagonist, imetelstat, efficiently targets glioblastoma tumor-initiating cells leading to decreased proliferation and tumor growth. Clin Cancer Res. 2010; 16:154–63. doi: 10.1158/1078-0432.CCR-09-2850.
20. d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003; 426:194–8. doi: 10.1038/nature02118.
21. Masutomi K1, Possemato R, Wong JM, Currier JL, Tothova Z, Manola JB, Ganesan S, Lansdorp PM, Collins K, Hahn WC. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc Natl Acad Sci. 2005; 102:8222–7. doi: 10.1073/pnas.0503095102.
22. Valerie K, Yacoub A, Hagan MP, Curiel DT, Fisher PB, Grant S, Dent P. Radiation induced cell signaling: inside-out and outside-in. Mol Cancer Ther. 2007; 6:789–801. doi: 10.1158/1535-7163.MCT-06-0596.
23. Wu X, Wanders A, Wardega P, Tinge B, Gedda L, Bergstrom S, Sooman L, Gullbo J, Bergqvist M, Hesselius P, Lennartsson J, Ekman S. Hsp90 is expressed and represents a therapeutic target in human oesophageal cancer using the inhibitor 17-allylamino-17-demethoxygeldanamycin. Br J Cancer. 2009; 100:334–43. doi: 10.1038/sj.bjc.6604855.
24. Ding YQ, Zhu HC, Chen XC, Sun XC, Yang X, Qin Q, Zhang H, Yang Y, Yang YH, Gao L, Luo JD, Zhou XF. Sunitinib modulates the radiosensitivity of esophageal squamous cell carcinoma cells in vitro. Dis Esophagus. 2015; 6. doi: 10.1111/dote.12440. [Epub ahead of print]