
Introduction
Twist1, a basic helix-loop-helix (bHLH) domain-containing transcription factor, was originally identified in Drosophila as an essential regulator during embryogenesis, particularly in mesoderm formation, specification, and differentiation [1, 2]. Drosophila embryos harboring Twist1 mutations fail to invaginate properly, resulting in embryos devoid of internal organs with a “Twisted” appearance [1, 3]. In humans, the Twist1 gene is located on 7q21.2 containing two exons and one intron [4]. Mutation of Twist1 in humans leads to Saethre-Chotzen syndrome, a disease of autosomal dominant inheritance characterized by manifestations such as craniosynostosis, ptosis, and hypertelorism [5-7]. Twist2 is another member of the Twist subfamily of bHLH protein in humans which shares great structural similarity with Twist1. Both Twist1 and Twist2 are key regulators in embryonic development and organogenesis. While a great number of studies have extensively demonstrated that Twist1 is implicated in tumor initiation, stemness, angiogenesis, dissemination, and chemoresistance in various carcinomas, sarcomas and hematological malignances, the biological functions of Twist2 in tumor are still highly controversial or unexplored [8-12]. Therefore, this review will focus mainly on Twist1.
The physiological and pathological contributions of Twist1 to the development and progression of different diseases have been widely reviewed. However, limited reviews have systematically summarized the network of signaling pathways and the fundamental molecular basis underlying Twist1’s multiple biological functions. Few reports have elaborated the independence and interdependency of Twist1’s multiple distinct pathological functions. Therefore, in the current review, we will first outline the diverse pathological functions of Twist1 in various cancers, then delineate the intricate underlying molecular mechanisms and network of signaling pathways (as illustrated in Figures 1 and 2), based on which we will further summarize current therapeutic strategies in cancer treatment that target or modulate Twist1-involved signaling pathways (Table 1). More importantly, we put special emphasis on revealing the molecular basis of the correlation between Twist1’s diversified biological functions, piecing together the whole delicate picture of Twist1’s multiple roles in various cancers and providing new directions for future research.
Table 1: Emerging role of Twist1 in drug resistance
Involved molecular pathways |
Drug resistance phenotypes |
Cancer types |
|
Twist1 promotes the expression of ABC transporters(e.g. MDR1) |
Induction of MDR |
||
Induction of resistance to anthracycline drugs |
|||
Induction of oxaliplatin resistance |
Colorectal cancer [37] |
||
Activation of PI3K/AKT pathways |
Induction of vincristine and paclitaxel resistance |
Nasopharyngeal cancer; bladder cancer; prostate cancer; ovarian cancer[107, 108] |
|
Twist1 induces AKT2 expression |
Induction of paclitaxel resistance |
Breast cancer [109] |
|
Twist1 down-regulates Bcl2 |
Reversion of NF-κB induced chemo-resistance |
Prostate cancer [110] |
|
Twist1 increases YB-1 expression |
Induction ofcsplatin and doxorubicin resistance |
Bladder cancer[112] |
|
Twist1 activates Jagged1/KLF4 pathway |
Induction of cisplatin and cetuximab resistance |
Head and neck cancer [101] |
|
Mitogen-activated protein kinases |
Down-regulation of Twist1 activates ERK pathway |
Reversion of radio-resistance |
Nasopharyngeal cancer [113] |
Twist1 increases p38 MAPK activity |
Induction of cisplatin resistance |
Pancreatic cancer [114] |
|
Twist1 silencing activates JNK pathway |
Reversion of cisplatin resistance |
Lung cancer [115] |
|
Twist1 depletion inhibits mTOR and reduces Mcl1 expression |
Reversion of cisplatin resistance |
Lung cancer[116] |
|
micro-RNAs |
miR-186 reduces Twist1 expression |
Reversion of cisplatin resistance |
Ovarian cancer [117] |
Inhibition of miR-106a up-regulates Twist1 expression |
Induction of gemcitabine resistance |
Hepatocellular carcinoma[118] |
|
miR-181a suppresses Twist1 expression |
Reversion of cisplatin resistance |
Tongue squamous cell carcinoma [119] |
|
miR-23a increases Twist1 expression |
Induction of cisplatin resistance |
Tongue squamous cell carcinoma cancer[120] |
|
Twist1 inhibits PI3K/AKT and down-regulates ET1/ETAR signaling |
Reversion of cisplatin resistance |
Osteosarcoma [121] |
|
miR-33a reduces Twist1 expression |
Induction of cisplatin resistance |
Osteosarcoma[122] |
|
Hypermethylation and over-expression of Twist1 as a prognostic marker
The notion that Twist1 is involved in cancer pathology triggered extensive analysis of Twist1 promoter hypermethylation, mRNA expression and protein level in a wide variety of tumors. Twist1 promoter hypermethylation, one of the most important factors in the epigenetic reprogramming of Twist1, has been identified in cancers of different origins such as breast, bladder, gastric, colon and rectum, lung, ovary, and uterine cervix cancer [10]. It is noteworthy that the hypermethylation of Twist1 promoter is not necessarily related to Twist1 mRNA or protein expression. Twist1 over-expression is commonly observed in breast cancer [13-18], bladder cancer [19-21], gastric cancer [22, 23], hepatocellular carcinoma [24], esophageal squamous cell carcinoma [25, 26], nasopharyngeal carcinoma [27, 28], head and neck cancer (HNC) [29], glioblastoma [30, 31], and chronic myelogenous leukemia [32]. Consistent with its significance in cancer biology, Twist1 over-expression is related to high grade or invasive aggressive cancer with lymph node involvement or distant metastasis indicating therapeutic failure, recurrence, and inferior prognosis [8]. As a result, Twist1 might serve as a useful diagnostic or prognostic factor predicting poor outcome, which has already been illustrated in breast cancer [15], bladder cancer [33], cervical cancer [34], esophageal squamous cell carcinoma [35], HNC [36], colorectal cancer [37], hepatocellular cancer [38, 39], melanoma [40], nasopharyngeal cancer [27], and ovarian cancer [41, 42]. The strength of the evidence supporting this notion ranges from cell lines, animal models to patient tissues, however, to date Twist1 detection is still far from clinical application. There are many obstacles hampering the clinical use of Twist1 as a diagnostic or prognostic marker, such as the lack of efficient, precise and economic detection methods and the unknown basal level of Twist1 expression in normal tissues.
Upstream regulators modulating Twist1 from the transcriptional to post-translational level
At the gene level, the expression of Twist1 is modulated by an array of different upstream regulators via multiple pathways depending on the cancer type and tissue context (Figure 1). Induction of these pathways results in activation of the Twist1 promoter by a variety of transcription factors in the nucleus. Firstly, signal transducer and activator of transcription 3 (STAT3) is implicated in Twist1 regulation by the observation that Twist1 expression is ablated upon STAT-3 knockdown in breast tumors in immunocompetent mice [43]. A subsequent study further confirmed that STAT3 directly binds to the second proximal STAT-3-binding site on the human Twist1 promoter and activates its transcriptional activity in human breast cancer cell lines [44]. This STAT-3/Twist1 axis has also been verified in hepatocellular carcinoma cells [45] and the Notch1/STAT3/Twist1 signaling axis has been identified in gastric cancer [46]. In addition to binding to Twist1 promoter directly, STAT3 is also demonstrated to regulate the transcription of Twist1 indirectly through HIF-1 stabilization in prostate cancer cells [47]. HIF-1, in turn, regulates expression of Twist1 by directly binding to the hypoxia-response element (HRE) in the Twist1 proximal promoter [36].
NF-κB is another transcription factor that can directly reprogram the gene expression of Twist1. In early Drosophila embryos, the NF-κB-like transcription factor dorsal (dl) activates Twist1 expression through binding to dl-binding sites in the Twist1 promoter [48]. This pattern of Twist1 regulation was later proven to be evolutionarily conserved in vertebrates by the observation that mammalian Twist1 expression is induced by TNF-α in an NF-κB dependent manner in immortalized fibroblasts derived from p65-/- mice [49]. In mammary epithelial cells, NF-κB and Twist1 expression are positively related and a functional NF-κB-binding site has been identified in the Twist1 promoter, further confirming the transcriptional regulation of Twist1 by NF-κB [50].
The steroid receptor co-activators (SRC, also known as nuclear receptor co-activators), in particular SRC1 and SRC3, also participate in tuning Twist1 expression. SRC3 can directly enhance Smad2 expression and subsequently increase Twist1 transcription. Knocking down SRC3 reduces the expression of Smad and Twist1 in vitro and suppresses tumor metastasis in vivo [51]. Alternatively, Qin et al. revealed that SRC1 is able to activate Twist1 transcription by physically interacting with the transcription factor polyoma enhancer activator 3 (PEA3) at the proximal Twist1 promoter by comparing SRC-1 wild-type (WT) with knockout (KO) cell lines [52]. PEA3 is also proposed to be involved in the Twist1 transcription regulation through the Wnt/β-catenin axis in mouse mammary cells [53]. The distal-less homeobox gene 4 (DLX4), a member of the DLX family widely expressed in various cancers, has also been shown to bind to regulatory regions of the Twist1 gene and enhances tumor migration, invasion, and metastasis in cell models and tumor tissues [54].
Moreover, Ras and TGF-β signaling pathways regulate Twist1 expression at both the transcriptional and protein levels. Specifically, the Msh homeobox protein (MSX2) is a downstream target of the Ras signaling pathway and is suggested to induce Twist1 expression in human pancreatic cancer cells [55]. TGF-β/Smad signaling induces high mobility group A2 (HMGA2) transcription and the latter can induce expression of Twist1 by directly associating with A:T-rich sequences and promoting transcription from the Twist1 promoter in mammary epithelial cells [56]. TGF-β can also induce fibulin 5 (FBLN5), which promotes tumor invasion and epithelial-mesenchymal transition (EMT) by elevating Twist1 transcription and reducing E-cadherin expression, although the precise mechanism by which FBLN5 regulates Twist1 transcription remains elusive [57].
Post-translational modifications are also crucial for the function of Twist1 protein in the cytosol. Phosphorylation and ubiquitination are the most important post-translational modifications that affect Twist1 function by regulating the stability of Twist1 protein [58]. Both Ras activation and TGF-β treatment are capable of activating the mitogen-activated protein kinases (MAPK) pathway, which significantly increases Ser68 phosphorylation of Twist1 and prevents Twist1 from undergoing E3-mediated ubiquitination and degradation without altering Twist1 mRNA expression in breast cancer cells [59]. Activation of PKB (AKT1) leads to a marked increase in Twist1 phosphorylation at Ser42 in the nucleus. Subsequent study further confirms that AKT1 physically associates with Twist1 and phosphorylation of Twist1 by AKT1 is required for Twist1 ubiquitination and degradation [60]. While AKT2 primarily phosphorylates Twist1 at Ser42, AKT1 phosphorylates Twist1 at Ser42 and Thr121 in vitro and at Ser42 in vivo [61]. Casein kinase 2 (CK2) interacts with Twist1 directly and phosphorylates Twist1 at Ser18 and Ser20, resulting in prolonged stability of Twist1 and enhancing the motility of HNC cells [62].
In contrast to increasing Twist1 stability, several publications have reported that the F-box protein Ppa, as well as its human homologue FBXL14, can mediate and induce the poly-ubiquitination and degradation of Twist1 protein in embryos and in HNC cells, respectively [63, 64].
Post-translational acetylation can also affect the function of the Twist1 protein. Evidence from basal-like breast cancer (BLBC) cells demonstrated that Twist1 can be diacetylated at K73 and K76 by Tip60. This diacetylation is critical for Twist-BRD4 interaction and the subsequent formation of activated Twist/BRD4/P-TEFb/RNA-Pol II complex at the WNT5A promoter and enhancer. The Twist-BRD4-WNT5a axis is crucial for the tumorigenicity of basal-like breast cancer both in vitro and in vivo. Disrupting the interaction of BRD4 with Twist1 by using BET-specific inhibitors (such as JQ1) may be a novel approach to indirectly suppress Twist1 function and provide a potential new target for the treatment of basal-like breast cancer [65].
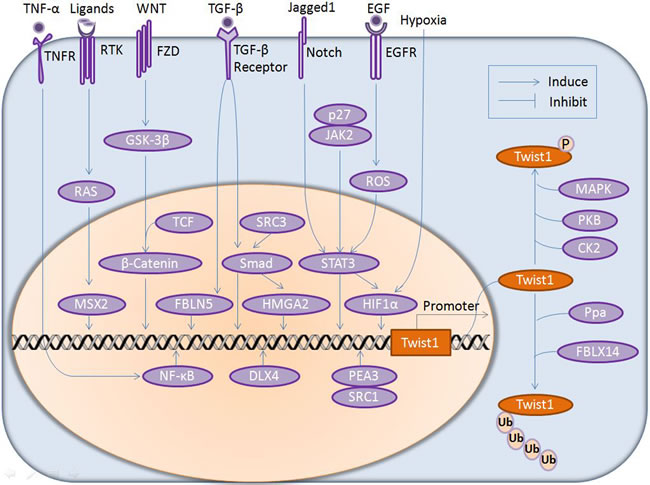
Figure 1: Modulation of Twist1 expression by upstream regulators at both gene and protein levels. Manifold extracellular insults (such as TNF-α, RTK ligands, WNT, TGF-β, Jagged1, EGF signaling and hypoxia) are transduced into the cell via trans-membrane receptors (TNFR, RTK, FZD, TGF-β, Notch and EGFR) and intracellular mediators in the cytosol (MAPK, AKT) and nucleus (NF-κB, MSX2, β-catenin, FBLN5, Smad, HMGA2, STAT3 and HIF-1α), thus regulating Twist1 expression at the gene level and Twist1 stability at the protein level, respectively. P, phosphrylation; Ub, Ubiquitination; RTK, receptor tyrosine kinases; EGF, epidermal growth factors; FZD, frizzed; GSK-3β, glycogen synthase kinase 3β; TCF, transcription factor; JAK2, janus kinase 2.
Downstream effectors involved in multiple functions of Twist1
Twist1 can exert multiple biological effects via various downstream pathways as either a transcriptional factor regulating the expression of an array of target genes or a functional modulator at the protein level (Figure 2). The most critical pathological function of Twist1 in cancer is facilitating tumor invasion and metastasis by promoting EMT [13]. Many studies have shown that Twist1 is also involved in other facets of tumor invasion and metastasis, such as the formation of invadopodia [66], intravascular migration, extravasation [67], and vasculogenic mimicry (VM) formation [68]. Furthermore, growing evidence indicates that Twist1 also plays a crucial role in supporting tumor initiation by evading p53 induced cell senescence and apoptosis, the well-known program to counter cell transformation [69-71]. In addition, evidence has shown that Twist1 can confer cancer cells stemness properties [72-73]. Moreover, the cross-talk between Twist1 and tumor angiogenesis has also attracted great attention [74]. Recently, many studies have also shed light on the connection between Twist1 and chemoresistance. At the same time, tremendous efforts have been made to modulate Twist1 as a therapeutic target for cancer treatment [10]. The multiple biological functions of Twist1 are elaborated in the following sections:
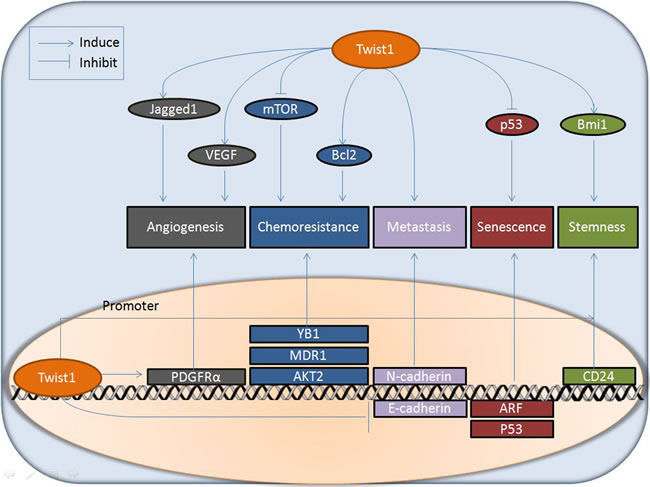
Figure2: Downstream effectors involved in multiple functions of Twist1 in cancer. Twist1 exerts its multiple biological effects (angiogenesis, chemo-resistance, metastasis, senescence, and stemness) via various downstream pathways, acting as a transcription factor regulating the expression of an array of target genes (such as PDGFRα, YB1, MDR1, AKT2, N-cadherin, E-cadherin, ARF, p53, and CD24) in the nucleus or modulating the function of effectors (e.g. Jagged1, VEGF, mTOR, Bcl2, p53, and Bmi1) at the protein level in the cytoplasm.
Contribution of Twist1 to tumor invasion and metastasis
Cancer metastasis consists of a sequence of distinct yet closely related steps: EMT, local invasion, intravasation, transit in blood or lymphatic circulation, extravasation, micro-metastasis, and colonization [75]. Twist1 has been well established as one of the master regulators of the EMT process. Additionally, extensive studies have demonstrated that Twist1 is also involved in other steps of the tumor invasion and metastasis process such as the formation of invadopodia [66], intravascular migration, extravasation [67] and vasculogenic mimicry (VM) formation [68, 76].
About a decade ago, Yang et al. first established the link between Twist1, EMT, and tumor metastasis by comparing the gene profile of different mouse tumor cell lines isolated from the same breast cancer [13]. Since then, the relationship between Twist1-induced EMT and cancer metastasis has been verified in a broad range of tumor types including hepatocellular carcinoma (HCC) [24], prostate cancer [77], gastric cancer [22], esophageal squamous cell carcinoma [25, 35], bladder cancer [19], pancreatic cancer [55], gliomas [30, 31], nasopharyngeal carcinoma [28], HNC [29], and epithelial ovarian carcinoma [78].
Although the up-stream regulators of Twist1 vary largely from one cancer to another during EMT, the down-stream effectors always involve the up-regulation of N-cadherin and/or down-regulation of E-cadherin. Twist1 has been demonstrated to induce N-cadherin at the mRNA level through the E-box cis-element located within the first intron of the N-cadherin gene in prostate cancer [79]. Furthermore, Twist1 can bind directly to the E-cadherin promoter, down-regulate promoter activity, and repress E-cadherin gene expression [80]. Further study reveals in detail that Twist1 interacts with several components of the Mi2/nucleosome remodeling and deacetylase (Mi2/NuRD) complex (MTA2, RbAp46, Mi2, HDAC2) and recruits them to the proximal regions of the E-cadherin promoter for transcriptional repression [81].
In addition to these cardinal EMT pathways, Twist1 also regulates other aspects of the tumor metastasis process via different signaling pathways. By using real-time intravital imaging of human tumor cells transplanted into transparent zebrafish, Stoletov et al. demonstrated that the expression of Twist1 in tumor cells increases their intravascular migration and extravasation through the vessel wall [67]. In addition, Twist1 promotes invadopodia formation via up-regulation of platelet-derived growth factor receptor (PDGFR) expression and activity [82]. Sun et al. discovered that Twist1 is frequently over-expressed in VM-positive HCCs, suggesting that Twist1 expression is likely to be associated with VM formation. This group further confirmed that over-expression of Twist1 significantly enhanced cell motility, invasiveness, and VM formation while Twist1 depletion substantially reduced cell migration, invasion, and VM formation [68].
Some microRNAs can also represent the target of Twist1 and mediate Twist1-induced EMT [83]. By binding to the putative promoter of miR-10b, Twist1 can induce the expression of miR-10b which inhibits the translation of homeobox D10, leading to the induction of pro-metastatic gene RHOC in breast cancer cells and a mouse model [84]. Additionally, Twist1 can suppress the expression of let-7i, resulting in the activation of RAC1 and enabling mesenchymal-mode movement in three-dimensional environments [85].
Twist1 as an oncoprotein promoting tumor initiation by evading senescence and apoptosis
Oncogenic insults usually induce p53 and/or retinoblastoma (Rb) expression and result in cell apoptosis or senescence, which are well-known defensive barriers against cell transformation and tumor initiation [86, 87]. Twist1 protein has been shown to override this safe-guard program to evade oncogene-induced senescence and apoptosis. Inactivation of Twist1 leads to the promotion of cellular senescence and cell growth arrest. In contrast, over-expression of Twist1 results in suppression of cellular senescence in response to genotoxic damage and promotion of cell proliferation with DNA damage accumulation [70]. Vichalkovski et al. demonstrated that PKB/AKT2 can phosphorylate Twist1 at Ser42 and inhibit p53 activity in response to DNA damage and this post-translational modification ensures functional activation of Twist1 after the promotion of survival during carcinogenesis [61]. Furthermore, by applying a functional screen for cDNAs that counteract the pro-apoptotic effects of the MYC oncogene, Maestro’s group discovered that Twist1 can bypass and inhibit p53-dependent cell death [88]. They proposed that Twist1 is capable of reducing expression of the ARF tumor suppressor and affecting p53 indirectly through modulation of the ARF/MDM2/p53 pathway. Consistent with this proposal, Kwok et al. demonstrated that Twist1-mediated cellular senescence was regulated through its negative effect on p14 (ARF) and subsequent suppression of MDM2/p53 and Chk1/2 DNA damage response pathways in prostate epithelial cells [70]. Similar results were observed by Valsesia et al. who found that Twist1 over-expression is responsible for inhibition of the ARF/p53 pathway involved in the Myc-dependent apoptotic response in neuroblastomas [71]. In addition to indirectly regulating p53 via ARF/MDM2, Paccinin et al. also demonstrated that Twist1 directly binds the p53 C-terminus through the Twist1 box which hinders the post-translational modification of p53 and facilitates its MDM2-mediated degradation [89]. Aside from regulating p53 at the post-transcription level, Twist1 was also shown to physically interact with HOXA-5 and negatively regulate p53 gene expression at the transcriptional level [90].
Conversely, Pinho et al. discovered that p53(-/-) pancreatic epithelial cells undergo EMT and express high levels of vimentin and of the transcriptional regulators Snai1, Snai2, Twist1, Zeb1 and Zeb2, implying that p53 inactivation in turn may promote Twist1 expression [91]. This study raised the possibility of mutual reciprocal regulation between Twist1 and p53, filling in the gap between p53 inactivation in tumor initiation and Twist1-induced tumor metastasis. This notion is supported by the study of Ansieau’s group who demonstrated that Twist1 overrides oncogene-induced premature senescence and simultaneously induces complete EMT, suggesting that some metastatic capabilities of cancer cells can be acquired during malignant conversion as a side effect of the inactivation of primary gatekeeper mechanisms [92]. However, Beck et al. established that Twist1 controls tumor initiation in a p53-dependent and -independent manner in the absence of EMT induction, suggesting Twist1-induced tumor initiation and EMT are not necessarily functionally linked [72]. This concept is discussed further in the perspectives and future directions section below.
Twist1 in a circuit of cancer stemness
Multiple lines of evidences have demonstrated the connection between EMT and cancer cell stemness [93]. One study reported that induction of EMT in immortalized human mammary epithelial cells (HMLEs) results in the expression of stem-cell markers and increased ability to form mammospheres, a property associated with mammary epithelial stem cells [94]. The same group further demonstrated that the EMT-derived cells are similar to mesenchymal stem cells (MSCs) in gene expression, multi-lineage differentiation, and migration ability. The functional connection between EMT and cancer stemness is molecularly mediated by the interdependency between Twist1 and Bmi-1 [95]. Bmi-1 is a polycomb-group protein that maintains self-renewal and stemness properties, which can be transcriptionally regulated by Twist1 via direct binding to the element on the Bmi-1 promoter. Bmi-1 knockdown leads to reversion of EMT while over-expression of Bmi-1 induces EMT, suggesting that Bmi-1 is critical mediator for Twist1-induced EMT. Conversely, silencing of Twist1 in Bmi1-overexpressing cells abolishes both EMT and stem-like properties. As a result, the interdependency between Twist1 and Bmi-1 provides the molecular connection between Twist1-induced EMT and stemness [73]. On the contrary, it is noteworthy that Twist1 can also promote the acquisition of stemness properties independent of EMT, as demonstrated by Vesuna et al. who established that the over-expression of Twist1 in breast cells can transcriptionally regulate CD24 expression and promote the generation of a breast cancer stem cell phenotype characterized by the high expression of CD44, and little or no expression of CD24 without inducing EMT [96]. In agreement with this study, Beck et al. also demonstrated that a low level of Twist1, which is insufficient to induce EMT, is able to endow skin tumor stemness [72]. Similarly, Schmidt et al. proposed that transient activation of Twist1, which is insufficient to induce EMT, is able to promote stem-cell-like properties such as mammosphere formation in human mammary epithelial cells. Persistent Twist1 could induce EMT but inhibits stem-cell-like properties and stemness only emerges and stably persists following Twist1 deactivation, implying the mutually exclusive nature of Twist1-induced EMT and stemness (see more detail in the perspectives and future directions section below) [97]. Taken together, this evidence suggests that it is the target genes down-stream of Twist1 (for example: CD24), rather than EMT, that confers cancer stem cell properties [98].
The association of Twist1 with tumor angiogenesis
Angiogenesis is a normal process that is transiently turned on under physiologic conditions such as female reproductive cycling and wound healing. In contrast, during tumor progression, angiogenesis is continually activated to help sustain expanding neoplastic growth [86]. Accumulating evidence has shown that Twist1 is positively associated with tumor angiogenesis [74]. Over-expression of Twist1 in breast cancer increases vascular endothelial growth factor (VEGF) expression and induces angiogenesis in vivo [99]. In agreement, Twist1 expression is positively correlated with up-regulation of VEGF in hepatocellular carcinoma cells and HCC specimens with positive Twist1 expression have a higher micro-vessel density than those without Twist1 expression [38]. Moreover, Twist1 is required for thrombin-induced angiogenesis and thrombin up-regulates Twist1, thus promoting endothelial cell migration, matrigel tubule formation, and tumor angiogenesis [100]. The same study shows that Twist1 mediates the thrombin induced up-regulation of angiogenesis growth factors and receptor proteins such as VEGF, GRO-α, KDR, Ang-2, MMP-1, and CD31 in both human breast cancer and murine melanoma cell lines. In addition to regulating vascular growth factors/receptors and activating endothelial cells in existing vessels, Twist1 can also enhance angiogenesis by inducing trans-differentiation of tumor cells into endothelial cells [101]. Twist1 can induce Jagged1 expression and subsequently activate KLF4, leading to endothelial differentiation in HNC cells. Interestingly, Bmi-1 is identified as another downstream effector of Twist1/Jagged-1 and this Twist1/Jagged-1/Bmi-1 axis confers stemness properties in HNC cells, indicating the molecular association between Twist1-induced angiogenesis and stemness [101].
Emerging role of Twist1 in drug resistance
Chemo-resistance is one of the biggest obstacles to the successful treatment of many cancers. Recently, emerging evidences has demonstrated that Twist1 can confer chemo-resistance in various cancer cell types. As summarized in Table 1, much effort has been made to explore the role of Twist1 in drug resistance.
Generally, the development of multidrug resistance is associated with increased expression of several ATP binding cassette transporters (ABC transporters) including ABCC1, ABCC3, ABCC4, ABCC5, and ABCC10. Saxena et al. found that Twist1 binds directly to the E-box elements of ABC transporters, identifying a molecular connection between Twist1 and multiple drug resistance [102]. Moreover, P-glycoprotein (P-gp, also known as MDR1) is a well-known member of the MDR/TAP subfamily of ABC transporters that pump many foreign substances (chemo-therapeutics) out of cells. Twist1 is co-expressed with P-gp in human bladder cancer cells and knockdown of Twist1 significantly sensitizes bladder cancer cells to anthracycline drugs via inhibiting P-gp expression [103]. The same group further demonstrated that DAB2IP can inhibit the phosphorylation and transactivation of STAT3, subsequently suppressing the expression of Twist1 and its target gene P-gp. This DAB2IP/STAT3/Twist1/P-gp axis is crucial for chemo-resistance to the anthracycline drugs (pirarubicin) and tumor re-growth of bladder cancer cells [104]. In agreement with this study, Li et al. demonstrated that Twist1-mediated EMT results in multidrug resistance and Twist1 depletion improves the efficacy of doxorubicin partially by suppression of drug-induced P-gp expression in breast cancer cells [105]. In addition, Deng et al. recently demonstrated that Twist1 and P-gp are also expressed correlatively in colorectal cancer cells (CRC) and confer CRC chemo-resistance to oxaliplatin [37]. Controversially, a recent study by Kong’s group suggests that the expression of ABC transporters (MDR1 and BCRP) is dispensable for the chemo-resistance mediated by Twist1 [106]. This seeming discrepancy might be explained by the use of cell lines with artificially elevated Hedgehog signaling pathway by Kong et al., which could enhance MDR1 and BCRP transcription, thus compensating for or even reversing the potential down-regulation of MDR1 and BCRP upon Twist1 knockdown.
In nasopharyngeal carcinoma, Twist1 is identified to play a central role in acquired resistance to paclitaxel. Up-regulation of Twist1 is associated with cellular resistance to microtubule-targeting anticancer drugs (vincristine and paclitaxel) but not to other drugs in nasopharyngeal, bladder, ovarian, and prostate cancer cells [107]. A subsequent mechanistic study by the same group suggests that Twist1-mediated paclitaxel resistance may be regulated through its positive involvement with the PI3K/AKT pathway [108]. In addition to the AKT pathway, Twist1 also positively regulates AKT2 expression by binding to E-box elements on the AKT2 promoter in breast cancer cells. AKT2 functions as a down-stream target of Twist1 and mediates Twist1-induced migration, invasion, and paclitaxel resistance [109]. In prostate carcinoma cells, Pham et al. demonstrated that Twist1 serves as a down-stream target of NF-κB, mediating NF-κB-induced chemo-resistance and TNF-α induced programmed cell death. Further detailed mechanistic study revealed that the protective activity of Twist1 seems to halt programmed cell death by controlling inhibitory Bcl-2 phosphorylation independently of interference with cytotoxic JNK, p53, and p19 (ARF) signaling [110]. In pancreatic cancer, deletion of mouse Twist1 did not halt the invasion, systemic dissemination or metastasis of pancreatic ductal adenocarcinoma (PDAC), but sensitized tumors to gemcitabine in vivo [111]. In bladder cancer, Twist1 regulates Y-box-binding protein-1 (YB1) expression and both Twist1 and YB1 are involved in cell growth, invasion, motility, and resistance to cisplatin and doxorubicin, but not to 5-fluorouracil (5-FU) [112]. As mentioned above, Twist1 can induce Jagged-1 expression and subsequently activate KLF4, not only inducing angiogenesis, but also conferring cisplatin and cetuximab resistance in HNC cells [101]. Of note, mitogen-activated protein kinases (ERK, JNK, and p38) are important down-stream mediators of Twist1-induced drug-resistance, but the specific effector varies from cancer to cancer. In nasopharyngeal carcinoma cells, down-regulation of Twist1 increases radio-sensitivity by inducing activation of the ERK pathway, but not the p-38 or JNK pathway [113]. However in pancreatic cancer cells, Twist1 promotes invasion and cisplatin resistance by inducing GDF15 expression by increasing p38 MAPK activity [114]. In non-small cell lung cancer (NSCLC), Zhuo et al. demonstrated that Twist1 silencing significantly sensitizes NSCLC cells to cisplatin by activating the JNK/mitochondrial pathway but not the ERK and p38 pathways [115]. Interestingly, Jin et al. proposed that Twist1 depletion sensitized NSCLC cells to cisplatin by stimulating AMPK-induced mTOR inhibition and subsequent reduction in Mcl-1 protein [116]. It is noteworthy that both groups used the same NSCLC cell line (A549) and drug (cisplatin) but obtained different results, implying that Twist1 might be able to mediate chemo-resistance through multiple down-stream pathways even in the same cancer type.
Recently, multiple studies suggest that various micro-RNAs are involved in chemo-resistance in a Twist1-dependent manner. Cisplatin-resistant ovarian cancer cell lines exhibit decreased miR-186 expression and increased Twist1 expression while the introduction of miR-186 can reverse drug resistance through Twist1 down-regulation [117]. Wang et al. demonstrated that PDGF-D markedly inhibited miR-106a expression and subsequently up-regulated Twist1 expression in gemcitabine-resistant hepatoma cells [118]. In tongue squamous cell carcinoma, Liu et al. found that miR-181a could reverse cisplatin resistance by directly targeting and repressing Twist1 expression [119]. In addition, miR-23a promoted cisplatin chemo-resistance and protected cisplatin-induced apoptosis through induction of Twist1 expression via a JNK-dependent mechanism in tongue squamous cell carcinoma cells [120].
Interestingly, down-regulation, rather than up-regulation of Twist1 is implicated in chemo-resistance in osteosarcoma. Zhou et al. demonstrated that Twist1 markedly decreases osteosarcoma cell survival following cisplatin treatment partially by down-regulating the endothelin-1 (ET1)/endothelin A receptor (ETAR) signaling via inhibition of the PI3K/AKT pathway [121]. Consistently, a subsequent study found that miR-33a promotes osteosarcoma cell resistance to cisplatin by down-regulating Twist1. Inhibition of miR-33a up-regulates Twist1 expression and enhances cisplatin-induced apoptosis [122].
Perspectives and future directions
Taken together, multiple studies show that Twist1 is a transcription factor that is over-expressed in a wide variety of carcinomas, and is implicated in many aspects of the carcinogenesis process including tumor initiation, stemness, angiogenesis, invasion, metastasis, and drug resistance. Among these varied functions, the contribution of Twist1 to EMT is the most thoroughly explored and extensively verified in various cancers. The involvement of Twist1 in tumor initiation, stemness, angiogenesis, and drug resistance and the underlying mechanisms remain largely undefined. Further detailed studies exploring how Twist1 participates in these processes will provide more comprehensive information about the crucial pathological functions of Twist1 in cancer. Moreover, instead of studying these seemingly independent pathological effects of Twist1 separately, there is growing interest in exploring the correlation and interdependency of these multiple biological functions. For example, Ansieau’s group discovered that Twist1 overrides oncogene-induced premature senescence and simultaneously induces complete EMT, indicating that Twist1-induced early escape from gatekeeper protection and the acquisition of invasive features are directly linked [69]. However, Beck et al. established that low levels of Twist1 control tumor initiation in both a p53-dependent and -independent manner without inducing EMT, suggesting that Twist1-induced tumor initiation and EMT are not necessarily functionally related [72]. This apparent inconsistency raises the notion that Twist1 function might be dictated by its expression level, namely, a low level of Twist1 promotes tumor initiation while a relatively higher level of Twist1 expression is a prerequisite for EMT induction. In agreement with this theory, Beck et al. demonstrated that a low level of Twist1, which is insufficient to induce EMT, is able to confer skin tumor stemness [72]. Continued efforts should be made to further explore the correlation between Twist1 expression level and its biological effects.
Twist1-induced EMT and stemness are suggested to be functionally associated by the molecular connection between Twist1 and Bmi-1 [73]. In contrast, Twist1 can also promote the acquisition of stemness properties in the absence of EMT induction. Vesuna et al. established that the over-expression of Twist1 in breast cells can transcriptionally regulate CD24 expression and promote the generation of a breast cancer stem cell phenotype independent of EMT [96]. This discrepancy may be explained by Schmidt’s observation that transient activation of Twist1, at a level insufficient to induce EMT, is able to promote stem-cell-like properties. Persistent Twist1 can induce EMT but inhibit stem-cell-like properties and stemness only emerges and stably persists following Twist1 deactivation, implying the mutually exclusive nature of Twist1-induced EMT and stemness [97]. As a result, in addition to regulating tumor biology in an expression level-dependent manner, the temporal regulation of Twist1 is also important for its distinctive biological functions. The “spatiotemporal” regulation of EMT by Twist1 was suggested by Tsai et al. who proposed that different Twist1 expression levels at different times in different places dictate EMT, tumor dissemination, proliferation and the formation of metastases [123]. Still, little is known about the temporal regulation of Twist1 and this field could be a promising direction for future research.
Twist1-induced angiogenesis and stemness are also functionally connected. Twist1 can induce Jagged-1 expression and subsequently activate KLF-4, leading to endothelial differentiation in HNC cells. Interestingly, Bmi-1 is identified as another downstream effector of Twist1/Jagged-1 and this Twist1/Jagged-1/Bmi-1 axis confers stemness properties in HNC cells, indicating the molecular connection between Twist1-induced angiogenesis and stemness [101]. Cancer stemness is well-established to be closely related to drug resistance [124, 125]. However, the detailed signaling pathways linking Twist1-induced drug resistance and cancer cell stemness remain elusive.
In addition to pursuing these many promising fields of Twist1 bench research, several areas can be further explored on the bed side. To date, little is known about the basal level of Twist1 in different tissues in healthy individuals and there is no efficient method for Twist1 detection. Quantitative information about the genetic mutation, amplification, and deletion of Twist1 is also lacking.
In summary, based on the significance of Twist1 in various hallmark properties of cancer, detailed studies are needed to delineate and unveil the underlying mechanisms of Twist1’s multiple pathological functions. A comprehensive understating of Twist1’s diversified roles in cancer biology will lay the foundation for further modulation of Twist1 as a diagnostic indicator, prognostic marker, and a therapeutic target for cancer treatment in the clinic.
Acknowledgments
We owe special thanks to Dr. Anthea Hammond for her critical comments and editorial review of this article.
Conflicts of Interest
No potential conflicts of interest were disclosed.
Grant Support
This work was supported by grants from the NCI of the NIH under award number U01CA151802.
References
1. Simpson P. Maternal-zygotic gene interactions during formation of the dorsoventral pattern in Drosophila embryos. Genetics. 1983; 105:615-632.
2. Thisse B, el MM, Perrin-Schmitt F. The twist gene: isolation of a Drosophila zygotic gene necessary for the establishment of dorsoventral pattern. Nucleic Acids Res. 1987; 15:3439-3453.
3. Nusslein-Volhard C, Wieschaus E, Kluding H. Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster. Roux’s Arch Dev Biol. 1984; 193:267-282.
4. Bourgeois P, Stoetzel C, Bolcato-Bellemin AL, Mattei MG, Perrin-Schmitt F. The human H-twist gene is located at 7p21 and encodes a B-HLH protein that is 96% similar to its murine M-twist counterpart. Mamm Genome. 1996; 7:915-917.
5. Howard TD, Paznekas WA, Green ED, Chiang LC, Ma N, Ortiz de Luna RI, Garcia Delgado C, Gonzalez-Ramos M, Kline AD, Jabs EW. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet. 1997; 15:36-41.
6. el Ghouzzi V, Le Merrer M, Perrin-Schmitt F. Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat Genet. 1997; 15:42-46.
7. Rose CS, Patel P, Reardon W, Malcolm S, Winter RM. The TWIST gene, although not disrupted in Saethre-Chotzen patients with apparently balanced translocations of 7p21, is mutated in familial and sporadic cases. Hum Mol Genet. 1997; 6:1369-1373.
8. Ansieau S, Morel AP, Hinkal G, Bastid J, Puisieux A. TWISTing an embryonic transcription factor into an oncoprotein. Oncogene. 2010; 29:3173-3184.
9. Qin Q, Xu Y, He T, Qin C, Xu J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012; 22:90-106.
10. Khan MA, Chen HC, Zhang D, Fu J. Twist: a molecular target in cancer therapeutics. Tumour Biol. 2013; 34:2497-2506.
11. Merindol N, Riquet A, Szablewski V, Eliaou JF, Puisieux A, Bonnefoy N. The emerging role of Twist proteins in hematopoietic cells and hematological malignancies. Blood Cancer J. 2014;4:e206.
12. Zhu QQ, Ma C, Wang Q, Song Y, Lv T. The role of TWIST1 in epithelial-mesenchymal transition and cancers. Tumour Biol. 2016; 37:185-197.
13. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004; 117:927-939.
14. Mehrotra J, Vali M, McVeigh M, Kominsky SL, Fackler MJ, Lahti-Domenici J, Polyak K, Sacchi N, Garrett-Mayer E, Argani P, Sukumar S. Very high frequency of hypermethylated genes in breast cancer metastasis to the bone, brain, and lung. Clin Cancer Res. 2004; 10:3104-3109.
15. Martin TA, Goyal A, Watkins G, Jiang WG. Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Ann Surg Oncol. 2005; 12:488-496.
16. Sahlin P, Windh P, Lauritzen C, Emanuelsson M, Gronberg H, Stenman G. Women with Saethre-Chotzen syndrome are at increased risk of breast cancer. Genes Chromosomes Cancer. 2007; 46:656-660.
17. Gort EH, Suijkerbuijk KP, Roothaan SM, Raman V, Vooijs M, van der Wall E, van Diest PJ. Methylation of the TWIST1 promoter, TWIST1 mRNA levels, and immunohistochemical expression of TWIST1 in breast cancer. Cancer Epidemiol Biomarkers Prev. 2008; 17:3325-3330.
18. Van der Auwera I, Bovie C, Svensson C, Trinh XB, Limame R, van Dam P, van Laere SJ, van Marck EA, Dirix LY, Vermeulen PB. Quantitative methylation profiling in tumor and matched morphologically normal tissues from breast cancer patients. BMC Cancer. 2010; 10:97.
19. Zhang Z, Xie D, Li X, Wong YC, Xin D, Guan XY. Significance of TWIST expression and its association with E-cadherin in bladder cancer. Hum Pathol. 2007; 38:598-606.
20. Yu Q, Zhang K, Wang X, Liu X, Zhang Z. Expression of transcription factors snail, slug, and twist in human bladder carcinoma. J Exp Clin Cancer Res. 2010; 29:119.
21. Renard I, Joniau S, van Cleynenbreugel B, Collette C, Naome C, Vlassenbroeck I, Nicolas H, de Leval J, Straub J, Van Criekinge W, Hamida W, Hellel M, Thomas A, et al. Identification and validation of the methylated TWIST1 and NID2 genes through real-time methylation-specific polymerase chain reaction assays for the noninvasive detection of primary bladder cancer in urine samples. Eur Urol. 2010; 58:96-104.
22. Luo GQ, Li JH, Wen JF, Zhou YH, Hu YB, Zhou JH. Effect and mechanism of the Twist gene on invasion and metastasis of gastric carcinoma cells. World J Gastroenterol. 2008; 14:2487-2493.
23. Schneider BG, Peng DF, Camargo MC, Piazuelo MB, Sicinschi LA, Mera R, Romero-Gallo J, Delgado AG, Bravo LE, Wilson KT, Peek RM, Jr., Correa P, El-Rifai W. Promoter DNA hypermethylation in gastric biopsies from subjects at high and low risk for gastric cancer. Int J Cancer. 2010; 127:2588-2597.
24. Lee TK, Poon RT, Yuen AP, Ling MT, Kwok WK, Wang XH, Wong YC, Guan XY, Man K, Chau KL, Fan ST. Twist overexpression correlates with hepatocellular carcinoma metastasis through induction of epithelial-mesenchymal transition. Clin Cancer Res. 2006; 12:5369-5376.
25. Yuen HF, Chan YP, Wong ML, Kwok WK, Chan KK, Lee PY, Srivastava G, Law SY, Wong YC, Wang X, Chan KW. Upregulation of Twist in oesophageal squamous cell carcinoma is associated with neoplastic transformation and distant metastasis. J Clin Pathol. 2007; 60:510-514.
26. Sasaki K, Natsugoe S, Ishigami S. Significance of Twist expression and its association with E-cadherin in esophageal squamous cell carcinoma. J Exp Clin Cancer Res. 2009; 28:158.
27. Song LB, Liao WT, Mai HQ, Zhang HZ, Zhang L, Li MZ, Hou JH, Fu LW, Huang WL, Zeng YX, Zeng MS. The clinical significance of twist expression in nasopharyngeal carcinoma. Cancer Lett. 2006; 242:258-265.
28. Horikawa T, Yang J, Kondo S, Yoshizaki T, Joab I, Furukawa M, Pagano JS. Twist and epithelial-mesenchymal transition are induced by the EBV oncoprotein latent membrane protein 1 and are associated with metastatic nasopharyngeal carcinoma. Cancer Res. 2007; 67:1970-1978.
29. Ou DL, Chien HF, Chen CL, Lin TC, Lin LI. Role of Twist in head and neck carcinoma with lymph node metastasis. Anticancer Res. 2008; 28:1355-1359.
30. Elias MC, Tozer KR, Silber JR, Mikheeva S, Deng M, Morrison RS, Manning TC, Silbergeld DL, Glackin CA, Reh TA, Rostomily RC. TWIST is expressed in human gliomas and promotes invasion. Neoplasia. 2005; 7:824-837.
31. Mikheeva SA, Mikheev AM, Petit A, Beyer R, Oxford RG, Khorasani L, Maxwell JP, Glackin CA, Wakimoto H, Gonzalez-Herrero I, Sanchez-Garcia I, Silber JR, Horner PJ, et al. TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol Cancer. 2010; 9:194.
32. Cosset E, Hamdan G, Jeanpierre S, Voeltzel T, Sagorny K, Hayette S, Mahon FX, Dumontet C, Puisieux A, Nicolini FE, Maguer-Satta V. Deregulation of TWIST-1 in the CD34+ compartment represents a novel prognostic factor in chronic myeloid leukemia. Blood. 2011; 117:1673-1676.
33. Fondrevelle ME, Kantelip B, Reiter RE, Chopin DK, Thiery JP, Monnien F, Bittard H, Wallerand H. The expression of Twist has an impact on survival in human bladder cancer and is influenced by the smoking status. Urol Oncol. 2009; 27:268-276.
34. Shibata K, Kajiyama H, Ino K, Terauchi M, Yamamoto E, Nawa A, Nomura S, Kikkawa F. Twist expression in patients with cervical cancer is associated with poor disease outcome. Ann Oncol. 2008; 19:81-85.
35. Xie F, Li K, Ouyang X. Twist, an independent prognostic marker for predicting distant metastasis and survival rates of esophageal squamous cell carcinoma patients. Clin Exp Metastasis. 2009; 26:1025-1032.
36. Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC, Wu KJ. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008; 10:295-305.
37. Deng JJ, Zhang W, Xu XM, Zhang F, Tao WP, Ye JJ, Ge W. Twist mediates an aggressive phenotype in human colorectal cancer cells. Int J Oncol. 2016; 48:1117-1124.
38. Niu RF, Zhang L, Xi GM, Wei XY, Yang Y, Shi YR, Hao XS. Up-regulation of Twist induces angiogenesis and correlates with metastasis in hepatocellular carcinoma. J Exp Clin Cancer Res. 2007; 26:385-394.
39. Yang MH, Chen CL, Chau GY, Chiou SH, Su CW, Chou TY, Peng WL, Wu JC. Comprehensive analysis of the independent effect of twist and snail in promoting metastasis of hepatocellular carcinoma. Hepatology. 2009; 50:1464-1474.
40. Hoek K, Rimm DL, Williams KR, Zhao H, Ariyan S, Lin A, Kluger HM, Berger AJ, Cheng E, Trombetta ES, Wu T, Niinobe M, Yoshikawa K, et al. Expression profiling reveals novel pathways in the transformation of melanocytes to melanomas. Cancer Res. 2004; 64:5270-5282.
41. Kajiyama H, Hosono S, Terauchi M, Shibata K, Ino K, Yamamoto E, Nomura S, Nawa A, Kikkawa F. Twist expression predicts poor clinical outcome of patients with clear cell carcinoma of the ovary. Oncology. 2006; 71:394-401.
42. Hosono S, Kajiyama H, Terauchi M, Shibata K, Ino K, Nawa A, Kikkawa F. Expression of Twist increases the risk for recurrence and for poor survival in epithelial ovarian carcinoma patients. Br J Cancer. 2007; 96:314-320.
43. Ling X, Arlinghaus RB. Knockdown of STAT3 expression by RNA interference inhibits the induction of breast tumors in immunocompetent mice. Cancer Res. 2005; 65:2532-2536.
44. Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola D, Mansour M, Xu LM, Costanzo C, Cheng JQ, Wang LH. Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J Biol Chem. 2008; 283:14665-14673.
45. Zhang C, Guo F, Xu G, Ma J, Shao F. STAT3 cooperates with Twist to mediate epithelial-mesenchymal transition in human hepatocellular carcinoma cells. Oncol Rep. 2015; 33:1872-1882.
46. Hsu KW, Hsieh RH, Huang KH, Fen-Yau Li A, Chi CW, Wang TY, Tseng MJ, Wu KJ, Yeh TS. Activation of the Notch1/STAT3/Twist signaling axis promotes gastric cancer progression. Carcinogenesis. 2012; 33:1459-1467.
47. Cho KH, Jeong KJ, Shin SC, Kang J, Park CG, Lee HY. STAT3 mediates TGF-beta1-induced TWIST1 expression and prostate cancer invasion. Cancer Lett. 2013; 336:167-173.
48. Jiang J, Kosman D, Ip YT, Levine M. The dorsal morphogen gradient regulates the mesoderm determinant twist in early Drosophila embryos. Genes Dev. 1991; 5:1881-1891.
49. Sosic D, Richardson JA, Yu K, Ornitz DM, Olson EN. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell. 2003; 112:169-180.
50. Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, Chao CH, Yamaguchi H, Yang NK, Ding Q, Wang Y, Lai YJ, LaBaff AM, et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012; 72:1290-1300.
51. Kajiro M, Hirota R, Nakajima Y, Kawanowa K, So-ma K, Ito I, Yamaguchi Y, Ohie SH, Kobayashi Y, Seino Y, Kawano M, Kawabe Y, Takei H, et al. The ubiquitin ligase CHIP acts as an upstream regulator of oncogenic pathways. Nat Cell Biol. 2009; 11:312-319.
52. Qin L, Liu Z, Chen H, Xu J. The steroid receptor coactivator-1 regulates twist expression and promotes breast cancer metastasis. Cancer Res. 2009; 69:3819-3827.
53. Howe LR, Watanabe O, Leonard J, Brown AM. Twist is up-regulated in response to Wnt1 and inhibits mouse mammary cell differentiation. Cancer Res. 2003; 63:1906-1913.
54. Zhang L, Yang M, Gan L, He T, Xiao X, Stewart MD, Liu X, Yang L, Zhang T, Zhao Y, Fu J. DLX4 upregulates TWIST and enhances tumor migration, invasion and metastasis. Int J Biol Sci. 2012; 8:1178-1187.
55. Satoh K, Hamada S, Kimura K, Kanno A, Hirota M, Umino J, Fujibuchi W, Masamune A, Tanaka N, Miura K, Egawa S, Motoi F, Unno M, et al. Up-regulation of MSX2 enhances the malignant phenotype and is associated with twist 1 expression in human pancreatic cancer cells. Am J Pathol. 2008; 172:926-939.
56. Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin CH, Moustakas A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J Cell Biol. 2006; 174:175-183.
57. Lee YH, Albig AR, Regner M, Schiemann BJ, Schiemann WP. Fibulin-5 initiates epithelial-mesenchymal transition (EMT) and enhances EMT induced by TGF-beta in mammary epithelial cells via a MMP-dependent mechanism. Carcinogenesis. 2008; 29:2243-2251.
58. Xue G, Hemmings BA. Phosphorylation of basic helix-loop-helix transcription factor Twist in development and disease. Biochem Soc Trans. 2012; 40:90-93.
59. Hong J, Zhou J, Fu J. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011; 71:3980-3990.
60. Li CW, Xia W, Lim SO, Hsu JL, Huo L, Wu Y, Li LY, Lai CC, Chang SS, Hsu YH, Sun HL, Kim J, Yamaguchi H, et al. AKT1 Inhibits Epithelial-to-Mesenchymal Transition in Breast Cancer through Phosphorylation-Dependent Twist1 Degradation. Cancer Res. 2016; 76:1451-1462.
61. Vichalkovski A, Gresko E, Hess D, Restuccia DF, Hemmings BA. PKB/AKT phosphorylation of the transcription factor Twist-1 at Ser42 inhibits p53 activity in response to DNA damage. Oncogene. 2010; 29:3554-3565.
62. Su YW, Xie TX, Sano D, Myers JN. IL-6 stabilizes Twist and enhances tumor cell motility in head and neck cancer cells through activation of casein kinase 2. PloS one. 2011; 6:e19412.
63. Lander R, Nordin K, LaBonne C. The F-box protein Ppa is a common regulator of core EMT factors Twist, Snail, Slug, and Sip1. J Cell Biol. 2011; 194:17-25.
64. Yang WH, Su YH, Hsu WH, Wang CC, Arbiser JL, Yang MH. Imipramine blue halts head and neck cancer invasion through promoting F-box and leucine-rich repeat protein 14-mediated Twist1 degradation. Oncogene. 2016; 35:2287-2298.
65. Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, Lin Y, Li J, Kang T, Tao M, Rusinova E, Zhang G, Wang C, et al., Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell. 2014;25:210-225.
66. Eckert MA, Lwin TM, Chang AT. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell. 2011; 19:372-386.
67. Stoletov K, Kato H, Zardouzian E, Kelber J, Yang J, Shattil S, Klemke R. Visualizing extravasation dynamics of metastatic tumor cells. J Cell Sci. 2010; 123:2332-2341.
68. Sun T, Zhao N, Zhao XL. Expression and functional significance of Twist1 in hepatocellular carcinoma: its role in vasculogenic mimicry. Hepatology. 2010; 51:545-556.
69. Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S, Maestro R, Voeltzel T, Selmi A, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008; 14:79-89.
70. Kwok WK, Ling MT, Yuen HF, Wong YC, Wang X. Role of p14ARF in TWIST-mediated senescence in prostate epithelial cells. Carcinogenesis. 2007; 28:2467-2475.
71. Valsesia-Wittmann S, Magdeleine M, Dupasquier S, Garin E, Jallas AC, Combaret V, Krause A, Leissner P, Puisieux A. Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell. 2004; 6:625-630.
72. Beck B, Lapouge G, Rorive S, Drogat B, Desaedelaere K, Delafaille S, Dubois C, Salmon I, Willekens K, Marine JC, Blanpain C. Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell. 2015; 16:67-79.
73. Yang MH, Hsu DS, Wang HW. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol. 2010; 12:982-992.
74. Tseng JC, Chen HF, Wu KJ. A twist tale of cancer metastasis and tumorangiogenesis. Histol Histopathol. 2015; 30:1283-94.
75. Cheng GZ, Zhang W, Wang LH. Regulation of cancer cell survival, migration, and invasion by Twist: AKT2 comes to interplay. Cancer Res. 2008; 68:957-960.
76. Sun B, Zhang D, Zhao N and Zhao X. Epithelial-to-endothelial transition and cancer stem cells: two cornerstones of vasculogenic mimicry in malignant tumors. Oncotarget. 2016; doi: 10.18632/oncotarget.8461.
77. Yuen HF, Chua CW, Chan YP, Wong YC, Wang X, Chan KW. Significance of TWIST and E-cadherin expression in the metastatic progression of prostatic cancer. Histopathology. 2007; 50:648-658.
78. Terauchi M, Kajiyama H, Yamashita M. Possible involvement of TWIST in enhanced peritoneal metastasis of epithelial ovarian carcinoma. Clin Exp Metastasis. 2007; 24:329-339.
79. Alexander NR, Tran NL, Rekapally H, Summers CE, Glackin C, Heimark RL. N-cadherin gene expression in prostate carcinoma is modulated by integrin-dependent nuclear translocation of Twist1. Cancer Res. 2006; 66:3365-3369.
80. Vesuna F, van Diest P, Chen JH, Raman V. Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem Biophys Res Commun. 2008; 367:235-241.
81. Fu J, Qin L, He T. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011; 21:275-289.
82. Eckert MA, Yang J. Targeting invadopodia to block breast cancer metastasis. Oncotarget. 2011; 2:562–568. doi: 10.18632/oncotarget.301.
83. Khanbabaei H, Teimoori A, Mohammadi M. The interplay between microRNAs and Twist1 transcription factor: a systematic review. Tumour Biol. 2016;37:7007-7019.
84. Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449(7163):682-688.
85. Yang WH, Lan HY, Huang CH, Tai SK, Tzeng CH, Kao SY, Wu KJ, Hung MC, Yang MH. RAC1 activation mediates Twist1-induced cancer cell migration. Nat Cell Biol. 2012;14:366-374.
86. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
87. Amin AR, Karpowicz PA, Carey TE, Arbiser J, Nahta R, Chen ZG, Dong JT, Kucuk O, Khan GN, Huang GS, Mi S, Lee HY, Reichrath J, et al. Evasion of anti-growth signaling: A key step in tumorigenesis and potential target for treatment and prophylaxis by natural compounds. Semin Cancer Biol. 2015; 35 Suppl:S55-77.
88. Maestro R, Dei Tos AP, Hamamori Y, Krasnokutsky S, Sartorelli V, Kedes L, Doglioni C, Beach DH, Hannon GJ. Twist is a potential oncogene that inhibits apoptosis. Genes Dev. 1999; 13:2207-2217.
89. Piccinin S, Tonin E, Sessa S, Demontis S, Rossi S, Pecciarini L, Zanatta L, Pivetta F, Grizzo A, Sonego M, Rosano C, Dei Tos AP, Doglioni C, et al. A “twist box” code of p53 inactivation: twist box: p53 interaction promotes p53 degradation. Cancer Cell. 2012; 22:404-415.
90. Stasinopoulos IA, Mironchik Y, Raman A, Wildes F, Winnard P, Jr., Raman V. HOXA5-twist interaction alters p53 homeostasis in breast cancer cells. J Biol Chem. 2005; 280:2294-2299.
91. Pinho AV, Rooman I, Real FX. p53-dependent regulation of growth, epithelial-mesenchymal transition and stemness in normal pancreatic epithelial cells. Cell Cycle. 2011; 10:1312-1321.
92. Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008; 14:818-829.
93. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009; 139(5):871-890.
94. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133:704-715.
95. Wu KJ, Yang MH. Epithelial-mesenchymal transition and cancer stemness: the Twist1-Bmi1 connection. Bioscience reports. 2011; 31:449-455.
96. Vesuna F, Lisok A, Kimble B, Raman V. Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia. 2009; 11:1318-1328.
97. Schmidt JM, Panzilius E, Bartsch HS, Irmler M, Beckers J, Kari V, Linnemann JR, Dragoi D, Hirschi B, Kloos UJ, Sass S, Theis F, Kahlert S, et al.. Stem-cell-like properties and epithelial plasticity arise as stable traits after transient Twist1 activation. Cell reports. 2015; 10)131-139.
98. Jung HY, Yang J. Unraveling the TWIST between EMT and cancer stemness. Cell Stem Cell. 2015; 16:1-2.
99. Mironchik Y, Winnard PT, Jr., Vesuna F. Twist overexpression induces in vivo angiogenesis and correlates with chromosomal instability in breast cancer. Cancer Res. 2005; 65:10801-10809.
100. Hu L, Roth JM, Brooks P, Ibrahim S, Karpatkin S. Twist is required for thrombin-induced tumor angiogenesis and growth. Cancer Res. 2008; 68:4296-4302.
101. Chen HF, Huang CH, Liu CJ, Hung JJ, Hsu CC, Teng SC, Wu KJ. Twist1 induces endothelial differentiation of tumour cells through the Jagged1-KLF4 axis. Nat Commun. 2014; 5:4697.
102. Saxena M, Stephens MA, Pathak H, Rangarajan A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011; 2:e179.
103. Chen Y, Li L, Zeng J, Wu K, Zhou J, Guo P, Zhang D, Xue Y, Liang L, Wang X, Chang LS, He D. Twist confers chemoresistance to anthracyclines in bladder cancer through upregulating P-glycoprotein. Chemotherapy. 2012; 58:264-272.
104. Wu K, Wang B, Chen Y, Zhou J, Huang J, Hui K, Zeng J, Zhu J, Zhang K, Li L, Guo P, Wang X, Hsieh JT, et al. DAB2IP regulates the chemoresistance to pirarubicin and tumor recurrence of non-muscle invasive bladder cancer through STAT3/Twist1/P-glycoprotein signaling. Cell Signal. 2015; 27:2515-2523.
105. Li QQ, Xu JD, Wang WJ, Cao XX, Chen Q, Tang F, Chen ZQ, Liu XP, Xu ZD. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin Cancer Res. 2009; 15:2657-2665.
106. Kong Y, Peng Y, Liu Y, Xin H, Zhan X, Tan W. Twist1 and Snail link Hedgehog signaling to tumor-initiating cell-like properties and acquired chemoresistance independently of ABC transporters. Stem Cells. 2015; 33:1063-1074.
107. Wang X, Ling MT, Guan XY, Tsao SW, Cheung HW, Lee DT, Wong YC. Identification of a novel function of TWIST, a bHLH protein, in the development of acquired taxol resistance in human cancer cells. Oncogene. 2004; 23:474-482.
108. Zhang X, Wang Q, Ling MT, Wong YC, Leung SC, Wang X. Anti-apoptotic role of TWIST and its association with Akt pathway in mediating taxol resistance in nasopharyngeal carcinoma cells. Int J Cancer. 2007; 120:1891-1898.
109. Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007; 67:1979-1987.
110. Pham CG, Bubici C, Zazzeroni F, Knabb JR, Papa S, Kuntzen C, Franzoso G. Upregulation of Twist-1 by NF-kappaB blocks cytotoxicity induced by chemotherapeutic drugs. Mol Cell Biol. 2007; 27:3920-3935.
111. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015; 527:525-530.
112. Shiota M, Yokomizo A, Itsumi M, Uchiumi T, Tada Y, Song Y, Kashiwagi E, Masubuchi D, Naito S. Twist1 and Y-box-binding protein-1 promote malignant potential in bladder cancer cells. BJU Int. 2011; 108(2 Pt 2):E142-149.
113. Zhuo X, Chang A, Huang C, Yang L, Zhao H, Wu Y, Zhou Q. Nanoparticle-mediated down-regulation of TWIST increases radiosensitivity of nasopharyngeal carcinoma cells via ERK pathway. Am J Cancer Res. 2015; 5:1571-1579.
114. Ji H, Lu HW, Li YM, Lu L, Wang JL, Zhang YF, Shang H. Twist promotes invasion and cisplatin resistance in pancreatic cancer cells through growth differentiation factor 15. Mol Med Rep. 2015; 12:3841-3848.
115. Zhuo WL, Wang Y, Zhuo XL, Zhang YS, Chen ZT. Short interfering RNA directed against TWIST, a novel zinc finger transcription factor, increases A549 cell sensitivity to cisplatin via MAPK/mitochondrial pathway. Biochem Biophys Res Commun. 2008; 369:1098-1102.
116. Jin HO, Hong SE, Woo SH, Lee JH, Choe TB, Kim EK, Noh WC, Lee JK, Hong SI, Kim JI, Park IC. Silencing of Twist1 sensitizes NSCLC cells to cisplatin via AMPK-activated mTOR inhibition. Cell Death Dis. 2012; 3:e319.
117. Zhu X, Shen H, Yin X, Long L, Xie C, Liu Y, Hui L, Lin X, Fang Y, Cao Y, Xu Y, Li M, Xu W, et al. miR-186 regulation of Twist1 and ovarian cancer sensitivity to cisplatin. Oncogene. 2016; 35:323-332.
118. Wang R, Li Y, Hou Y, Yang Q, Chen S, Wang X, Wang Z, Yang Y, Chen C, Wang Z, Wu Q. The PDGF-D/miR-106a/Twist1 pathway orchestrates epithelial-mesenchymal transition in gemcitabine resistance hepatoma cells. Oncotarget. 2015; 6:7000–7010. doi: 10.18632/oncotarget.3193.
119. Liu M, Wang J, Huang H, Hou J, Zhang B and Wang A. miR-181a-Twist1 pathway in the chemoresistance of tongue squamous cell carcinoma. Biochem Biophys Res Commun. 2013; 441:364-370.
120. Peng F, Zhang H, Du Y, Tan P. miR-23a promotes cisplatin chemoresistance and protects against cisplatin-induced apoptosis in tongue squamous cell carcinoma cells through Twist. Oncol Rep. 2015; 33:942-950.
121. Zhou Y, Zang X, Huang Z, Zhang C. TWIST interacts with endothelin-1/endothelin A receptor signaling in osteosarcoma cell survival against cisplatin. Oncol Lett. 2013; 5:857-861.
122. Zhou Y, Huang Z, Wu S, Zang X, Liu M, Shi J. miR-33a is up-regulated in chemoresistant osteosarcoma and promotes osteosarcoma cell resistance to cisplatin by down-regulating TWIST. J Exp Clin Cancer Res. 2014; 33:12.
123. Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012; 22:725-736.
124. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005; 5:275-284.
125. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001; 414:105-111.