
1. The SP/KLF-family of transcription factors
Specificity proteins (SP) and Krüppel-like factors (KLFs) are collectively referred to as the SP1-like/KLF or SP/KLF family of transcription factors. SP1 was first identified in the early 1980s as a protein that was able to bind GC- and related GT-rich regions or CACCC elements in the SV40 promoter and, therefore, could serve as a transcriptional regulator [1]. Several SP-like factors have been found since and, to date, this subfamily contains 9 members (SP1 – 9). The DNA-binding region of SPs consists of three highly conserved Cys2/His2 zinc fingers, localized near the C-terminal end of the protein [2]. The interfinger linking sequences, called “H/C links”, containing a stretch of seven amino acids also show a high degree of conservation (TGE(R/K)P(Y/F)X) within the family and between species [3]. The presence of this typical structure in the Drosophila melanogaster gap gene Krüppel [4] has given rise to the association with the Krüppel-like part of this transcription factor family [5].
The first mammalian homologue to Drosophila Krüppel was discovered in a murine erythroleukemic cell line [6] and named E-KLF (Erythroid Krüppel-like factor, KLF1). This factor was shown to trans-activate ß-globin expression by binding the CACCC element within its promoter [7]. Loss-of-function studies demonstrated that homozygous E-KLF-/- mice developed fatal ß-thalassaemia during early fetal liver erythropoiesis [8] Numerous closely related human proteins have been identified since and collectively named Krüppel-like factors (KLFs), preceded by an index letter of the tissue or origin of enriched expression (X-KLF, Table 1 and Fig 1a).
Table 1: Classification and summary of the SP/KLF family in humans.
HGNC |
Alias |
Gene accession # |
UniProt KB entry # |
# amino acids |
Molecular weight (kDa) |
SP1 |
TFSP1 |
BC062539 |
P08047 |
785 |
80.7 |
SP2 |
KIAA0048 |
NM_003110 |
Q02086 |
613 |
64.9 |
SP3 |
SPR-2 |
AY070137 |
Q02447 |
781 |
81.9 |
SP4 |
SPR-1,HF1B, MGC130008, MGC130009 |
NM_003112 |
Q02446 |
784 |
82 |
SP5 |
AB096175 |
Q6BEB4 |
398 |
42 |
|
SP7 |
OSX |
BC113613 |
Q8TDD2 |
431 |
45 |
SP8 |
BC038669 |
Q8IXZ3 |
490 |
48.7 |
|
SP9 |
ZNF990 |
NM_001145250 |
P0CG40 |
484 |
48.9 |
KLF1 |
E-KLF |
JX877554 |
Q13351 |
362 |
38.2 |
KLF2 |
L-KLF |
EF078888 |
Q9Y5W3 |
355 |
37.4 |
KLF3 |
B-KLF, TEF-2 |
NM_016531 |
P57682 |
345 |
38.8 |
KLF4 |
G-KLF, EZF |
DQ658241 |
O43474 |
513 |
54 |
KLF5 |
I-KLF, C-KLF, BTEB2 |
AF287272 |
Q13887 |
457 |
50.8 |
KLF6 |
BCD1, COBEP, CBPB, ST12, GBF |
AF284036 |
Q99612 |
283 |
31.9 |
KLF7 |
U-KLF |
O75840 |
302 |
33.4 |
|
KLF8 |
BKLF3, ZNF741 |
NM_007250 |
O95600 |
359 |
39.3 |
KLF9 |
BTEB, BTEB1 |
NM_001206 |
Q13886 |
244 |
27.2 |
KLF10 |
TIEG, TIEG1, EGRα |
NM_005655 |
Q13118 |
480 |
52.6 |
KLF11 |
F-KLF, TIEG2, MODY7 |
O14901 |
512 |
55.1 |
|
KLF12 |
AP2rep, HSPC122 |
Q9Y4X4 |
402 |
44.2 |
|
KLF13 |
BTEB3, NSLP1, RFLAT-1 |
NM_015995 |
Q9Y2Y9 |
288 |
31.2 |
KLF14 |
BTEB5, SP6, EPFN |
DQ534757 |
Q8TD94 |
323 |
33.1 |
KLF15 |
K-KLF |
NM_014079 |
Q9UIH9 |
416 |
44 |
KLF16 |
BTEB4, NSLP2, DRRF |
NM_031918 |
Q9BXK1 |
252 |
25.4 |
KLF17 |
ZNF393 |
NM_173484 |
Q5JT82 |
389 |
42.6 |
Abbreviations: AP2rep AP2 repressor, B basic, BCD B-cell derived protein, BTEB basic transcription element binding, C colon, COBEP core promoter element binding protein, CPBP core promoter binding protein, DRRF dopamine receptor regulating factor, E erythroid, EGRα early growth response gene α, EPFN epiprofin, EZF epithelial zinc finger, F embryonic/fetal ß-like globin gene-activating, G gut, GBF GC-rich binding factor, I intestinal, K kidney, L lung, MODY7 maturity-onset diabetes of the young 7, NSLP Novel SP1-Like Protein, OSX osterix, RFLAT RANTES factor of late activated T-lymphocytes, SP specificity protein / SV40-promoter protein, ST suppressor of tumorigenicity, TFSP transcription factor SP, SPR SP1-related factor, TEF transcriptional enhancer factor, TIEG TGFß-inducible early gene, U ubiquitous, Z(N)F zinc finger
Sources: HUGO Gene Nomenclature Committee (www.genenames.org) and [9]. Protein molecular weights were retrieved from The Human Protein Atlas (www.proteinatlas.org) [149].
The homology between SP/KLF proteins is mainly restricted to the zinc finger and linker domains, situated at the C-terminal, and underlines the importance of this structure in transcription biology (Fig 1b). The major difference distinguishing SPs from KLFs is the absence of a Buttonhead box CXCPXC preceding the triple zinc finger region in the latter [9]. Furthermore, several SP/KLFs (KLF1, 2, 4, 9, 13, 16) share a nuclear localization signal (NLS), necessary for post-translational transport towards the nucleus [10]. More variability exists within the N-terminal regions of KLFs, containing both activator and/or repressor domains that interact with specific coactivators and -repressors, providing unique functions to each family member.
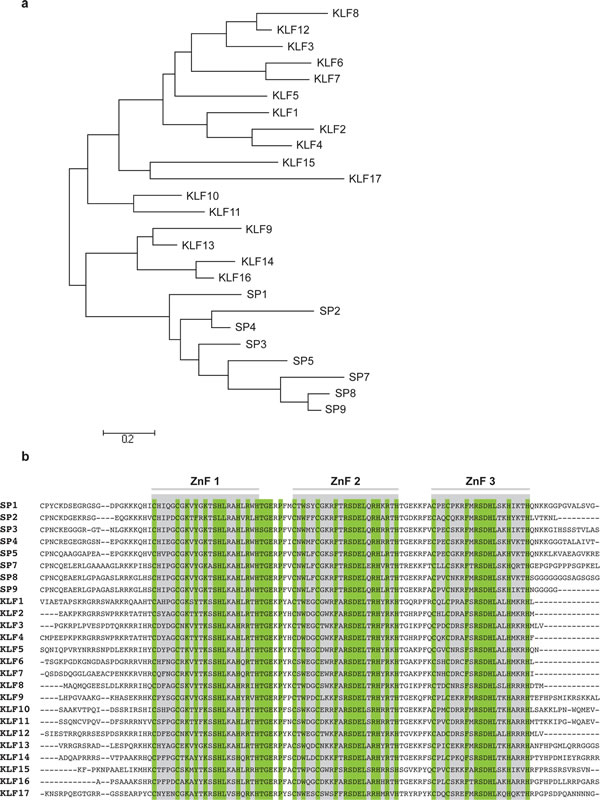
Fig 1: a). Phylogenetic tree resulting from a molecular phylogenetic analysis, based on the protein sequences of all human SP/KLF members using the Mega5 (v5.1) built-in Maximum Likelihood method and Nearest Neighbor-Joining algorithm [150]. b). Amino acid sequence alignment of the zinc finger C-terminal region of all known SPs and KLFs, as determined by ClustalW using Mega5 (v5.1) software. Indicated are the three separate zinc finger sequences (grey boxes, ZnF) and conserved amino acids (green).
As of currently, the KLF family of transcription factors comprises 17 identified members (KLF1 - 17) with diverse regulatory roles in differentiation, survival, proliferation and development. To avoid confusion because of alternative names, a straightforward SP/KLF nomenclature has been generated by the Human Genome Organization Gene Nomenclature Committee (HGNC, www.genenames.org) in which all SPs and KLFs have been numbered sequentially in order of their discovery (Table 1) [5,9]. This nomenclature will be followed throughout this review.
For reasons of conciseness, this review will further handle current knowledge on the KLF subfamily within the epithelial-mesenchymal transition (EMT), carcinoma progression and metastasis, added with uncovered roles in induced pluripotency and self renewal biology. More extended structural and functional information regarding SPs can be found in previous reviews [11,12].
2. KLFs in EMT and invasion
2.1 EMT
EMT constitutes a transdifferentiation program whereby cells shift from an apical-basal to front-back polarity. The existence of EMT was first established within embryonic development [13]. This transition characteristically involves the loss of adherens junctions, typically E-cadherin (CDH1), that guarantee the lateral cell-cell contacts in epithelial layers. In parallel, desmosomes, cytoplasmic ß-catenin (CTNNB1), tight junctions (claudins, occludins and ZO-1/TJP1) and epithelial cytokeratins (CK18/KRT18) become downregulated. On the other hand, the expression of vimentin (VIM), part of the mesenchymal intermediate cytoskeleton, is induced together with N-cadherin (CDH2). In addition, the increased deposition of cellular fibronectin (FN1) and the subsequent activation of integrins facilitate cell migration and extracellular matrix (ECM) invasion [14,15].
EMT provides a mechanism that enables cancer cells to invade individually into the surrounding stroma. In addition, the morphotype switch has been demonstrated to be reversible, with the existence of mesenchymal-epithelial transition (MET) as an inverse mechanism, necessary during metastatic colonization [13,16].
Several master transcription factors have been identified in conferring EMT (Fig 2): Snail (SNAI1) [17,18], Slug (SNAI2) [19], Twist (TWIST1) [20], ZEB1/δEF1 (ZEB1) [21,22], ZEB2/SIP1 (ZEB2) [23], E12/E47 (TCF3) [24], Hey1 (HEY1) [25] and HMGA2 (HMGA2) [26]. Most of these factors primarily share the direct repression of CDH1 through binding of E-boxes in its promoter.
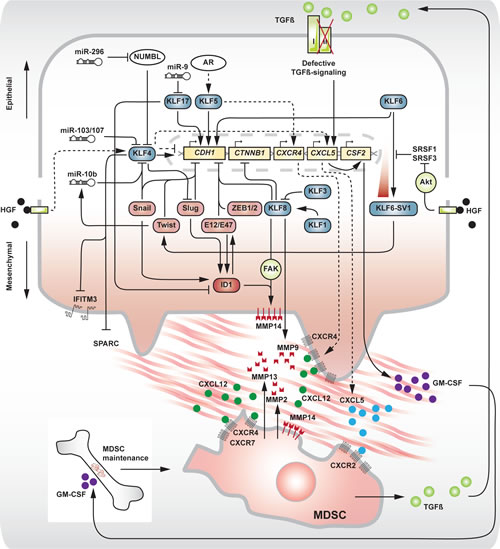
Fig 2: Involvement of different KLFs in the molecular circuitry of EMT and invasion in a single cell and interaction with a recruited myeloid-derived suppressor cell (MDSC). Upper and lower half of the main image represent epithelial and mesenchymal states respectively. Arrows and perpendicular symbols indicate promoting and inhibitory interactions respectively. Dotted arrows indicate pro-invasive interactions of KLF4 and KLF5. Boxes: KLF (blue), EMT master transcription factors (red), pro-metastatic factors (deep red), genes (yellow), kinase (green). Filled circles represent different secreted chemokines/growth factors, as indicated.
KLF8 is a potent inducer of EMT through CDH1 repression [27]. KLF8 was shown to directly bind to a GT box within the CDH1 promoter, hereby uncovering the presence of another consensus element that is targeted by the distinct family of KLF zinc finger transcription factors, next to the known triple E-box motif. As the DNA binding zinc finger regions are highly conserved between KLFs, similar target sequences may be recognized by different members. The variable nature of the N-terminal domains, on the other hand, may give rise to opposite trans-regulatory effects. Despite the ambiguous reporting on the tumor suppressing yet oncogenic roles of KLF4 in epithelial cancer biology (see invasion and metastasis and Fig 1), its function in EMT/MET has increasingly become clear. In cancer-related EMT as well as in EMT processes that contribute to the reprogramming route, KLF4 is a potent inducer of epithelial differentiation and antagonizes the switch to a mesenchymal phenotype. Transcriptional regulation by KLF4 increases the expression of CDH1 and forces adult human fibroblasts into an epithelial state that proved a prerequisite for successful reprogramming to pluripotency [28]. Indeed, in MCF-10A normal mammary epithelium, KLF4 was shown to activate CDH1 transcription through binding of CACCC consensus sequences in the proximal promoter of the CDH1 gene [29]. KLF4 silencing resulted in a cadherin switch (loss of CDH1 with concomitant gain of CDH2 [30]) and a decrease of cytosolic ß-catenin. Consequently, overexpression of KLF4 in the metastatic MDA-MB-231 breast cancer cell line dramatically increased CDH1 and KRT18 expression, indicating the restoration of an epithelial phenotype and loss of metastasis. The mechanism of EMT impairment by KLF4 can be explained by its repressive action on the EMT transcription factors SNAI1 [31] and SNAI2 [28,32]. Conversely, in EMT-induced colon cancer cells, Snail was shown to repress the expression of KLF4 [33]. This finding fits within a previously described role of KLF4 in specific aspects of epithelial cell differentiation [34] and provides a rationale for loss of this factor in cancer. Direct binding sites for KLF4 have been detected in the promoter sequences of vimentin (VIM), VEGF-A (VEGFA), endothelin-1 (EDN1) and JNK-1 (MAPK8), next to E-cadherin (CDH1), N-cadherin (CDH2) and CTNNB1, indicating a central role for KLF4 within the EMT program [35]. These findings can at least partly explain the downregulation of KLF4 as reported for several epithelial cancer types and the inverse correlation of KLF4 expression with clinical outcome [32,36,37].
Nevertheless, in HepG2 hepatocellular carcinoma and Madin-Darby canine kidney (MDCK) cells, a direct KLF4-mediated downregulation of CDH1 was observed, downstream of hepatocyte growth factor (HGF, also known as scatter factor/SF)-induced cell scattering (Fig 2). Moreover, KLF4 expression was activated by early growth response-1 (EGR1) under stimulation of HGF and sustained itself through a transcriptional auto-activation loop [38]. HGF-mediated cell scattering is largely dependent on SNAI1 activation, downstream of HGF signaling [39], implying an important EMT transcription factor in this process. These apparent contradictions might be explained using a contextual view on these cellular events. KLF4 is able to act as an activator or repressor of downstream genes, depending on the availability of co-activators or co-repressors in its environment.
KLF5 activates CDH1 expression and downregulation of VIM in non-small cell lung cancer cells [40]. Both KLF4 and KLF5 show competition for promoter binding sites with antagonizing effects regarding proliferation [41]. In intestinal cells, KLF4 represents a marker for differentiated villus cells whereas KLF5 positively regulates proliferation in the crypt cells [41]. The latter has been confirmed by providing evidence of increased cyclin-D1 (CCND1) transcription, colony formation and cell growth in normal ileal cells (IEC-18) and Immorto-Min Colon Epithelial (IMCE) cells overexpressing KLF5 [42]. Contrastingly however, the same group demonstrated inhibition of proliferation in KLF5-overexpressing DLD-1 colon adenocarcinoma cells. Interestingly, in the context of oncogenic Ras, KLF5 became significantly downregulated by at least two mechanisms, namely reduced mRNA transcription and proteasomal degradation. Reduced expression of KLF5 in intestinal cancer when compared to normal epithelium could also indirectly promote EMT by relieving the promotional activity upon the CDH1 promoter.
KLF6, originally identified as a tumor suppressor in prostate carcinoma [43], also activates the CDH1 promoter [44]. The tumor suppressor function of KLF6 was further demonstrated in hepatic [45] and gastric cancer [46,47] and linked to a role in cellular differentiation. Several mechanisms leading to KLF6 loss have been described, including promoter hypermethylation [48], somatic mutations and loss of heterozygosity in prostate [43], gastric [47] and ovarian cancer [49]. Interestingly, an alternative mechanism to downregulate KLF6 was found to involve the generation of three splice variants (SV1-3) with antagonistic effects on the wild type KLF6 tumor suppressor function [50]. KLF6-SV1, one of these splice variants, is able to drive breast cancer cells into an EMT-like phenotype, with loss of CDH1 and increased expression of CDH2 and FN1 [51]. This was associated with enhanced metastatic potential, however without significant morphological changes. Expression of KLF6-SV1 was found to indicate poor prognosis in several epithelial cancer types, including breast cancer [51], prostate cancer [52] and lung cancer [53]. Primordial findings on KLF6-SV2 point in a similar direction [45]. The origin of this deregulated splicing activity was traced to a germline single nucleotide polymorphism (SNP) in the KLF6 allele, generating a novel binding site for the SRFS5/SRp40 splicing factor [50]. HGF-dependent phosphorylation of Akt potentiates KLF6-SV1 signaling through subsequent inactivation of the splicing regulators SRSF3 and SRSF1 [54] (Fig 2).
In breast cancer, KLF17 was identified as a metastasis suppressor, counteracting EMT in the 168FARN murine breast cancer cell line, normal murine (nMuMG) and human (HMLE) breast epithelium [55]. Knockdown of KLF17 significantly reduced CDH1, TJP1 and CTNNB1 expression with concomitant increase of CDH2, VIM and FN1. In a cohort of human breast cancers, KLF17 was found to be decreased in lymph node positive when compared to lymph node negative tumors, hereby indicating a prognostic value. The authors further identified inhibitor-of-differentiation protein ID1 as a pro-metastatic regulator downstream of KLF17, which becomes expressed in breast cancers due to loss of its repressor (Fig 2). Indeed, an inverse correlation between KLF17 and ID1 was noted, specifically high KLF17 – low ID1 and low KLF17 – high ID1 in node negative and node positive breast tumors respectively. ID factors (ID1 – 3) are helix-loop-helix (HLH) transcription factors lacking a basic domain and unable to bind cognate DNA sequences. They act as dominant-negative regulators of the basic HLH transcription factor E47 [19,56]. E47 is an EMT promoting transcription factor capable of directly repressing CDH1 [24]. On a transient level, the formation of a complex with ID1 inhibits E47 from binding the CDH1 proximal promoter sequence, hereby switching on E-cadherin expression. However, introduction of ID1 in E47-induced mesenchymal MDCK cells was insufficient to restore E-cadherin transcription or an epithelial phenotype. Moreover, ID1 expression maintains a stable EMT phenotype and preserves cell viability [57]. The versatility of ID proteins is illustrated by their crucial role in early-phase metastatic colonization of the lung [58].
Associations between loss of KLF17 and reduced survival have been reported for lung and hepatic cancer [59,60]. In HepG2 hepatic cancer cells, KLF17 was found to be under post-transcriptional regulation by miR-9, implying an oncogenic and pro-metastatic role for this miR through repression of KLF17 [61].
2.2 TGFß-induced EMT
Epithelial cells can be driven to EMT under the influence of signaling events resulting from upstream extracellular cues (Fig 3). Transforming growth factor ß-1 (TGFß1/TGFB1) is known to drive cells towards a mesenchymal state through smad2/3-dependent transcription of SNAI1 [62,63]. Additionally, HMGA2 and Hey1 have been identified acting downstream of smad2/3 and similarly blocking CDH1 expression [25,26]. In a mouse model of progressive prostate cancer (Pten/TP53 null) with stem cell and EMT characteristics, TGFß-induced EMT mainly acted through SNAI2 and to a lesser extent through SNAI1 [64] (Fig 3). In this setting, KLF4 inhibits TGFß-driven EMT by directly repressing SNAI2. KLF4 may sustain a positive feedback loop involving TGFß ligand and receptors through binding of GC-boxes in the proximal promoters, as shown in vascular smooth muscle cells (VSMC) [65]. A similar mechanism had previously been demonstrated for KLF6 [66].
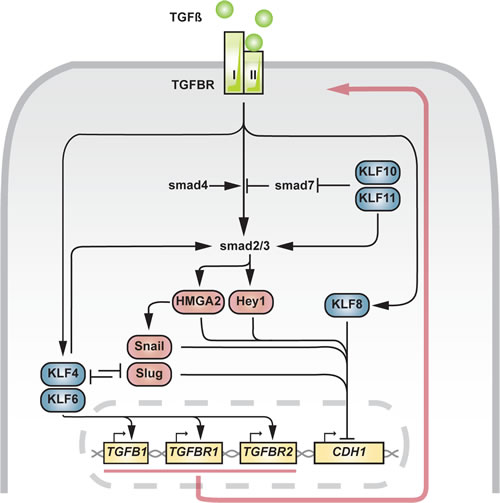
Fig 3: Involvement of KLFs in TGFß-induced EMT. Blue: KLF, red: EMT master transcription factors, yellow: genes. Red arrow indicates positive feedback mechanism.
Given the reciprocal repression between KLF4 and SNAI2 [64], the final output generated by TGFß, be it differentiation or EMT, could be determined by the intracellular balance KLF4/SNAI2. When other channels including signals from the surrounding microenvironment trigger SNAI2 in excess to KLF4, EMT through TGFß-signaling could become the dominant process, sustained by KLF4-dependent positive feedback signaling.
KLF8, a potent EMT-regulator, is induced by TGFß1 and acts as an indispensable player in the TGFß-mediated EMT in gastric cancer cells [67]. Elimination of KLF8 led to attenuation of EMT and inhibition of the associated capacity of migration and invasion.
Two members of the KLF family, currently known as KLF10 and KLF11, are deeply embedded in TGFß signaling and have originally been named thereafter, respectively TGFß inducible early gene 1 (TIEG1) and 2 (TIEG2) (Table 1). Initially described as directly regulated by TGFß1, KLF10 [68] and KLF11 [69] provide supporting actions in the TGFß signaling pathway, exerting anti-proliferative and pro-apoptotic effects in epithelial cells. Both factors show a strong structural similarity, specifically within the zinc finger regions (91%, [69]). In PANC-1, MIA Paca2 and Colo357 pancreatic adenocarcinoma cells, KLF11 was found to sustain TGFß signaling both by terminating the inhibitory smad7 loop and through activation of smad3 [70,71]. Abrogating effects on smad7 have also been demonstrated for KLF10 in hepatic and breast cancer cells [72]. Phosphorylation of KLF11 by MAPK in pancreatic cancer cells abrogates the inhibition of smad7, leading to decreased TGFß-mediated growth inhibition [70]. A transcriptomic screen in TGFß- and EGF-stimulated kidney proximal tubular cells for cis-regulatory elements in the differentially expressed gene pool identified, among others, the KLF-targeted GC-boxes, and KLF10 as a principal factor in the EMT-program, mediated by TGFß1 [73]. In agreement, KLF10 protein expression correlated inversely with disease stage in a collection of 95 tissue samples of pancreatic adenocarcinoma and independently predicted progression-free and overall survival in pancreatic cancer [74].
2.3 Invasion and metastasis
Local tissue invasion marks the first step of carcinoma progression towards the systemic dissemination of cancer cells and metastatic colonization of distant organs, a multi-step process named the invasion-metastasis cascade [75-77]. Next to important roles of KLFs in cell cycle-associated regulation of proliferation (reviewed in [78]), influences on the progression of epithelial cancers towards invasive and metastatic states have been described, often with effects overlapping both fields. This section will describe current knowledge of KLFs in invasiveness pathways, apart from direct associations with EMT processes. In addition, upstream triggering events and potential roles of KLFs in metastatic dissemination of epithelial cancer cells will be discussed.
Within this scope however, several EMT-modulating KLFs regulate invasion. KLF4 has yielded ambiguous results regarding its oncogenic yet tumor suppressing role (Fig 4). Initially, KLF4 was identified as a transforming oncogene in oral squamous epithelia [79] and subsequently, increased expression of KLF4 was found in ductal carcinoma in situ of the breast and invasive breast cancers when compared to normal breast epithelium [80]. KLF4 was a marker of terminally differentiated epithelial cells that became deregulated in dysplastic epithelium. Alternatively, KLF4 transcription was essential in the maintenance of a breast cancer stem cell (CSC) population [81]. Knockdown of KLF4 in the MCF-7 and MDA-MB-231 breast cancer cell lines drastically decreased the proportional number of CSC-like cells as defined by ALDH1 expression, side population and in vitro mammosphere formation capacity. Furthermore, downregulation of KLF4 inhibited breast cancer cell migration and invasion through Notch (NOTCH1)-mediated activity. In addition, the same group recently revealed a correlation between KLF4 expression and production of the C-X-C motif chemokine CXCL5 (CXCL5) by primary cancer cells [82]. Chemotactic CXCL5 stimuli recruit C-X-C motif chemokine receptor (CXCR2)-positive myeloid-derived suppressor cells (MDSC) to the primary tumor. Interestingly, MDSCs residing at the invasive tumor front express high levels of TGFB1, further leading to increased production of CXCL5 in TGFBR2-deficient primary breast cancer cells [83]. Although the link between defective TGFß-signaling and CXCL5 expression remains elusive, this mechanism is able to contribute to the chemotactic recruitment of MDSCs. Moreover, enhanced production of CXCL5 by cancer cells gives rise to the systemic secretion of granulocyte/monocyte colony stimulating factor (GM-CSF), a cytokine that contributes to the maintenance of the MDSC pool in the bone marrow. These results may, in part, explain the decreased in vivo tumorigenicity and reduced occurrence of pulmonary metastases in a BALB/c mouse model, orthotopically inoculated with stable Klf4 knockdown 4T1 murine breast cancer cells. The outcome of early-stage breast cancer patients, defined as death by breast cancer, was linked to the immunohistochemical staining pattern of KLF4 in their primary tumor. Strong nuclear and low cytoplasmic presence of KLF4 was highly indicative for poor prognosis [84]. Additionally, in squamous cell carcinoma of the head and neck (HNSCC), persistent expression of KLF4 correlated with poor prognosis, specifically in patients with advanced disease [85]. On a cell line level, ectopic overexpression of KLF4 increased the tumorigenicity, migration and invasion of HNSCC cells. Corroborating evidence in the CSC domain of breast cancer was established in a study of cancer cell dissemination to the brain [86]. The authors found that, in a mammary CSC population defined as being CD44+, CD24- and EpCAM+, successful invasion and colonization of brain tissue correlated with high expression of KLF4 and loss of a microRNA, miR-7, that targets KLF4 mRNA. This was opposed to CSCs metastasizing to bone, where KLF4 was expressed at lower levels. In metastatic brain lesions from patients, KLF4 and miR-7 expression were shown to correlate inversely, indicating a role for KLF4 transcriptional activity in the establishment of lesions in the brain parenchyma. Interestingly, 9 out of a set of 17 genes previously identified as correlated with brain relapse [87] contain putative binding sites for KLF4.
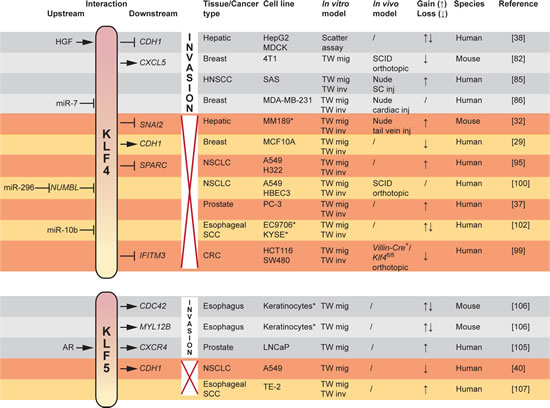
Fig 4: Functional duality in promotion or inhibition (red cross) of invasion by KLF4 and KLF5 as dictated by contextual and microenvironmental conditions. Upstream and downstream interactions are indicated left and right to the KLF symbol respectively, added with experimental modalities and associated references per finding. CRC colorectal carcinoma, HNSCC head and neck squamous cell carcinoma, NSCLC non-small cell lung cancer, SC subcutaneous, SCC squamous cell carcinoma, SCID severe combined immunodeficient, TW transwell. * primary cultures.
These findings, related to both tumorigenicity and invasive behavior, are in sharp contrast with other reports on KLF4 function, specifically within the EMT framework as discussed above. Yori and colleagues have extensively described an inverse correlation between expression of KLF4 and invasiveness, mechanistically based on CDH1 induction and thus in an EMT context [31]. MCF-10A normal mammary epithelium showed increased migration after knockdown of KLF4. In specimens of lung, gastric and prostate cancer, KLF4 also demonstrated decreased levels of expression when compared with normal tissue counterparts [37,88-92]. Restoration of KLF4 expression in vitro impaired migration and invasion of prostate cancer cells [37]. In the context of epithelial differentiation, the authors have identified the cell cycle inhibitor p27KIP1 (CDKN1B) as a target of KLF4, through which proliferation of pancreatic cancer cells was blocked [93]. KLF4-mediated G1/S cell cycle arrest was previously shown to imply activation of p21CIP1 (CDKN1A) [94]. However, loss of KLF4 also proved to be stimulating invasion, independent of CDKN1B, by the increased deposition of secreted protein acidic and rich in cysteine (SPARC, also known as osteonectin) [95]. The involvement of SPARC, a small calcium-binding glycoprotein that modifies cell-matrix adhesion, was already shown in breast cancer and melanoma progression due to its ability to induce MMP2 and SNAI1 and repress CDH1, hereby promoting ECM invasion and EMT [96,97]. These results coincide with the theory of KLF4 as a suppressor of EMT and invasion. The antagonistic effect of KLF4 on tumorigenicity and disease progression was further demonstrated in colorectal carcinoma (CRC). IFITM3, an interferon-inducible gene overexpressed in CRC [98], is directly repressed by KLF4 [99]. Also recently, repression of KLF4 by the cell polarity protein Numb-like (Numbl/NUMBL) was reported from knockdown experiments in the A549 lung cancer cell line. Downregulation of miR-296 causes aberrant expression of its target NUMBL leading to reduced KLF4-expression and increased random cell migration, invasion and in vivo metastasis [100]. This mechanism may be more general since loss of miR-296 is described in several cancers [101]. Another microRNA, miR-10b, had been associated with metastasis by directly targeting KLF4 [102] or indirectly inducing the expression of RHOC, a prominent pro-metastatic gene [103].
In prostate cancer, KLF5 was found to become expressed downstream of androgen receptor (AR) signaling. The chemokine receptor CXCR4, a direct transcriptional target of KLF5, is subsequently activated and binding of its ligand CXCL12 (CXCL12, also known as SDF-1α) underlies the preferential chemotactic migration of prostate cancer cells to organ sites with elevated levels of CXCL12, for example bone [104,105]. Furthermore, from a study of keratinocyte migration, it is reported that KLF5 activates the expression of cell-division-cycle 42 (CDC42), myosin light chain (MYL12B) and their upstream regulator integrin-linked kinase (ILK), hereby directly driving cell migration [106]. However, in esophageal cancer cells, KLF5 inhibited proliferation and invasive behavior [107]. This is in agreement with the more recently identified association between siRNA-mediated loss of KLF5 and increased expression of CDH1 in A549 lung cancer cells [40]. Similar to KLF4, KLF5 seems to act in a context-dependent fashion, partially determined by the genetic background (Fig 4).
KLF8 activates invasion in cooperation with focal adhesion kinase (FAK) by increased transcription of matrix metalloproteinase-14 (MMP14, also known as MT1-MMP) in gastric cancer cells (Fig 2). In parallel, nuclear transportation of ß-catenin and expression of T-cell factor 1 (TCF1), participate in the initiation of MMP14 transcription, indirectly effectuated by KLF8 [108]. In addition, KLF8 directly activated MMP9 expression [109]. Conversely, knockdown of KLF8 drastically inhibited lung metastasis in nude mice. In human breast and gastric cancer, overexpression of KLF8 has been shown to predict poor prognosis [109,110]. Similar conclusions resulted from a study in hepatocellular carcinoma, attributing a pro-invasive role to KLF8, conferring early relapse in human HCC [111].
Emerging data, although still limited, have identified KLF9 as a marker of differentiation in glioblastoma neurosphere cells. Knockdown of KLF9 was sufficient to rescue differentiating neurosphere cells as exposed to retinoic acid [112]. Current knowledge points toward a regulatory role of KLF9 in the proliferation and differentiation of diverse cell types [113,114]. KLF10 is known as a supportive player in the TGFβ signaling cascade and is able to initiate apoptosis [115]. Next to its role in TGFβ-mediated EMT, KLF10 repressed the EGFR gene through potential binding sites in its proximal promoter. Reduced expression of KLF10, as reported for breast and pancreatic cancer [74,116], can promote invasive and metastatic behaviour by enhancing EGFR expression [117]. This may be considered in contrast with its supporting activity in TGFß-induced EMT, however depletion of KLF10 may be substituted by KLF11, which exerts identical effects on smad proteins. In a sense, invasion can then be propelled through intact EGFR signaling while preserving the EMT mechanism as driven by TGFß.
3. KLFs in stem cell transcriptional circuitries
Consequent to their structural properties and involvements in diverse cellular processes like proliferation, differentiation, apoptosis, EMT and motility, the activity of the KLF family shows a high degree of context dependence, related to both tissue and cellular backgrounds. A counterintuitive finding would be the absolute requirement of KLF4/Klf4 in somatic cell reprogramming [118,119]. It has been demonstrated that induction of adult somatic cells to pluripotent cells (induced pluripotent stem cells, iPSCs), with characteristics similar to embryonic stem cells (ESCs), can be managed through the introduction of KLF4 (human KLF4, mouse Klf4), SOX2 (human SOX2, mouse Sox2), OCT4 (human OCT4, mouse Oct4) and c-Myc (MYC), also referred to as the Yamanaka factors. From this set of transcriptional regulators, c-Myc has been regarded the only dispensable factor, leading to a less efficient yet more specific induction process [120]. In these studies, adult fibroblasts, murine or human, are often used as a somatic source to be redirected to a pluripotent state. Klf4 has been proven necessary for both induction and maintenance of pluripotency and self renewal. Interestingly, Klf4 is able to bind a distal enhancer of the central reprogramming factor Nanog [121], as well as its proximal promoter element [122,123]. Moreover, a mechanism of redundancy between different Klfs has been revealed in the maintenance of self renewal capacity. Klf4, initially considered as dispensable for sustaining the stem cell state, acts in cooperation with Klf2 and Klf5 to regulate the expression of Nanog through its distal enhancer element [121]. Only the coordinated and simultaneous depletion of Klf2, Klf4 and Klf5 drives ESCs into differentiation, indicating an important function of these Klfs in phenotype maintenance of embryonic/induced stem cells. In addition, it was noted that forced downregulation of Klf2, Klf4 and Klf5 gave rise to cells solely expressing typical ectodermal, and not endodermal, markers (Fgf5, Nes, Cxcl12), suggesting downregulation of lineage specific genes by certain Klfs.
Apart from the identified redundancy, Klf5 by itself was shown to exhibit specific functions in the commitment of ESCs to an undifferentiated state. Chromatin immunoprecipitation experiments demonstrated binding of Klf5 on the promoters of both Nanog and Oct4, and vice versa. Nanog cis-regulatory elements were detected in the Klf5 genomic region, indicating a feedback loop in ESCs [124]. Klf5 was found to act independently of Klf4, underlining its potential importance as a sole factor in maintaining the ESC state. Elimination of Klf5 impaired maintenance of the undifferentiated state of ESCs and induced the expression of several early differentiation markers. This effect was due to a deregulation of at least eight genes, targeted by Klf5, that contributed to the maintenance of ESCs [125].
Another study uncovered the link between the induction of pluripotency and EMT. Inducing mouse embryonic fibroblasts (MEFs) towards a pluripotent state essentially implied a MET program in the early stage of reprogramming [28,126]. Fibroblasts, showing a mesenchymal phenotype and expressing high levels of Snai1, needed the introduction of Klf4 to activate the epithelial marker E-cadherin (Fig 5). Repression of mesenchymal markers (Snai1, Fn1, Vim) is mediated by Sox2 and Oct4. The early and temporary transition to an epithelial-like state showed a prerequisite for successful reprogramming of fibroblasts to iPSCs. As mammary epithelial cells highly express endogenous Klf4 and the initial MET step thus becoming obsolete, the authors showed that reprogramming could be performed by introducing only Sox2 and Oct4. It was therefore postulated that epithelial cells are more amenable to somatic cell reprogramming than their fibroblastic counterparts [127]. Indeed, previous research revealed significantly higher efficiencies using primary keratinocytes instead of fibroblasts [128,129].
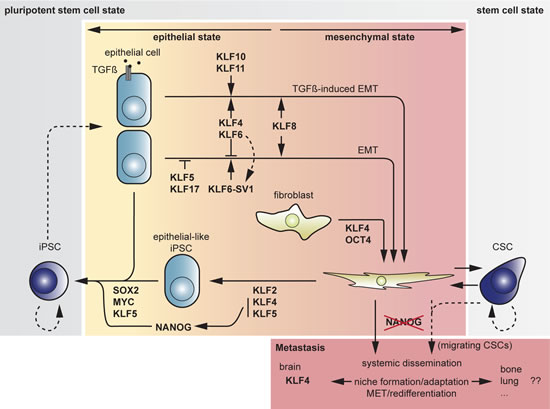
Fig 5: Overview of transitions between cell states and associated actions or counteractions by KLFs. The center field represents somatic cells, distributed in an epithelial (left) and a mesenchymal section (right) according to their respective properties. The outer field (grey) represents the stem cell state, divided into a pluripotent area (dark grey, left) and a general stem cell area (light grey, right) containing the CSC compartment. The iPSC reprogramming route departs from the somatic fibroblast through the “hypermesenchymal” state via an “epithelial-like” state to iPSC. Dotted arrows indicate putative actions.
Recently, another approach was applied to increase reprogramming efficiencies. By performing sequential, rather than simultaneous introduction of the reprogramming factors, Liu and colleagues obtained a significantly higher yield of iPSCs [130]. The sequential introduction of the Yamanaka factors, starting with Klf4 and Oct4, then Myc and finally Sox2 revealed an EMT-MET program in the early phase of reprogramming. Oct4 and Sox2 are known to stimulate, respectively inhibit the expression of Snai2, a mediator of EMT. Introducing Sox2 at a later stage during reprogramming allows Oct4 to regulate an EMT step, hereby possibly homogenizing the fibroblastic cell population to a “hyper-mesenchymal” state [131]. Klf4 activates Cdh1 transcription, eventually leading to the subsequent MET step, which is enhanced by the later introduction of Sox2 with a blocking effect on Snai2 [130]. Yet, driving Cdh1 expression is not the only function of Klf4 as forced expression of Cdh1 without Klf4 introduction did not give rise to iPSCs. The early introduction of Klf4 may be necessitized by its downstream activity upon Nanog transcription, as described in murine ESCs, and its ability to prevent ESCs from ensuing a path to differentiation [123]. In a recent study, the heterogeneity among MEFs was considered as another possible cause of the usual poor yield of iPSCs. It was revealed that different MEF subpopulations, based on surface marker expression, showed different degrees of reprogrammability. The CD90-/Sca-1+ cell populations could be reprogrammed to a pluripotent state using only two of the Yamanaka factors, namely Oct4 combined with either Sox2 or Klf4. From this approach, a partial functional redundancy between Sox2 and Klf4 was derived. Moreover, with respect to the intra-population heterogeneity, the rate-limiting MET as observed previously during reprogramming, could be considered only one out of many possible transient stages in a cell between a somatic and embryonic state [132].
4. Context dependence and redundancy: lessons from proliferation
Certain studies yielded evidence for pro-tumoral effects, others for tumor suppressing effects by the same KLF member (Fig 4). Such duality has been demonstrated for the role of KLF4 in the regulation of cell proliferation [94]. In an untransformed cell, KLF4 decreased p53 (TP53) levels by repressively binding a PE21 element located in the proximal promoter region of the TP53 gene. Simultaneously, KLF4 induced expression of CDKN1A leading to cell cycle arrest. The latter effect seemed dominant in untransformed cells, illustrating the tumor suppressor role associated with KLF4. Changing the cell’s genetic background by introducing an oncogenic RasV12 allele not only abolished the cytostatic function of KLF4 through bypassing CDKN1A induction, but switched it into an oncogenic effect, with KLF4 showing its dominant action on repressing TP53 and thus preventing apoptosis. It was concluded that the output mediated by KLF4 was dependent on the genetic background.
This phenomenon is reminiscent of the dual activity of TGFß signaling on tumor propagation, where TGFß was reported to act as a tumor suppressor in early-stage cancers, and as an oncogene in progressive disease [94,133]. Remarkably, both the TGFß pathway and KLF4 seem to converge on CDKN1A which, on its turn, acts as a gatekeeper preventing oncogenic transformation [94]. KLF5, a transcriptional opponent of KLF4 and a driver of proliferation in intestinal cells [41], was shown to be a key factor in TGFß-mediated inhibition of proliferation. In unstimulated keratinocytes (HaCaT epidermal epithelial cell line), KLF5 activated cell cycle progression and proliferation by blocking the cell cycle inhibitor CDKN2B (p15) [134]. On the other hand, in the presence of TGFß1, KLF5 became a coactivator in TGFß-induced expression of CDKN2B. It was revealed that p300/CBP (CREB-binding protein, CREBBP), initially recruited to the Smad2/3/4 complex upon TGFß treatment, reversed the function of KLF5 by acetylation. In its acetylated state, KLF5 was able to bind three sites in the proximal region of the CDKN2B promoter and activate transcription. Contextual modulation of KLF5 function was also found in its interaction with MYC, a proto-oncogene with proliferation-promoting activity. In similar experiments with keratinocytes, KLF5 stimulated MYC transcription through binding of both a KLF5-binding element (KBE) and a TGFß-inhibitory element (TIE) in the proximal promoter region of MYC, thus confirming the activating effect on proliferation [135]. Conversely, when TGFß was applied to induce inhibition of proliferation, binding to KBE decreased significantly as KLF5 was recruited to the TIE sequence by TGFß in order to block MYC transcription. These results strongly point to the reversibility of KLF transcription function as a tool for balancing between proliferation and differentiation in the maintenance of epithelial homeostasis. This model adds a dimension to the conventional view of changing the levels of different transcription factors in order to mediate opposing outcomes. The dependency of KLF5-mediated effects on cellular backgrounds has been further illustrated in intestinal epithelial cells. Ectopic overexpression of KLF5 indeed stimulated proliferation of normal intestinal epithelium through activation of CCND1, yet in colon cancer cells, proliferation and colony formation capacity were reduced through failure of KLF5 to induce CCND1 [42]. This tumor suppressing role of KLF5 has been suggested in clinical breast [136] and prostate carcinomas [137], where loss of KLF5 was observed when compared to normal tissue counterparts.
Despite being derived mainly from processes related to proliferation, the identified mechanisms comprise molecular modifications that alter the function of the same transcription factor in a context-dependent fashion. These alterations may also be present within an EMT- and/or invasion-related context, mediating cancer progression to a metastatic state. The expression of different KLFs is often tissue-specific and existence of redundant subgroups within the KLF family is emerging [121,138]. Redundancy enables certain tissue-specific KLF family members to exert identical actions in different tissues where expression of companion KLFs is limited or absent. Moreover, due to their highly similar cis-acting properties combined with trans-acting variability, redundant KLFs may compete with one another for an identical cognate DNA binding site and impose opposite effects on transcription. KLF4 and KLF5 are known for binding site competition as well as for transcriptional autoregulation. KLF4 activates its own transcription, thus sustaining its presence, and explicitly blocks cell cycle progression [139]. KLF5, a stimulator of proliferation, represses the auto-activation loop of KLF4. However, KLF4 is able to inhibit transcription of KLF5 in order to maintain a dominant role in preventing proliferation [41]. In the intestine, differential expression of both these KLFs was demonstrated between crypt and villus cells. Crypt cells with an intense proliferative activity indeed expressed KLF5 at higher levels, whereas terminally differentiated villus cells showed the opposite, with higher expression of KLF4. This implies a KLF4/KLF5 balance shift occurring during the movement of cells from the crypts towards the villi in intestinal tissue homeostasis. Similar implications for KLF4 in terminal epithelial differentiation have been found earlier in epidermal epithelial cells [140]. This mechanism is engaged by two related KLFs in close competition to yield different cellular outcomes, without chemical modulation.
5. KLFs in metastasis: attempt to an integrated view
The epithelial state is profoundly influenced by the KLF family of transcription factors, based on studies using a diverse set of cell types and tissues. As their presence and activity has been demonstrated in different epithelial tissues and derived carcinoma types, added with their fundamental role in somatic reprogramming and its associated transit through an epithelial phase (Fig 5), KLFs are thoroughly embedded in a regulatory system that participates in cell fate determination. Nevertheless, the integration of the various mechanisms that have been identified remains a complex challenge.
It has been agreed that the dissemination of cancer cells from the primary site to distant organs and the subsequent successful colonization leading to overt metastasis, requires a significant degree of cellular plasticity [141].
The EMT/MET program and the cancer “stemness” program, provide a dynamic framework in which different KLFs appear to contribute a significant role. The presence of both E-boxes as CACCC elements in the promoters of EMT genes allows Krüppel-like transcriptional regulators to participate in the EMT program, in parallel with the previously recognized zinc finger E-box binding and bHLH factors. Moreover, it has been demonstrated that cellular state changes are mediated, in part, by specific KLFs in a time-dependent manner. It has become clear that the functioning of KLFs, individually and in concert, is dependent of three main determinants: (1) the tissue of origin, (2) the cellular genetic background (context) and (3) the surrounding microenvironment. The final output, as triggered by KLF transcription, will often be the result of the interplay between these three features. Furthermore, this also illustrates the intra- and inter-tumor heterogeneity that has increasingly been recognized in several epithelial cancer types [142]. Given the versatility of their functioning, the KLF circuitry seems to fit in this plasticity that provides a cancer cell with the ability to proceed to invasiveness and to overcome the multiple barriers towards metastasis.
KLF8 unambigously drives cancer cells into the mesenchymal state [27], hereby facilitating invasion. However, maintenance of this phenotype is unfavorable in the long term as ectopic survival requires a switch back to a more epithelial state resembling the primary tumor [143]. The contrasting findings regarding KLF4 and KLF5 have been shown to originate from both the cellular and microenvironmental contexts. KLF4 has been reported to have a role in epithelial differentiation and inhibition of growth [29], and thus often becomes repressed in cancerous tissue. Interestingly, the presence of HGF in the proximity of cancer cells, triggers KLF4 to inhibit CDH1 transcription [38]. In addition, by modulating splice factor expression, HGF mediated an intracellular shift from the tumor suppressor KLF6 to the pro-metastatic KLF6-SV1 ([54], see also Fig 2 and 5). These examples illustrate microenvironmental properties igniting invasion and metastasis. An altered genetic background, for example oncogenic Ras, can lead to phosphorylation by extracellular signal-regulated kinase (ERK), hereby inversing the functioning of particular KLFs. Within this context, KLF5 has been shown to lose its proliferation promoting effect that is typical for normal intestinal crypt cells [42]. A similar outcome was observed in normal keratinocytes under exposure to TGFß [134], indicating that Ras-transformed cells have acquired an intrinsic competence to modulate KLF function whereas normal cells need the extrinsic influence from the TGFß cytokine. As KLF5 also directly activates transcription of CDH1, downregulation may indirectly play in favor of a progressive phenotype of certain cancer types, but not all. AR signaling was shown to activate CXCR4 protein expression through KLF5 in prostate cancer cells, propelling chemotactic migration in response to CXCL12, a chemokine abundantly present in the microenvironment and specific sites of metastasis [104,105]. In the latter case, KLF5 becomes a tool to assist invasive behavior.
TGFß acts as a master regulatory cytokine, being able to manipulate transcriptional functions in early and advanced-stage cancers. Within this scope, TGFß has the potency to induce EMT, partly through KLF8 [67]. KLF10 and KLF11 sustain the TGFß signaling node by inhibiting smad7 and supporting the smad2/3 complex formation. The recent association of low expression levels of KLF10 with advanced disease in pancreatic cancer [74], may in part be explained by the repressive action of KLF10 on EGFR expression [117]. In a way, TGFß signaling may be maintained by KLF11, hereby driving an EMT program, while downregulation of KLF10 favors the expression of EGFR, triggering other pro-invasive signaling cascades. KLF4 was reported as an opponent of TGFß-induced EMT due to its repression of SNAI2 [32]. However, as this repression is reciprocal, i.e. SNAI2 itself represses KLF4 as well, a balance between both factors can determine a transit towards a mesenchymal state, with a positive feedback on expression of the central TGFß components, mediated by KLF4. Similarly, the extensively described tumor suppressor KLF6 antagonizes EMT, but activates transcription of TGFß ligand and receptors 1 and 2 [66]. In an EMT context, propelled by TGFß, KLF6 may play a supporting role, favoring cancer cell progression. Moreover, cancer cells with disabled TGFß machinery produce high levels of CXCL5 when confronted with persistent TGFß ligand, both in an autocrine and a paracrine fashion [83]. KLF4 is also known to activate CXCL5 transcription [82], corroborating the establishment of a chemotactic gradient that guides MDSC recruitment to the invasive front. As reported most recently, MDSCs induce stemness and tumorigenicity in proximal cancer cells through miR-101-mediated repression of C-terminal binding protein 2 (CtBP2), a corepressor of stemness core genes [144].
When considering the circuitries involving KLFs in cancer progression, their involvement in the establishment and maintenance of stem cell phenotypes must not be overlooked. KLF4, and KLF5, have a fundamental role in the reprogramming of somatic cells and the maintenance of self renewal capability. In this light, it might be speculated that these factors may become upregulated in a subset of cancer cells with stemness properties. This is reasonably supported by the upregulation of KLF4 in breast cancer stem cells as defined by marker expression, and the observed loss in bulky cancerous tissues. Furthermore, KLF4 is necessary in the adaptation of metastatic breast cancer cells in the brain niche [86]. On the other hand, EMT confers cancer cells with stem cell properties [145], a finding that apparently contrasts with the indispensable MET step as initiated by KLF4 during somatic reprogramming [146]. Nevertheless, in order to successfully reprogram cells towards a pluripotent state, KLF4 needs to be introduced at an early phase, probably in line with its ability to activate NANOG and OCT4 expression, in parallel with CDH1 [121,122].
It must be noted, however, that migrating cancer cells exhibit EMT in combination with self renewal capabilities and anoikis resistance, thus traits of CSCs, yet without pluripotency. Logically, the latter is an unnecessary property as CSCs only need to recapitulate the original epithelial phenotype upon arrival at a distant organ site. Altogether, whether CSCs can be assigned a pluripotent state remains unclear. These features have been merged in a concept involving so-called migrating cancer stem cells [147]. Metastasizing cancer cells should have obtained both EMT and CSC characteristics that enable them to locally invade the surrounding stroma, survive in the circulation and establish metastatic progeny. As NANOG represents the cornerstone transcription factor driving cells into a ground pluripotent state [148] and given that KLF4 and KLF5 are known NANOG inducers, a strict regulation of their expression would be needed to avoid passing irreversibly towards a pluripotent state. However, a certain degree of (transient) expression may be needed to instigate the potential of self renewal. As reported recently, MDSCs are able to contribute to the induction of stemness in cancer cells [144]. Pursuing this hypothetic path, colonization of the metastatic site de facto implies proliferation and, therefore, may require downregulation of KLF5 in Ras-transformed cancer cells. In certain metastatic niches, this may also be the case for KLF4, with the preliminary exception for the cerebral tissue environment [86]. As the survival of metastasized cancer cells is dependent on the expression of ID genes [58], KLF17 may need to remain downregulated as well in order to maintain the released repression of ID1 [55] (Fig 6).
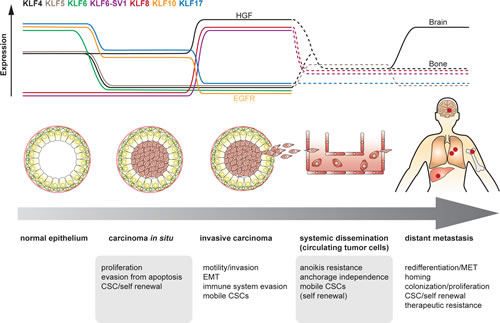
Fig 6: Hypothetic view on the plasticity of KLF expression as a contributor to cellular plasticity, shown from normal epithelium through different stages to metastasis. Putative expression levels of different KLFs vary depending on the requirements per phase. Dotted lines represent putative expression changes based on indicative findings, yet without reported direct evidence. HGF: upregulation of KLF4 under stimulation of HGF. EGFR: release of EGFR-inhibition through downregulation of KLF10. In the metastatic setting, KLF4 became abundantly expressed in brain metastatic, but not bone metastatic cancer cells.
We wish to illustrate the plasticity of KLFs in their upstream regulation and their subsequent downstream effects on cellular plasticity as proposed for the progression of cancer to metastatic disease (Fig 4 – 6). This also reflects the potential suitability of KLFs to be used as markers participating in cancer phenotype definition and adding to the dissection of inter-tumor heterogeneity. As stated recently, future therapeutic approaches against cancers in a metastatic or metastasizing state, will most probably need to comprise a cocktail of compounds interacting with various processes, in an individually tailored strategy [141]. An activity profile containing KLFs and downstream target genes may indeed prove to substantially contribute to the individual definition of cancer characteristics at different stages. Furthermore, the gene pool downstream of KLF transcription factors can hide novel invasion and metastasis suppressor genes that may be amenable as potential therapeutic targets.
References
1. Dynan WS, Tjian R. The promoter-specific transcription factor Sp1 binds to upstream sequences in the SV40 early promoter. Cell. 1983;35:79–87.
2. Kadonaga JT, Carner KR, Masiarz FR, Tjian R. Isolation of cDNA encoding transcription factor Sp1 and functional analysis of the DNA binding domain. Cell. 1987;51:1079–90.
3. Schuh R, Aicher W, Gaul U, Côté S, Preiss A, Maier D, Seifert E, Nauber U, Schröder C, Kemler R. A conserved family of nuclear proteins containing structural elements of the finger protein encoded by Krüppel, a Drosophila segmentation gene. Cell. 1986;47:1025–32.
4. Preiss A, Rosenberg UB, Kienlin A, Seifert E, Jäckle H. Molecular genetics of Krüppel, a gene required for segmentation of the Drosophila embryo. Nature. 1985;313:27–32.
5. Turner J, Crossley M. Mammalian Krüppel-like transcription factors: more than just a pretty finger. Trends Biochem Sci. 1999;24:236–40.
6. Miller IJ, Bieker JJ. A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the Krüppel family of nuclear proteins. Mol Cell Biol. 1993;13:2776–86.
7. Nuez B, Michalovich D, Bygrave A, Ploemacher R, Grosveld F. Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature. 1995;375:316–8.
8. Perkins AC, Sharpe AH, Orkin SH. Lethal beta-thalassaemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature. 1995;375:318–22.
9. Suske G, Bruford E, Philipsen S. Mammalian SP/KLF transcription factors: bring in the family. Genomics. 2005;85:551–6.
10. Shields JM, Yang VW. Two potent nuclear localization signals in the gut-enriched Krüppel-like factor define a subfamily of closely related Krüppel proteins. J Biol Chem. 1997;272:18504–7.
11. Philipsen S, Suske G. A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucleic Acids Res. 1999;27:2991–3000.
12. Kaczynski J, Cook T, Urrutia R. Sp1- and Krüppel-like transcription factors. Genome Biol. 2003;4:206.
13. Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-Mesenchymal Transitions in Development and Disease. Cell. 2009;139:871–90.
14. Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–29.
15. Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–72.
16. Gunasinghe NPAD, Wells A, Thompson EW, Hugo HJ. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. 2012;31:469–78.
17. Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, García De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–9.
18. Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83.
19. Bolós V, Peinado H, Pérez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci. 2003;116:499–511.
20. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–39.
21. Guaita S, Puig I, Franci C, Garrido M, Dominguez D, Batlle E, Sancho E, Dedhar S, De Herreros AG, Baulida J. Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J Biol Chem. 2002;277:39209–16.
22. Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, Berx G, Cano A, Beug H, Foisner R. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene. 2005;24:2375–85.
23. Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7:1267–78.
24. Pérez-Moreno MA, Locascio A, Rodrigo I, Dhondt G, Portillo F, Nieto MA, Cano A. A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem. 2001;276:27424–31.
25. Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004;23:1155–65.
26. Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin C-H, Moustakas A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J Cell Biol. 2006;174:175–83.
27. Wang X, Zheng M, Liu G, Xia W, McKeown-Longo PJ, Hung M-C, Zhao J. Krüppel-like factor 8 induces epithelial to mesenchymal transition and epithelial cell invasion. Cancer Res. 2007;67:7184–93.
28. Li R, Liang J, Ni S, Zhou T, Qing X, Li H, He W, Chen J, Li F, Zhuang Q, Qin B, Xu J, Li W et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell. 2010;7:51–63.
29. Yori JL, Johnson E, Zhou G, Jain MK, Keri RA. Kruppel-like factor 4 inhibits epithelial-to-mesenchymal transition through regulation of E-cadherin gene expression. J Biol Chem. 2010;285:16854–63.
30. Maeda M, Johnson KR, Wheelock MJ. Cadherin switching: essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J Cell Sci. 2005;118:873–87.
31. Yori JL, Seachrist DD, Johnson E, Lozada KL, Abdul-Karim FW, Chodosh LA, Schiemann WP, Keri RA. Krüppel-like factor 4 inhibits tumorigenic progression and metastasis in a mouse model of breast cancer. Neoplasia. 2011;13:601–10.
32. Lin Z-S, Chu H-C, Yen Y-C, Lewis BC, Chen Y-W. Krüppel-like factor 4, a tumor suppressor in hepatocellular carcinoma cells reverts epithelial mesenchymal transition by suppressing slug expression. PLoS ONE. 2012;7:e43593.
33. De Craene B, Gilbert B, Stove C, Bruyneel E, van Roy F, Berx G. The transcription factor snail induces tumor cell invasion through modulation of the epithelial cell differentiation program. Cancer Res. 2005;65:6237–44.
34. Segre JA, Bauer C, Fuchs E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet. 1999;22:356–60.
35. Tiwari N, Meyer-Schaller N, Arnold P, Antoniadis H, Pachkov M, van Nimwegen E, Christofori G. Klf4 is a transcriptional regulator of genes critical for EMT, including Jnk1 (Mapk8). PLoS ONE. 2013;8:e57329.
36. Akaogi K, Nakajima Y, Ito I, Kawasaki S, Oie S-H, Murayama A, Kimura K, Yanagisawa J. KLF4 suppresses estrogen-dependent breast cancer growth by inhibiting the transcriptional activity of ERalpha. Oncogene. 2009;28:2894–902.
37. Wang J, Place RF, Huang V, Wang X, Noonan EJ, Magyar CE, Huang J, Li L-C. Prognostic value and function of KLF4 in prostate cancer: RNAa and vector-mediated overexpression identify KLF4 as an inhibitor of tumor cell growth and migration. Cancer Res. 2010;70:10182–91.
38. Lai J-K, Wu H-C, Shen Y-C, Hsieh H-Y, Yang S-Y, Chang C-C. Krüppel-like factor 4 is involved in cell scattering induced by hepatocyte growth factor. J Cell Sci. 2012;125:4853–64.
39. Grotegut S, Schweinitz von D, Christofori G, Lehembre F. Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail. EMBO J. 2006;25:3534–45.
40. Shimamura T, Imoto S, Shimada Y, Hosono Y, Niida A, Nagasaki M, Yamaguchi R, Takahashi T, Miyano S. A novel network profiling analysis reveals system changes in epithelial-mesenchymal transition. PLoS ONE. 2011;6:e20804.
41. Dang DT, Zhao W, Mahatan CS, Geiman DE, Yang VW. Opposing effects of Krüppel-like factor 4 (gut-enriched Krüppel-like factor) and Krüppel-like factor 5 (intestinal-enriched Krüppel-like factor) on the promoter of the Krüppel-like factor 4 gene. Nucleic Acids Res. 2002;30:2736–41.
42. Bateman NW, Tan D, Pestell RG, Black JD, Black AR. Intestinal tumor progression is associated with altered function of KLF5. J Biol Chem. 2004;279:12093–101.
43. Narla G, Heath KE, Reeves HL, Li D, Giono LE, Kimmelman AC, Glucksman MJ, Narla J, Eng FJ, Chan AM, Ferrari AC, Martignetti JA, Friedman SL. KLF6, a candidate tumor suppressor gene mutated in prostate cancer. Science. 2001;294:2563–6.
44. DiFeo A, Narla G, Camacho-Vanegas O, Nishio H, Rose SL, Buller RE, Friedman SL, Walsh MJ, Martignetti JA. E-cadherin is a novel transcriptional target of the KLF6 tumor suppressor. Oncogene. 2006;25:6026–31.
45. Zhenzhen Z, De’an T, Limin X, Wei Y, Min L. New candidate tumor-suppressor gene KLF6 and its splice variant KLF6 SV2 counterbalancing expression in primary hepatocarcinoma. Hepatogastroenterology. 2012;59:473–6.
46. Zhang Q, Tan X-P, Yuan Y-S, Hu C-M, He C-H, Wang W-Z, Li J-C, Zhao Q, Liu N-Z. Decreased expression of KLF6 and its significance in gastric carcinoma. Med. Oncol. 2010;27:1295–302.
47. Cho YG, Kim CJ, Park CH, Yang YM, Kim SY, Nam SW, Lee SH, Yoo NJ, Lee JY, Park WS. Genetic alterations of the KLF6 gene in gastric cancer. Oncogene. 2005;24:4588–90.
48. Yamashita K, Upadhyay S, Osada M, Hoque MO, Xiao Y, Mori M, Sato F, Meltzer SJ, Sidransky D. Pharmacologic unmasking of epigenetically silenced tumor suppressor genes in esophageal squamous cell carcinoma. Cancer Cell. 2002;2:485–95.
49. DiFeo A, Narla G, Hirshfeld J, Camacho-Vanegas O, Narla J, Rose SL, Kalir T, Yao S, Levine A, Birrer MJ, Bonome T, Friedman SL, Buller RE et al. Roles of KLF6 and KLF6-SV1 in ovarian cancer progression and intraperitoneal dissemination. Clin Cancer Res. 2006;12:3730–9.
50. Narla G, DiFeo A, Reeves HL, Schaid DJ, Hirshfeld J, Hod E, Katz A, Isaacs WB, Hebbring S, Komiya A, McDonnell SK, Wiley KE, Jacobsen SJ et al. A germline DNA polymorphism enhances alternative splicing of the KLF6 tumor suppressor gene and is associated with increased prostate cancer risk. Cancer Res. 2005;65:1213–22.
51. Hatami R, Sieuwerts AM, Izadmehr S, Yao Z, Qiao RF, Papa L, Look MP, Smid M, Ohlssen J, Levine AC, Germain D, Burstein D, Kirschenbaum A et al. KLF6-SV1 Drives Breast Cancer Metastasis and Is Associated with Poor Survival. Sci Transl Med. 2013;5:169ra12–2.
52. Narla G, DiFeo A, Fernandez Y, Dhanasekaran S, Huang F, Sangodkar J, Hod E, Leake D, Friedman SL, Hall SJ, Chinnaiyan AM, Gerald WL, Rubin MA et al. KLF6-SV1 overexpression accelerates human and mouse prostate cancer progression and metastasis. J Clin Invest. 2008;118:2711–21.
53. DiFeo A, Feld L, Rodriguez E, Wang C, Beer DG, Martignetti JA, Narla G. A functional role for KLF6-SV1 in lung adenocarcinoma prognosis and chemotherapy response. Cancer Res. 2008;68:965–70.
54. Muñoz Ú, Puche JE, Hannivoort R, Lang UE, Cohen-Naftaly M, Friedman SL. Hepatocyte growth factor enhances alternative splicing of the Kruppel-like factor 6 (KLF6) tumor suppressor to promote growth through SRSF1. Mol Cancer Res. 2012;10:1216–27.
55. Gumireddy K, Li A, Gimotty PA, Klein-Szanto AJ, Showe LC, Katsaros D, Coukos G, Zhang L, Huang Q. KLF17 is a negative regulator of epithelial|[ndash]|mesenchymal transition and metastasis in breast cancer. Nat Cell Biol. 2009;11:1297–304.
56. Perk J, Iavarone A, Benezra R. Id family of helix-loop-helix proteins in cancer. Nat Rev Cancer. 2005;5:603–14.
57. Cubillo E, Diaz-Lopez A, Cuevas EP, Moreno-Bueno G, Peinado H, Montes A, Santos V, Portillo F, Cano A. E47 and Id1 interplay in epithelial-mesenchymal transition. PLoS ONE. 2013;8:e59948.
58. Gupta GP, Perk J, Acharyya S, de Candia P, Mittal V, Todorova-Manova K, Gerald WL, Brogi E, Benezra R, Massague J. ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc Natl Acad Sci USA. 2007;104:19506–11.
59. Cai X-D, Zhou Y-B, Huang L-X, Zeng Q-L, Zhang L-J, Wang Q-Q, Li S-L, Feng J-Q, Han A-J. Reduced expression of Krüppel-like factor 17 is related to tumor growth and poor prognosis in lung adenocarcinoma. Biochem Biophys Res Commun. 2012;418:67–73.
60. Liu F-Y, Deng Y-L, Li Y, Zeng D, Zhou Z-Z, Tian D-A, Liu M. Down-regulated KLF17 expression is associated with tumor invasion and poor prognosis in hepatocellular carcinoma. Med. Oncol. 2013;30:425.
61. Sun Z, Han Q, Zhou N, Wang S, Lu S, Bai C, Zhao RC. MicroRNA-9 enhances migration and invasion through KLF17 in hepatocellular carcinoma. Mol Oncol. 2013;7:884-94.
62. Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994;127:2021–36.
63. Peinado H, Quintanilla M, Cano A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem. 2003;278:21113–23.
64. Liu Y-N, Abou-Kheir W, Yin JJ, Fang L, Hynes P, Casey O, Hu D, Wan Y, Seng V, Sheppard-Tillman H, Martin P, Kelly K. Critical and reciprocal regulation of KLF4 and SLUG in transforming growth factor β-initiated prostate cancer epithelial-mesenchymal transition. Mol Cell Biol. 2012 ;32:941–53.
65. Li H-X, Han M, Bernier M, Zheng B, Sun S-G, Su M, Zhang R, Fu J-R, Wen J-K. Krüppel-like factor 4 promotes differentiation by transforming growth factor-beta receptor-mediated Smad and p38 MAPK signaling in vascular smooth muscle cells. J Biol Chem. 2010;285:17846–56.
66. Kim Y, Ratziu V, Choi SG, Lalazar A, Theiss G, Dang Q, Kim SJ, Friedman SL. Transcriptional activation of transforming growth factor beta1 and its receptors by the Kruppel-like factor Zf9/core promoter-binding protein and Sp1. Potential mechanisms for autocrine fibrogenesis in response to injury. J Biol Chem. 1998;273:33750–8.
67. Zhang H, Liu L, Wang Y, Zhao G, Xie R, Liu C, Xiao X, Wu K, Nie Y, Zhang H, Fan D. KLF8 involves in TGF-beta-induced EMT and promotes invasion and migration in gastric cancer cells. J Cancer Res Clin Oncol 2013;139:1033–42.
68. Tachibana I, Imoto M, Adjei PN, Gores GJ, Subramaniam M, Spelsberg TC, Urrutia R. Overexpression of the TGFbeta-regulated zinc finger encoding gene, TIEG, induces apoptosis in pancreatic epithelial cells. J Clin Invest. 1997;99:2365–74.
69. Cook T, Gebelein B, Mesa K, Mladek A, Urrutia R. Molecular cloning and characterization of TIEG2 reveals a new subfamily of transforming growth factor-beta-inducible Sp1-like zinc finger-encoding genes involved in the regulation of cell growth. J Biol Chem. 1998;273:25929–36.
70. Ellenrieder V, Buck A, Harth A, Jungert K, Buchholz M, Adler G, Urrutia R, Gress TM. KLF11 mediates a critical mechanism in TGF-beta signaling that is inactivated by Erk-MAPK in pancreatic cancer cells. Gastroenterology. 2004;127:607–20.
71. Buck A, Buchholz M, Wagner M, Adler G, Gress T, Ellenrieder V. The tumor suppressor KLF11 mediates a novel mechanism in transforming growth factor beta-induced growth inhibition that is inactivated in pancreatic cancer. Mol Cancer Res. 2006;4:861–72.
72. Johnsen SA, Subramaniam M, Janknecht R, Spelsberg TC. TGFbeta inducible early gene enhances TGFbeta/Smad-dependent transcriptional responses. Oncogene. 2002;2:5783–90.
73. Venkov C, Plieth D, Ni T, Karmaker A, Bian A, George AL, Neilson EG. Transcriptional networks in epithelial-mesenchymal transition. PLoS ONE. 2011;6:e25354.
74. Chang VHS, Chu P-Y, Peng S-L, Mao T-L, Shan Y-S, Hsu C-F, Lin C-Y, Tsai KKC, Yu WCY, Ch’ang H-J. Krüppel-like factor 10 expression as a prognostic indicator for pancreatic adenocarcinoma. Am J Pathol. 2012;181:423–30.
75. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–72.
76. Sahai E. Illuminating the metastatic process. Nat Rev Cancer. 2007;7:737–49.
77. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–92.
78. Tetreault M-P, Yang Y, Katz JP. Krüppel-like factors in cancer. Nat Rev Cancer. 2013;13:701–13.
79. Foster KW, Ren S, Louro ID, Lobo-Ruppert SM, McKie-Bell P, Grizzle W, Hayes MR, Broker TR, Chow LT, Ruppert JM. Oncogene expression cloning by retroviral transduction of adenovirus E1A-immortalized rat kidney RK3E cells: transformation of a host with epithelial features by c-MYC and the zinc finger protein GKLF. Cell Growth Differ. 1999;10:423–34.
80. Foster KW, Frost AR, McKie-Bell P, Lin CY, Engler JA, Grizzle WE, Ruppert JM. Increase of GKLF messenger RNA and protein expression during progression of breast cancer. Cancer Res. 2000;60:6488–95.
81. Yu F, Li J, Chen H, Fu J, Ray S, Huang S, Zheng H, Ai W. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30:2161–72.
82. Yu F, Shi Y, Wang J, Li J, Fan D, Ai W. Deficiency of kruppel-like factor KLF4 in mammary tumor cells inhibits tumor growth and pulmonary metastasis and is accompanied by compromised recruitment of myeloid-derived suppressor cells. Int J Cancer. 2013.
83. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC, Moses HL. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35.
84. Pandya AY, Talley LI, Frost AR, Fitzgerald TJ, Trivedi V, Chakravarthy M, Chhieng DC, Grizzle WE, Engler JA, Krontiras H, Bland KI, LoBuglio AF, Lobo-Ruppert SM et al. Nuclear localization of KLF4 is associated with an aggressive phenotype in early-stage breast cancer. Clin Cancer Res. 2004;10:2709–19.
85. Tai S-K, Yang M-H, Chang S-Y, Chang Y-C, Li W-Y, Tsai T-L, Wang Y-F, Chu P-Y, Hsieh S-L. Persistent Krüppel-like factor 4 expression predicts progression and poor prognosis of head and neck squamous cell carcinoma. Cancer Sci. 2011;102:895–902.
86. Okuda H, Xing F, Pandey PR, Sharma S, Watabe M, Pai SK, Mo Y-Y, Iiizumi-Gairani M, Hirota S, Liu Y, Wu K, Pochampally R, Watabe K. miR-7 suppresses brain metastasis of breast cancer stem-like cells by modulating KLF4. Cancer Res. 2013;73:1434–44.
87. Bos PD, Zhang XHF, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA, eacute JM. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–9.
88. Dang DT, Bachman KE, Mahatan CS, Dang LH, Giardiello FM, Yang VW. Decreased expression of the gut-enriched Krüppel-like factor gene in intestinal adenomas of multiple intestinal neoplasia mice and in colonic adenomas of familial adenomatous polyposis patients. FEBS Lett. 2000;476:203–7.
89. Zhao W, Hisamuddin IM, Nandan MO, Babbin BA, Lamb NE, Yang VW. Identification of Krüppel-like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene. 2004;23:395–402.
90. Wei D, Gong W, Kanai M, Schlunk C, Wang L, Yao JC, Wu T-T, Huang S, Xie K. Drastic down-regulation of Krüppel-like factor 4 expression is critical in human gastric cancer development and progression. Cancer Res. 2005;65:2746–54.
91. Hu W, Hofstetter WL, Li H, Zhou Y, He Y, Pataer A, Wang L, Xie K, Swisher SG, Fang B. Putative Tumor-Suppressive Function of Kruppel-Like Factor 4 in Primary Lung Carcinoma. Clin Cancer Res. 2009;15:5688–95.
92. Nagata T, Shimada Y, Sekine S, Hori R, Matsui K, Okumura T, Sawada S, Fukuoka J, Tsukada K. Prognostic significance of NANOG and KLF4 for breast cancer. Breast Cancer. 2012 Apr 17.
93. Wei D, Kanai M, Jia Z, Le X, Xie K. Kruppel-like factor 4 induces p27Kip1 expression in and suppresses the growth and metastasis of human pancreatic cancer cells. Cancer Res. 2008;68:4631–9.
94. Rowland BD, Bernards R, Peeper DS. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol. 2005;7:1074–82.
95. Zhou Y, Hofstetter WL, He Y, Hu W, Pataer A, Wang L, Wang J, Zhou Y, Yu L, Fang B, Swisher SG. KLF4 inhibition of lung cancer cell invasion by suppression of SPARC expression. Cancer Biol Ther. 2010;9:507–13.
96. Gilles C, Bassuk JA, Pulyaeva H, Sage EH, Foidart J-M, Thompson EW. SPARC/osteonectin induces matrix metalloproteinase 2 activation in human breast cancer cell lines. Cancer Res. 1998;58:5529–36.
97. Robert G, Gaggioli C, Bailet O, Chavey C, Abbe P, Aberdam E, Sabatié E, Cano A, Garcia de Herreros A, Ballotti R, Tartare-Deckert S. SPARC represses E-cadherin and induces mesenchymal transition during melanoma development. Cancer Res. 2006;66:7516–23.
98. Fan J, Peng Z, Zhou C, Qiu G, Tang H, Sun Y, Wang X, Li Q, Le X, Xie K. Gene-expression profiling in Chinese patients with colon cancer by coupling experimental and bioinformatic genomewide gene-expression analyses: identification and validation of IFITM3 as a biomarker of early colon carcinogenesis. Cancer. 2008;113:266–75.
99. Li D, Peng Z, Tang H, Wei P, Kong X, Yan D, Huang F, Li Q, Le X, Li Q, Xie K. KLF4-mediated negative regulation of IFITM3 expression plays a critical role in colon cancer pathogenesis. Clin Cancer Res. 2011;17:3558–68.
100. Vaira V, Faversani A, Martin NM, Garlick DS, Ferrero S, Nosotti M, Kissil JL, Bosari S, Altieri DC. Regulation of Lung Cancer Metastasis by Klf4-Numb-like Signaling. Cancer Res. 2013;73:2695–705.
101. Vaira V, Faversani A, Dohi T, Montorsi M, Augello C, Gatti S, Coggi G, Altieri DC, Bosari S. miR-296 regulation of a cell polarity-cell plasticity module controls tumor progression. Oncogene. 2012;3:27–38.
102. Tian Y, Luo A, Cai Y, Su Q, Ding F, Chen H, Liu Z. MicroRNA-10b promotes migration and invasion through KLF4 in human esophageal cancer cell lines. J Biol Chem. 2010;285:7986–94.
103. Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–8.
104. Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verástegui E et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6.
105. Frigo DE, Sherk AB, Wittmann BM, Norris JD, Wang Q, Joseph JD, Toner AP, Brown M, McDonnell DP. Induction of Kruppel-like factor 5 expression by androgens results in increased CXCR4-dependent migration of prostate cancer cells in vitro. Mol Endocrinol. 2009;23:1385–96.
106. Yang Y, Tetreault M-P, Yermolina YA, Goldstein BG, Katz JP. Krüppel-like factor 5 controls keratinocyte migration via the integrin-linked kinase. J Biol Chem. 2008;283:18812–20.
107. Yang Y, Goldstein BG, Chao H-H, Katz JP. KLF4 and KLF5 regulate proliferation, apoptosis and invasion in esophageal cancer cells. Cancer Biol Ther. 2005;4:1216–21.
108. Lu H, Hu L, Yu L, Wang X, Urvalek AM, Li T, Shen C, Mukherjee D, Lahiri SK, Wason MS, Zhao J. KLF8 and FAK cooperatively enrich the active MMP14 on the cell surface required for the metastatic progression of breast cancer. Oncogene. 2013 Jul 1.
109. Wang X, Lu H, Urvalek AM, Li T, Yu L, Lamar J, DiPersio CM, Feustel PJ, Zhao J. KLF8 promotes human breast cancer cell invasion and metastasis by transcriptional activation of MMP9. Oncogene. 2010;30:1901–11.
110. Wang W-F, Li J, Du L-T, Wang L-L, Yang Y-M, Liu Y-M, Liu H, Zhang X, Dong Z-G, Zheng G-X, Wang C-X. Krüppel-like factor 8 overexpression is correlated with angiogenesis and poor prognosis in gastric cancer. World J Gastroenterol. 2013;19:4309–15.
111. Li J-C, Yang X-R, Sun H-X, Xu Y, Zhou J, Qiu S-J, Ke A-W, Cui Y-H, Wang Z-J, Wang W-M, Liu K-D, Fan J. Up-regulation of Krüppel-like factor 8 promotes tumor invasion and indicates poor prognosis for hepatocellular carcinoma. Gastroenterology. 2010;139:2146–2157.e12.
112. Ying M, Sang Y, Li Y, Guerrero-Cazares H, Quinones-Hinojosa A, Vescovi AL, Eberhart CG, Xia S, Laterra J. Krüppel-like family of transcription factor 9, a differentiation-associated transcription factor, suppresses Notch1 signaling and inhibits glioblastoma-initiating stem cells. Stem Cells. 2011;29:20–31.
113. Spörl F, Korge S, Jürchott K, Wunderskirchner M, Schellenberg K, Heins S, Specht A, Stoll C, Klemz R, Maier B, Wenck H, Schrader A, Kunz D et al. Krüppel-like factor 9 is a circadian transcription factor in human epidermis that controls proliferation of keratinocytes. Proc Natl Acad Sci USA. 2012;109:10903–8.
114. Simmen FA, Xiao R, Velarde MC, Nicholson RD, Bowman MT, Fujii-Kuriyama Y, Oh SP, Simmen RCM. Dysregulation of intestinal crypt cell proliferation and villus cell migration in mice lacking Kruppel-like factor 9. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1757–69.
115. Chalaux E, López-Rovira T, Rosa JL, Pons G, Boxer LM, Bartrons R, Ventura F. A zinc-finger transcription factor induced by TGF-beta promotes apoptotic cell death in epithelial Mv1Lu cells. FEBS Lett. 1999;457:478–82.
116. Reinholz MM, An M-W, Johnsen SA, Subramaniam M, Suman VJ, Ingle JN, Roche PC, Spelsberg TC. Differential gene expression of TGF beta inducible early gene (TIEG), Smad7, Smad2 and Bard1 in normal and malignant breast tissue. Breast Cancer Res Treat. 2004;86:75–88.
117. Jin W, Chen B-B, Li J-Y, Zhu H, Huang M, Gu S-M, Wang Q-Q, Chen J-Y, Yu S, Wu J, Shao Z-M. TIEG1 inhibits breast cancer invasion and metastasis by inhibition of epidermal growth factor receptor (EGFR) transcription and the EGFR signaling pathway. Mol Cell Biol. 2012;32:50–63.
118. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76.
119. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72.
120. Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, Takizawa N, Yamanaka S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–6.
121. Jiang J, Chan Y-S, Loh Y-H, Cai J, Tong G-Q, Lim C-A, Robson P, Zhong S, Ng H-H. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat Cell Biol. 2008;10:353–60.
122. Chan KK-K, Zhang J, Chia N-Y, Chan Y-S, Sim HS, Tan KS, Oh SK-W, Ng H-H, Choo AB-H. KLF4 and PBX1 directly regulate NANOG expression in human embryonic stem cells. Stem Cells. 2009;27:2114–25.
123. Zhang P, Andrianakos R, Yang Y, Liu C, Lu W. Kruppel-like factor 4 (Klf4) prevents embryonic stem (ES) cell differentiation by regulating Nanog gene expression. J Biol Chem. 2010;285:9180–9.
124. Parisi S, Passaro F, Aloia L, Manabe I, Nagai R, Pastore L, Russo T. Klf5 is involved in self-renewal of mouse embryonic stem cells. J Cell Sci. 2008;121:2629–34.
125. Parisi S, Cozzuto L, Tarantino C, Passaro F, Ciriello S, Aloia L, Antonini D, De Simone V, Pastore L, Russo T. Direct targets of Klf5 transcription factor contribute to the maintenance of mouse embryonic stem cell undifferentiated state. BMC Biol. 2010;8:128.
126. Samavarchi-Tehrani P, Golipour A, David L, Sung H-K, Beyer TA, Datti A, Woltjen K, Nagy A, Wrana JL. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell. 2010;7:64–77.
127. Polo JM, Hochedlinger K. When fibroblasts MET iPSCs. Cell Stem Cell. 2010;7:5–6.
128. Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, Vassena R, Bilić J, Pekarik V, Tiscornia G, Edel M, Boué S, Izpisua Belmonte JC. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26:1276–84.
129. Maherali N, Ahfeldt T, Rigamonti A, Utikal J, Cowan C, Hochedlinger K. A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell. 2008;3:340–5.
130. Liu X, Sun H, Qi J, Wang L, He S, Liu J, Feng C, Chen C, Li W, Guo Y, Qin D, Pan G, Chen J et al. Sequential introduction of reprogramming factors reveals a time-sensitive requirement for individual factors and a sequential EMT-MET mechanism for optimal reprogramming. Nat Cell Biol. 2013;15:829–38.
131. Gaeta X, Xie Y, Lowry WE. Sequential addition of reprogramming factors improves efficiency. Nat Cell Biol. 2013;15:725–7.
132. Nemajerova A, Kim SY, Petrenko O, Moll UM. Two-factor reprogramming of somatic cells to pluripotent stem cells reveals partial functional redundancy of Sox2 and Klf4. Cell Death Differ. 2012;19:1268–76.
133. Siegel PM, Massagué J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–21.
134. Guo P, Dong X-Y, Zhang X, Zhao K-W, Sun X, Li Q, Dong J-T. Pro-proliferative factor KLF5 becomes anti-proliferative in epithelial homeostasis upon signaling-mediated modification. J Biol Chem. 2009;284:6071–8.
135. Guo P, Dong X-Y, Zhao K, Sun X, Li Q, Dong J-T. Opposing effects of KLF5 on the transcription of MYC in epithelial proliferation in the context of transforming growth factor beta. J Biol Chem. 2009;284:28243–52.
136. Chen C, Bhalala HV, Qiao H, Dong J-T. A possible tumor suppressor role of the KLF5 transcription factor in human breast cancer. Oncogene. 2002;21:6567–72.
137. Chen C, Bhalala HV, Vessella RL, Dong J-T. KLF5 is frequently deleted and down-regulated but rarely mutated in prostate cancer. Prostate. 2003;55:81–8.
138. Eaton SA, Funnell APW, Sue N, Nicholas H, Pearson RCM, Crossley M. A network of Krüppel-like Factors (Klfs). Klf8 is repressed by Klf3 and activated by Klf1 in vivo. J Biol Chem. 2008;283:26937–47.
139. Chen X, Johns DC, Geiman DE, Marban E, Dang DT, Hamlin G, Sun R, Yang VW. Krüppel-like factor 4 (gut-enriched Krüppel-like factor) inhibits cell proliferation by blocking G1/S progression of the cell cycle. J Biol Chem. 2001;276:30423–8.
140. Garrett-Sinha LA, Eberspaecher H, Seldin MF, de Crombrugghe B. A gene for a novel zinc-finger protein expressed in differentiated epithelial cells and transiently in certain mesenchymal cells. J Biol Chem. 1996;271:31384–90.
141. Brabletz T, Lyden D, Steeg PS, Werb Z. Roadblocks to translational advances on metastasis research. Nat Med. 2013;19:1104–9.
142. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92.
143. Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, Knuechel R, Kirchner T. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci USA. 2001;98:10356–61.
144. Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, Vatan L, Szeliga W, Mao Y, Thomas DG, Kotarski J, Tarkowski R, Wicha M et al. Myeloid-Derived Suppressor Cells Enhance Stemness of Cancer Cells by Inducing MicroRNA101 and Suppressing the Corepressor CtBP2. Immunity. 2013;39:611–21.
145. Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15.
146. Ocaña OH, Nieto MA. Epithelial plasticity, stemness and pluripotency. Cell Res. 2010;20:1086–8.
147. Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nature Reviews Cancer. 2005;5:744–9.
148. Silva J, Nichols J, Theunissen TW, Guo G, van Oosten AL, Barrandon O, Wray J, Yamanaka S, Chambers I, Smith A. Nanog is the gateway to the pluripotent ground state. Cell. 2009;138:722–37.
149. Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, Wernerus H, Björling L, Ponten F. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol. 2010;28:1248–50.
150. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9.