
INTRODUCTION
Inflammatory bowel disease (IBD) is an idiopathic, chronic relapsing inflammatory disorder of the gastrointestinal tract, comprising of ulcerative colitis (UC) and Crohn’s disease (CD). Patients with IBD have an increased risk of developing colorectal cancer (CRC) as early as 8-10 years after the diagnosis [1, 2]. A meta-analysis demonstrates a 4.5-fold increase in risk of CRC in patients with IBD compared to the general population, and CRC accounts for approximately 15% of all deaths in patients with IBD [3-5].
Inflammation and genetic instability contribute to the development of IBD-associated CRC (IBD-CRC) [6-8]. Series of alterations in oncogenes and tumor suppressor genes typically observed in sporadic CRC (S-CRC) are also important in the carcinogenesis of IBD-CRC. KRAS, a proto-oncogene, and TP53, a tumor suppressor gene, are strongly implicated in S-CRC. Mutations in KRAS and TP53 have demonstrated strong association with tumor progression in S-CRC [9-11]. Notably, KRAS mutational status determines the efficacy of epidermal growth factor receptor (EGFR) inhibitor, a potent therapy for patients with CRC in clinical practice [12, 13]. However, involved gene sequences and mutation frequencies of KRAS and TP53 may differ between S-CRC and IBD-CRC [14]. For example, TP53 mutations occur in the early stages of oncogenesis in IBD-CRC compared to late stages in S-CRC [15-17]. Furthermore, the loss of heterozygosity for TP53 is associated with progression of dysplasia, and TP53 mutation can also occur before the loss of heterozygosity in patients with UC. In contrast, KRAS mutation occurs in the later stages and less frequently in the oncogenesis of IBD-CRC compared to S-CRC [16, 18, 19].
Understanding differences in tumorigenesis between IBD-CRC and S-CRC may provide opportunity for targeted therapy for patients with IBD-CRC. In addition, evaluation of KRAS and TP53 mutation status can be used as potential biomarkers for dysplasia and cancer screening in patients with IBD. The aim of the meta-analysis was to compare the frequency of KRAS and TP53 mutation among patients with IBD-CRC, S-CRC, and IBD without dysplasia.
RESULTS
Literature search and description of included studies
An initial search retrieved 410 published studies. After a careful selection process, 17 case-control studies and two cohort studies were included in this meta-analysis (Figure 1) [20-38]. Quality scores of the 19 selected studies ranged from seven to nine indicating moderate to high quality. All studies were considered acceptable for inclusion in the meta-analysis. Quality scores of the included studies were summarized in Table 1.
A total of 482 patients with IBD-CRC, 4,222 with S-CRC, and 281 with IBD without dysplasia were included in our analysis. Twelve studies were conducted in the U.S., which is a high-prevalence area for IBD. Six studies were conducted in Europe, and one study was conducted in India [39-41]. The mean sample size of patients with IBD-CRC in all the studies was 24 (range 6 to 47). Thirteen studies described TP53 mutation, and 12 studies reported KRAS mutation, respectively. The most frequently reported mutational sites of TP53 and KRAS were exon 4-8 and exon 2, respectively. Codon 12 located in exon 2 had the highest mutation rate of KRAS. The demographic and clinical characteristics of included studies were shown in Table 1.
Table 1: Demographic and clinical characteristics of included studies.
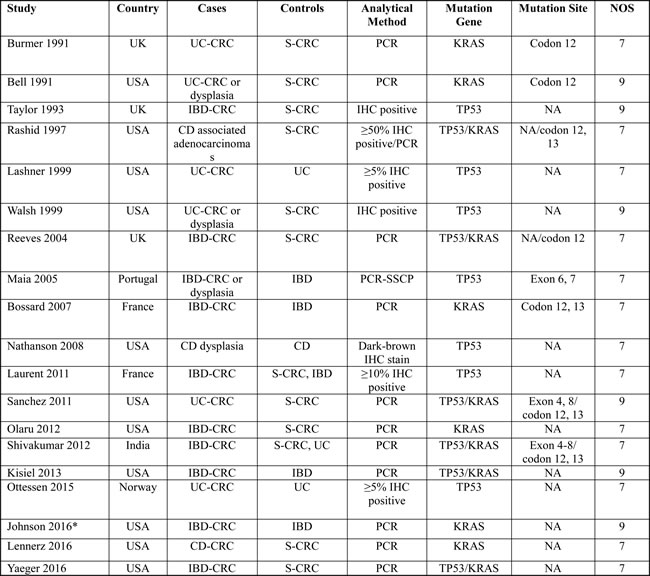
IBD: inflammatory bowel disease; UC: ulcerative colitis; CD: Crohn’s disease; CRC: colorectal cancer; PCR: polymerase chain reaction; SSCP: single-strand conformation polymorphism; S-CRC: sporadic colorectal cancer; NOS: Newcastle-Ottawa Scale.
*The study by Johnson et al 2016 contained two independent phases, and the data of one cohort phase was first presented in 2014
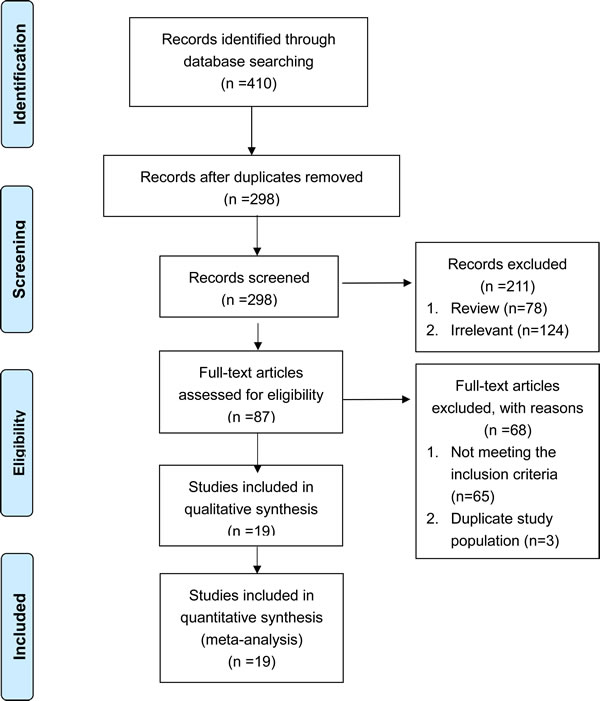
Figure 1: Flow chart of the selection process for the included studies.
KRAS mutation in IBD-CRC
KRAS mutation occurred more frequently in patients with IBD-CRC compared to IBD patients without dysplasia (RR = 3.09; 95%CI 1.47-6.51, P = 0.003). Neither significant heterogeneity (I2 = 8.4%, P = 0.36) nor publication bias (Begg’s test P = 0.09, Egger’s test P = 0.08) was detected (Figure 2A and 2B).
However, KRAS mutation occurred less commonly in IBD-CRC compared to S-CRC (RR = 0.71; 95% CI: 0.56-0.90, P = 0.004). Neither significant heterogeneity (I2 = 42.9%, P = 0.08) nor publication bias (Begg’s test P = 0.60, Egger’s test P = 0.65) was detected (Figure 2C and 2D).
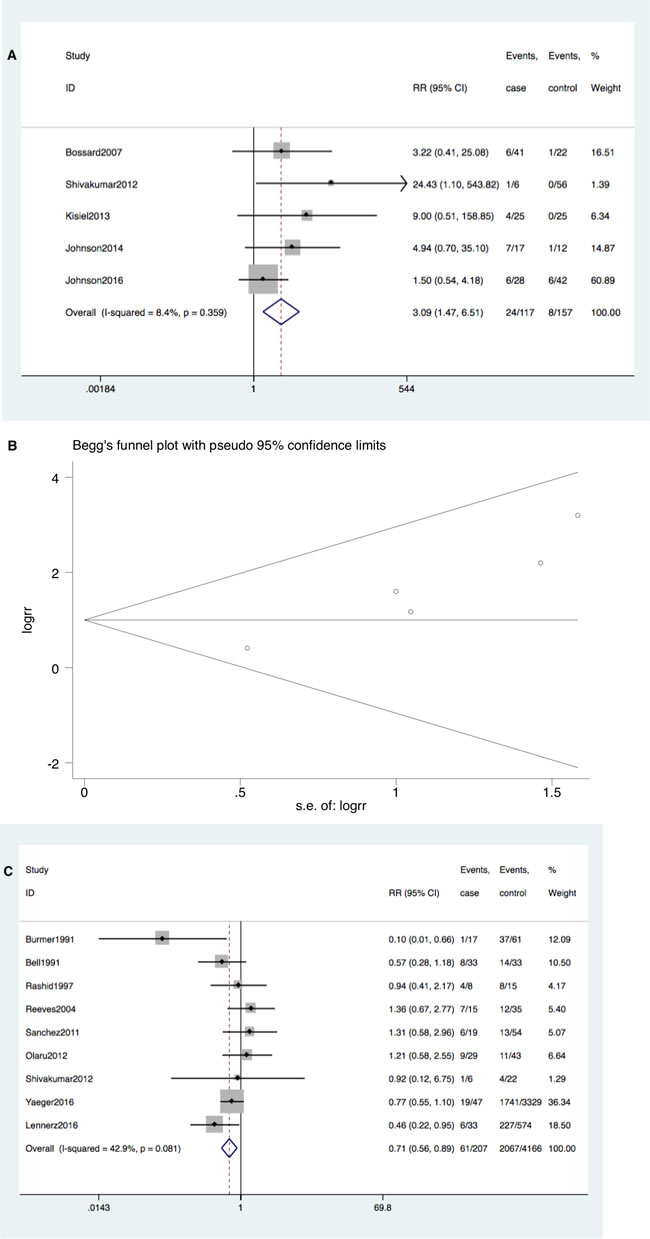
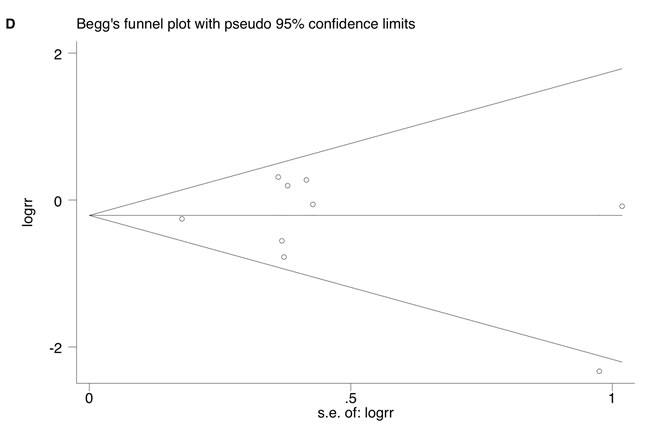
Figure 2: A. Forrest plot of risk ratio (RR) for KRAS mutation comparing IBD-CRC and IBD without dysplasia. B. Begg’s funnel plot of enrolled studies. C. Forrest plot of RR for KRAS mutation comparing IBD-CRC and S-CRC. D. Begg’s funnel plot of enrolled studies.
TP53 mutation in IBD-CRC
TP53 mutation also occurred more frequently in patients to IBD-CRC compared with IBD patients without dysplasia (RR = 2.15; 95% CI: 1.07-4.31, P = 0.03). Significant heterogeneity among studies (I2 = 87.8%, P < 0.001) but no publication bias (Begg’s test P = 0.368, Egger’s test P = 0.131) was detected (Figure 3A and 3B). However, no single publication was found to be significantly biasing the results using sensitivity analysis (Figure 3C).
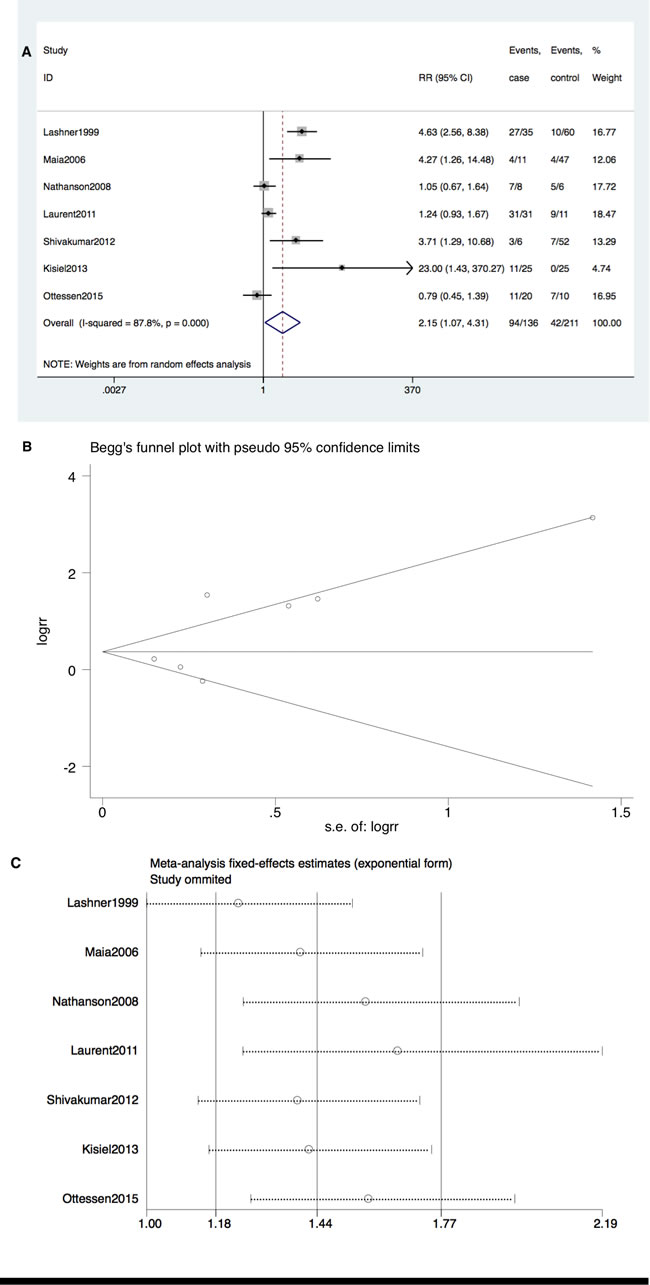
Figure 3: A. Forrest plot of RR for TP53 mutation comparing IBD-CRC and IBD without dysplasia. B. Begg’s funnel plot of enrolled studies. C. sensitivity analysis.
Furthermore, TP53 mutation occurred at a higher rate in IBD-CRC compared to S-CRC (RR = 1.24; 95% CI: 1.10-1.39, P < 0.001). Neither significant heterogeneity (I2 = 11.8%, P = 0.34) nor publication bias (Begg’s test P = 0.23, Egger’s test P = 0.91) was detected (Figure 4A and 4B).
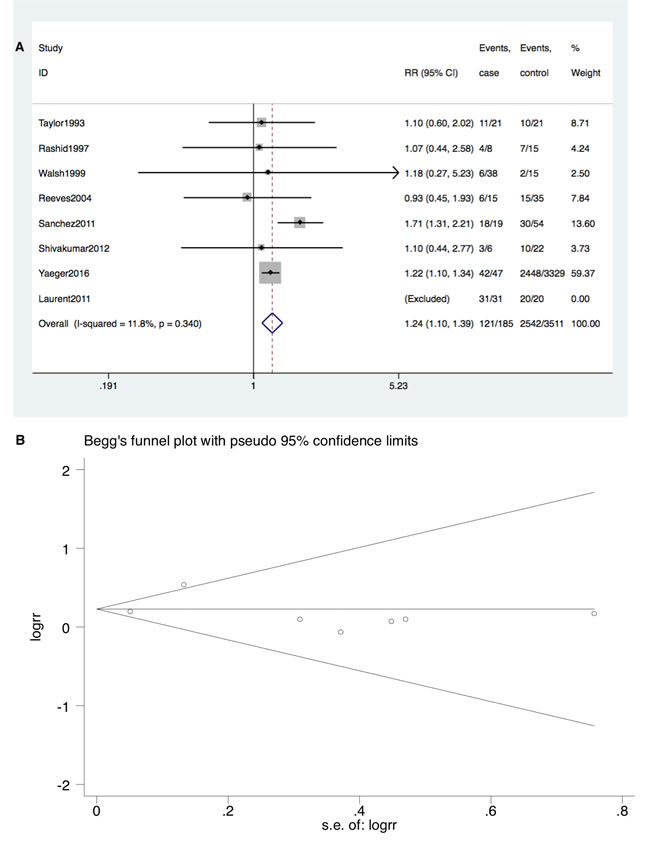
Figure 4: A. Forrest plot of RR for TP53 mutation comparing IBD-CRC and S-CRC. B. Begg’s funnel plot of enrolled studies.
Carcinogenesis in IBD patients with TP53 mutation
Of the 19 studies, only two studies reported the incidence of IBD-CRC as an outcome among IBD patients with or without TP53 mutation. As shown in Figure 5, there was a significant association between incidence of IBD-CRC and TP53 mutation among patients with IBD without dysplasia (RR = 5.28; 95% CI: 2.80-10.0, P < 0.001).
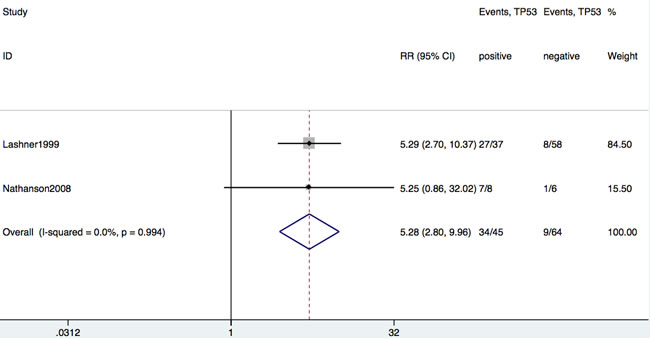
Figure 5: Forrest plot of RR for developing CRC in patients with IBD with or without TP53 mutation.
DISCUSSION
To our knowledge, this is the first meta-analysis comparing the frequency of KRAS and TP53 gene mutations among patients with IBD-CRC, S-CRC, and IBD without dysplasia. The meta-analysis demonstrated that TP53 mutation is more frequent while KRAS mutation is less prevalent in patients with IBD-CRC compared to S-CRC. Furthermore, both TP53 and KRAS mutations are more common in patients with IBD-CRC compared to IBD without dysplasia. Finally, IBD patients with TP53 mutation are more likely to develop IBD-CRC.
Observational studies have demonstrated no appreciable difference in clinical outcome including tumor recurrence, disease-free survival, and overall survival among patients with IBD-CRC compared to S-CRC patients [42, 43]. Possible reasons for the lack of difference in outcome despite distinct demographic and clinical patient characteristics may be related to utilizing conventional CRC therapy for patients with IBD-CRC, without accounting for the difference in molecular pathway. However, there are a number of important differences in carcinogenesis and outcome among patients with IBD-CRC and S-CRC, which may provide opportunity for targeted therapy and surveillance. Specifically, KRAS and TP53 status have been shown to be associated with progression-free survival and overall survival in patients with CRC [44, 45]. Previous molecular studies have highlighted the differences between KRAS and TP53 in the prevalence and onset of molecular events in between IBD-CRC and S-CRC [46]. Although KRAS mutations commonly have been reported among patients with S-CRC (18%-61%) and IBD-CRC/dysplasia (6%-50%) [21, 23, 33], KRAS mutation may occur later in IBD-CRC/dysplasia compared to S-CRC. Furthermore although TP53 mutations also are common in S-CRC (13%-100%) and IBD-CRC/dysplasia (16%-100%), TP53 mutation may occur early in IBD-CRC/dysplasia compared to S-CRC [25, 30]. However, a wide variation in mutational rate have been reported between different studies likely related to the differences in the study population.
The results of this meta-analysis provides support for possible genomic differences between IBD-CRC and S-CRC. Furthermore, the anatomic location of the tumors as well as the frequency and sequence of the molecular events differ between IBD-CRC and S-CRC according to previous evidence. IBD-CRC develops in chronically inflamed mucosa typically from absence of dysplasia, to indefinite dysplasia, to low-grade dysplasia, to high-grade dysplasia, and to carcinoma. This sequence is different from S-CRC that generally develops from a polypoid adenoma [15, 47]. The differences in expression of mutational KRAS and TP53 genes in IBD-CRC may account for the phenotypical differences of the two groups of CRC.
Some studies demonstrated that KRAS gene mutation is less frequent in IBD-CRC compared to S-CRC, consistent with the result of the meta-analysis [48, 49]. KRAS is a membrane bound proto-oncogene and functions as GDP-GTP-regulated binary on-off switch that regulates cytoplasmic signaling pathways and controls wide range of physiologic cellular processes [50]. KRAS is a key downstream effector of EGFR, and permanent activation of KRAS as a result of mutation causes cells to grow without exogenous stimulation and drives tumor initiation [51, 52]. Thus, tumors with mutant KRAS tend to be unresponsive to anti-EGFR therapy with increased risk of relapse and death compared with patients who have tumors characterized by a wide-type KRAS status [53]. A meta-analysis that includes patients with metastatic CRC demonstrated that KRAS mutations are highly specific, negative predictors of anti-EGFR therapy [54]. Given that KRAS status is a robust predictor of clinical response in multiple clinical trials, National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines have recommended genotyping KRAS status upon diagnosis of stage IV disease and also prior to administering EGFR-targeted treatment [55, 56]. Furthermore, presence of codon 12 mutations, the most common KRAS mutation, is associated with worsening relapse-free survival across all stages of CRC [57]. Therefore, KRAS mutation status is an important predictor of therapeutic efficacy in CRC.
Tumor suppressor genes are important guardians preventing expansion of mutant cells. Thus, the genetic abnormalities are associated with poor prognosis in patients with S-CRC. The important role of wide-type TP53, a tumor suppressor gene, is to halt the progression of cell cycle if DNA damage has occurred. Thus, a mutant p53 protein shows loss of growth-inhibitory function [58]. Furthermore, the wild-type p53 proteins have an extremely short half-life, whereas mutated p53 protein products are relatively stable and can be used as markers of mutated TP53 [59, 60]. Therefore, immunohistochemistry (IHC) can be used to determine the expression and location of p53 proteins that have accumulated in the cell nuclei of cancer tissues. Previous studies have shown that IHC and polymerase chain reaction (PCR) have a 95% concordance of measuring genetic mutations or protein accumulation in the nucleus [61]. A possible reason for the heterogeneity among studies comparing TP53 between IBD-CRC and IBD without dysplasia may be related with differing methodologies for evaluating TP53. Although determining TP53 status, unlike KRAS status, was not recommended by the NCCN as a prognostic predictor, the high frequency of TP53 mutations observed in many sequenced cancers highlight the important role of TP53 in the inhibition of cancer development [62, 63]. For example, TP53 mutation or loss of heterozygosity is associated with a higher cancer stage at presentation, a higher rate of recurrence, and a higher mortality [64-66]. Burmer and colleagues [67] found that TP53 loss of heterozygosity was detected in 6% of the biopsy specimens of UC patients without dysplasia, 9% with indefinite dysplasia, 33% with low grade dysplasia, 63% with high grade dysplasia and 85% with cancer. The results of our meta-analysis indicate that both KRAS and TP53 mutations occur more frequently in patients with IBD-CRC compared to IBD without dysplasia, consistent with previous studies. Furthermore, we found that TP53 mutation is associated with more advanced stage (Duke’s Class A, B, C) in patients with IBD-CRC (data not shown). Given our findings, TP53 status may potentially be used as a biomarker to improve cancer and dysplasia screening among patients with IBD.
There are limitations to our meta-analysis mainly related to the small number of published studies in full-text and English including some studies with a small sample size leading to the possibility of bias. For example, our findings demonstrating increased risk of CRC among IBD patients with TP53 mutation compared to those without TP53 mutation will require validation, given the inclusion of only two studies for the analysis. In addition, heterogeneity among the studies including study design and methods assessing KRAS and TP53 mutation may potentially affect the results of the meta-analysis.
In conclusion, our meta-analysis suggests that IBD-CRC and S-CRC may have different molecular pathways given the higher prevalence of TP53 but lower prevalence of KRAS mutation in the patients with IBD-CRC compared to S-CRC. Our findings suggest possible roles of TP53 and KRAS as candidate biomarkers for cancer and dysplasia screening among patients with IBD. Furthermore, elucidating the molecular pathway unique to IBD-CRC may provide potential targeted therapy. Further clinical studies are needed to validate our findings.
MATERIALS AND METHODS
Search strategy
A comprehensive literature search was conducted using Pubmed, EMBASE, and The Cochrane Library databases with an end date of June 2016. The main search strategies were as follows: “KRAS OR TP53” AND “mutation OR mutational analysis” AND “inflammatory bowel disease OR ulcerative colitis OR Crohn’s disease” AND “colorectal OR colon OR intestinal” AND “cancer OR adenocarcinoma OR carcinoma OR neoplasm”.
Study selection
Studies were required to meet the following inclusion criteria: (1) case-control or cohort studies; (2) provided a confirmed diagnosis of IBD-CRC in humans; (3) explicitly reported the detection methods for KRAS and TP53 alterations including PCR-SSCP, DNA sequencing, or other specific approaches for identifying gene mutation; and (4) written in English. In addition, reviews, animal studies, case reports, and studies lacking relevant data were excluded. Two investigators (L.D., B.C.) independently read the titles and abstracts of candidate studies. Afterwards, the two investigators analyzed the full texts of selected studies to determine whether the studies met the inclusion criteria. For duplicate studies based on identical or overlapping patient populations, the most recent or complete study was included in this meta-analysis. Any disagreement was resolved by a third investigator (N.D.).
Data extraction and quality evaluation
Two investigators (L.D., J.S.) independently extracted the data including the first author, publication year, geographic location, analytical method (protein/gene), cut-off values, and detected exons. The quality of the studies were assessed by using the Newcastle-Ottawa Scale (NOS) based on three perspectives: selection, comparability, and ascertainment of outcome [68]. Full score is nine stars, and studies with more stars were considered to be of higher quality.
Study endpoints and statistical analysis
The mutational status of KRAS and TP53 in IBD-CRC compared to IBD without dysplasia and S-CRC based on data from case-control and cohort studies were assessed. The endpoints of interest were expressed by risk ratios (RRs) with 95% CI. If the study was homogeneous (I2 < 50%), the fixed-effects model was used; otherwise (I2 > 50%), the random-effects model was chosen [69]. A P-value < 0.05 was considered statistically significant. If the study was heterogeneous, a sensitivity analysis was performed to examine the impact on the overall results. Publication bias was assessed by Egger’s and Begg’s test. All data were analyzed with Stata 12.0.
Conflicts of interest
This study has no conflicts of interest.
Author contributions
LJD and ND designed the research; LJD, JHS and BRC performed the research; LJD analyzed the data; LJD wrote the paper; JJK and ND provided critical feedback and revised the manuscript. All authors have approved the final draft of the manuscript.
References
1. Bernstein CN, Blanchard JF, Kliewer E, Wajda A. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer. 2001;91:854-62.
2. Jess T, Rungoe C, Peyrin-Biroulet L. Risk of colorectal cancer in patients with ulcerative colitis: a meta-analysis of population-based cohort studies. Clin Gastroenterol Hepatol. 2012;10:639-45.
3. Castano-Milla C, Chaparro M, Gisbert JP. Systematic review with meta-analysis: the declining risk of colorectal cancer in ulcerative colitis. Aliment Pharmacol Ther. 2014;39:645-59.
4. Canavan C, Abrams KR, Mayberry J. Meta-analysis: colorectal and small bowel cancer risk in patients with Crohn’s disease. Aliment Pharmacol Ther. 2006;23:1097-104.
5. Vagefi PA, Longo WE. Colorectal cancer in patients with inflammatory bowel disease. Clin Colorectal Cancer. 2005;4:313-9.
6. van der Woude CJ, Kleibeuker JH, Jansen PL, Moshage H. Chronic inflammation, apoptosis and (pre-)malignant lesions in the gastro-intestinal tract. Apoptosis. 2004;9:123-30.
7. Jaiswal M, LaRusso NF, Gores GJ. Nitric oxide in gastrointestinal epithelial cell carcinogenesis: linking inflammation to oncogenesis. American journal of physiology Gastrointestinal and liver physiology. 2001;281:G626-34.
8. Scarpa M, Castagliuolo I, Castoro C, Pozza A, Scarpa M, Kotsafti A, Angriman I. Inflammatory colonic carcinogenesis: a review on pathogenesis and immunosurveillance mechanisms in ulcerative colitis. World journal of gastroenterology. 2014;20:6774-85.
9. Munro AJ, Lain S, Lane DP. P53 abnormalities and outcomes in colorectal cancer: a systematic review. British journal of cancer. 2005;92:434-44.
10. Bournet B, Buscail C, Muscari F, Cordelier P, Buscail L. Targeting KRAS for diagnosis, prognosis, and treatment of pancreatic cancer: Hopes and realities. European journal of cancer (Oxford, England : 1990). 2016;54:75-83.
11. Pan W, Yang Y, Zhu H, Zhang Y, Zhou R, Sun X. KRAS mutation is a weak, but valid predictor for poor prognosis and treatment outcomes in NSCLC: A meta-analysis of 41 studies. Oncotarget. 2016;7:8373-88. doi: 10.18632/oncotarget.7080.
12. Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, Schilsky RL. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. Journal of clinical oncology. 2009;27:2091-6.
13. Meyerhardt JA, Mayer RJ. Systemic therapy for colorectal cancer. The New England journal of medicine. 2005;352:476-87.
14. Wang ZH, Fang JY. Colorectal Cancer in Inflammatory Bowel Disease: Epidemiology, Pathogenesis and Surveillance. Gastrointestinal tumors. 2014;1:146-54.
15. Itzkowitz SH. Molecular biology of dysplasia and cancer in inflammatory bowel disease. Gastroenterology clinics of North America. 2006;35:553-71.
16. Feagins LA, Souza RF, Spechler SJ. Carcinogenesis in IBD: Potential targets for the prevention of colorectal cancer. Nature Reviews Gastroenterology and Hepatology. 2009;6:297-305.
17. Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, vanTuinen P, Ledbetter DH, Barker DF, Nakamura Y, White R, Vogelstein B. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science (New York, NY). 1989;244:217-21.
18. Chen J, Compton C, Cheng E, Fromowitz F, Viola MV.c-Ki-ras mutations in dysplastic fields and cancers in ulcerative colitis. Gastroenterology. 1992;102:1983-7.
19. Umetani N, Sasaki S, Watanabe T, Shinozaki M, Matsuda K, Ishigami H, Ueda E, Muto T. Genetic alterations in ulcerative colitis-associated neoplasia focusing on APC, K-ras gene and microsatellite instability. Japanese journal of cancer research. 1999;90:1081-7.
20. Bell SM, Kelly SA, Hoyle JA, Lewis FA, Taylor GR, Thompson H, Dixon MF, Quirke P. c-Ki-ras gene mutations in dysplasia and carcinomas complicating ulcerative colitis. 1991(0007-0920 (Print)).
21. Burmer GC, Rabinovitch PS, Loeb LA. Frequency and spectrum of c-Ki-ras mutations in human sporadic colon carcinoma, carcinomas arising in ulcerative colitis, and pancreatic adenocarcinoma. Environmental health perspectives. 1991;93:27-31.
22. Taylor HW, Boyle M, Smith SC, Bustin S, Williams NS. Expression of p53 in colorectal cancer and dysplasia complicating ulcerative colitis. The British journal of surgery. 1993;80:442-4.
23. Rashid A, Hamilton SR. Genetic alterations in sporadic and Crohn’s-associated adenocarcinomas of the small intestine. Gastroenterology. 1997;113(1):127-35.
24. Lashner BA, Shapiro BD, Husain A, Goldblum JR. Evaluation of the usefulness of testing for p53 mutations in colorectal cancer surveillance for ulcerative colitis. Am J Gastroenterol. 1999;94:456-62.
25. Walsh SV, Loda M, Torres CM, Antonioli D, Odze RD. P53 and beta catenin expression in chronic ulcerative colitis—associated polypoid dysplasia and sporadic adenomas: an immunohistochemical study. The American journal of surgical pathology. 1999;23:963-9.
26. Reeves HL, Narla G, Ogunbiyi O, Haq AI, Katz A, Benzeno S, Hod E, Harpaz N, Goldberg S, Tal-Kremer S, Eng FJ, Arthur MJ, Martignetti JA, et al. Kruppel-like factor 6 (KLF6) is a tumor-suppressor gene frequently inactivated in colorectal cancer. Gastroenterology. 2004;126:1090-103.
27. Maia L, Dinis J, Cravo M, Claro I, Baltazar C, Fonseca I, Veloso T, Capelinha AF, Carneiro F, Nobre-Leitao C. Who takes the lead in the development of ulcerative colitis-associated colorectal cancers: mutator, suppressor, or methylator pathway? Cancer genetics and cytogenetics. 2005;162:68-73.
28. Bossard C, Denis MG, Bezieau S, Bach-Ngohou K, Bourreille A, Laboisse CL, Mosnier JF. Involvement of the serrated neoplasia pathway in inflammatory bowel disease-related colorectal oncogenesis. Oncology reports. 2007;18:1093-7.
29. Nathanson JW, Yadron NE, Farnan J, Kinnear S, Hart J, Rubin DT. p53 mutations are associated with dysplasia and progression of dysplasia in patients with Crohn’s disease. Digestive Diseases and Sciences. 2008;53:474-80.
30. Laurent C, Svrcek M, Flejou JF, Chenard MP, Duclos B, Freund JN, Reimund JM. Immunohistochemical expression of CDX2, beta-catenin, and TP53 in inflammatory bowel disease-associated colorectal cancer. Inflammatory bowel diseases. 2011;17:232-40.
31. Sanchez JA, Dejulius KL, Bronner M, Church JM, Kalady MF. Relative role of methylator and tumor suppressor pathways in ulcerative colitis-associated colon cancer. Inflammatory bowel diseases. 2011;17:1966-70.
32. Olaru AV, Cheng Y, Agarwal R, Yang J, David S, Abraham JM, Yu W, Kwon JH, Lazarev M, Brant SR, Marohn MR, Hutcheon DF, Harpaz N, et al. Unique patterns of CpG island methylation in inflammatory bowel disease-associated colorectal cancers. Inflammatory bowel diseases. 2012;18:641-8.
33. Shivakumar BM, Kumar BL, Bhat G, Suvarna D, Rao L, Pai CG, Satyamoorthy K. Molecular alterations in colitis-associated colorectal neoplasia: study from a low prevalence area using magnifying chromo colonoscopy. Journal of Crohn’s & colitis. 2012;6:647-54.
34. Kisiel JB, Yab TC, Nazer Hussain FT, Taylor WR, Garrity-Park MM, Sandborn WJ, Loftus EV, Wolff BG, Smyrk TC, Itzkowitz SH, Rubin DT, Zou H, Mahoney DW, et al. Stool DNA testing for the detection of colorectal neoplasia in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2013;37:546-54.
35. Friis-Ottessen M, Burum-Auensen E, Schjolberg AR, Ekstrom PO, Andersen SN, Clausen OP, De Angelis PM. TP53/p53 alterations and Aurora A expression in progressor and non-progressor colectomies from patients with longstanding ulcerative colitis. International journal of molecular medicine. 2015;35:24-30.
36. Johnson DH, Taylor WR, Aboelsoud MM, Foote PH, Yab TC, Cao X, Smyrk TC, Loftus EV, Jr., Mahoney DW, Ahlquist DA, Kisiel JB. DNA Methylation and Mutation of Small Colonic Neoplasms in Ulcerative Colitis and Crohn’s Colitis: Implications for Surveillance. Inflammatory bowel diseases. 2016;22:1559-67.
37. Lennerz JK, van der Sloot KWJ, Le LP, Batten JM, Han JY, Fan KC, Siegel CA, Srivastava A, Park DY, Chen JH, Sands BE, Korzenik JR, Odze RD, et al. Colorectal cancer in Crohn’s colitis is comparable to sporadic colorectal cancer. International journal of colorectal disease. 2016;31:973-82.
38. Yaeger R, Shah MA, Miller VA, Kelsen JR, Wang K, Heins ZJ, Ross JS, He Y, Sanford E, Yantiss RK, Balasubramanian S, Stephens PJ, Schultz N, et al. Genomic Alterations Observed in Colitis-Associated Cancers Are Distinct From Those Found in Sporadic Colorectal Cancers and Vary by Type of Inflammatory Bowel Disease. Gastroenterology. 2016.
39. Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, Kaplan GG. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46-54.e42; quiz e30.
40. Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140:1785-94.
41. Ng SC. Epidemiology of inflammatory bowel disease: focus on Asia. Best practice & research Clinical gastroenterology. 2014;28:363-72.
42. Leowardi C, Schneider ML, Hinz U, Harnoss JM, Tarantino I, Lasitschka F, Ulrich A, Buchler MW, Kadmon M. Prognosis of Ulcerative Colitis-Associated Colorectal Carcinoma Compared to Sporadic Colorectal Carcinoma: A Matched Pair Analysis. Ann Surg Oncol. 2016;23:870-6.
43. Dugum M, Lin J, Lopez R, Estfan B, Manilich E, Stocchi L, Shen B, Liu X. Recurrence and survival rates of inflammatory bowel disease-associated colorectal cancer following postoperative chemotherapy: a comparative study. Gastroenterology report. 2016.
44. Di Bartolomeo M, Pietrantonio F, Perrone F, Dotti KF, Lampis A, Bertan C, Beretta E, Rimassa L, Carbone C, Biondani P, Passalacqua R, Pilotti S, Bajetta E. Lack of KRAS, NRAS, BRAF and TP53 mutations improves outcome of elderly metastatic colorectal cancer patients treated with cetuximab, oxaliplatin and UFT. Targeted oncology. 2014;9:155-62.
45. Sclafani F, Gonzalez D, Cunningham D, Hulkki Wilson S, Peckitt C, Tabernero J, Glimelius B, Cervantes A, Dewdney A, Wotherspoon A, Brown G, Tait D, Oates J, et al. TP53 mutational status and cetuximab benefit in rectal cancer: 5-year results of the EXPERT-C trial. Journal of the National Cancer Institute. 2014;106.
46. Fogt F, Vortmeyer AO, Goldman H, Giordano TJ, Merino MJ, Zhuang Z. Comparison of genetic alterations in colonic adenoma and ulcerative colitis-associated dysplasia and carcinoma. Human pathology. 1998;29:131-6.
47. Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011;140:1807-16.
48. Burmer GC, Levine DS, Kulander BG, Haggitt RC, Rubin CE, Rabinovitch PS. c-Ki-ras mutations in chronic ulcerative colitis and sporadic colon carcinoma. Gastroenterology. 1990;99:416-20.
49. Burmer GC, Loeb LA. Mutations in the KRAS2 oncogene during progressive stages of human colon carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:2403-7.
50. Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. Journal of cell science. 2016;129:1287-92.
51. Cox AD, Der CJ. Ras history: The saga continues. Small GTPases. 2010;1:2-27.
52. Tan C, Du X. KRAS mutation testing in metastatic colorectal cancer. World journal of gastroenterology. 2012;18:5171-80.
53. Vauthey JN, Zimmitti G, Kopetz SE, Shindoh J, Chen SS, Andreou A, Curley SA, Aloia TA, Maru DM. RAS mutation status predicts survival and patterns of recurrence in patients undergoing hepatectomy for colorectal liver metastases. Annals of surgery. 2013;258:619-26; discussion 26-7.
54. Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, Papadimitriou CA, Murray S. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. The Lancet Oncology. 2008;9:962-72.
55. Engstrom PF, Arnoletti JP, Benson AB, 3rd, Chen YJ, Choti MA, Cooper HS, Covey A, Dilawari RA, Early DS, Enzinger PC, Fakih MG, Fleshman J, Jr., Fuchs C, et al. NCCN Clinical Practice Guidelines in Oncology: colon cancer. Journal of the National Comprehensive Cancer Network. 2009;7:778-831.
56. Engstrom PF, Arnoletti JP, Benson AB, 3rd, Chen YJ, Choti MA, Cooper HS, Covey A, Dilawari RA, Early DS, Enzinger PC, Fakih MG, Fleshman J, Jr., Fuchs C, et al. NCCN Clinical Practice Guidelines in Oncology: rectal cancer. Journal of the National Comprehensive Cancer Network. 2009;7:838-81.
57. Andreyev HJ, Norman AR, Cunningham D, Oates J, Dix BR, Iacopetta BJ, Young J, Walsh T, Ward R, Hawkins N, Beranek M, Jandik P, Benamouzig R, et al. Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. British journal of cancer. 2001;85:692-6.
58. Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413-31.
59. Midgley CA, Lane DP. p53 protein stability in tumour cells is not determined by mutation but is dependent on Mdm2 binding. Oncogene. 1997;15:1179-89.
60. Finlay CA, Hinds PW, Tan TH, Eliyahu D, Oren M, Levine AJ. Activating mutations for transformation by p53 produce a gene product that forms an hsc70-p53 complex with an altered half-life. Mol Cell Biol. 1988;8:531-9.
61. Costa A, Marasca R, Valentinis B, Savarino M, Faranda A, Silvestrini R, Torelli G. p53 gene point mutations in relation to p53 nuclear protein accumulation in colorectal cancers. The Journal of pathology. 1995;176:45-53.
62. Cancer Genome Atlas Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061-8.
63. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330-7.
64. Pricolo VE, Finkelstein SD, Wu TT, Keller G, Bakker A, Swalsky PA, Bland KI. Prognostic value of TP53 and K-ras-2 mutational analysis in stage III carcinoma of the colon. American journal of surgery. 1996;171:41-6.
65. Auvinen A, Isola J, Visakorpi T, Koivula T, Virtanen S, Hakama M. Overexpression of p53 and long-term survival in colon carcinoma. British journal of cancer. 1994;70:293-6.
66. Yamaguchi A, Kurosaka Y, Fushida S, Kanno M, Yonemura Y, Miwa K, Miyazaki I. Expression of p53 protein in colorectal cancer and its relationship to short-term prognosis. Cancer. 1992;70:2778-84.
67. Burmer GC, Rabinovitch PS, Haggitt RC, Crispin DA, Brentnall TA, Kolli VR, Stevens AC, Rubin CE. Neoplastic progression in ulcerative colitis: histology, DNA content, and loss of a p53 allele. Gastroenterology. 1992;103:1602-10.
68. Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. European journal of epidemiology. 2010;25:603-5.
69. Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Statistics in medicine. 2002;21:1539-58.