
Introduction
Fms-like tyrosine kinase 3 (FLT3) is a member of the class III family of receptor tyrosine kinases, including platelet-derived growth factor receptors α and β, c-kit and fms. [1, 2] Their general structure consists of five extracellular immunoglobin-like domains, a single transmembrane domain, a short juxtamembrane region and an interrupted kinase domain. FLT3 expression is limited primarily to primitive hematopoietic stem/progenitor cells and dendritic cells. It plays roles in the processes of differentiation, survival and proliferation. The kinase activity of FLT3 is normally stimulated upon binding of FLT3 ligand (FL), which leads to receptor dimerization, autophosphorylation, and phosphorylation of other signaling proteins. [3, 4] This, in turn, leads to activation of downstream signaling cascades, including signal transducers of activation and transcription (STATs), mitogen-activated protein kinases (MAP kinases) and phosphatidyl inositol-3 kinase/AKT pathways. Mutation of FLT3 induces constitutive kinase activation which is independent of ligand and occurs in about one-third of AML cases. [5, 6] About 20-25% of AML patients express FLT3 mutations as in-frame internal tandem duplications (ITDs) of varying length in the juxtamembrane domain. An additional 7-10% of patients harbor point mutations of the kinase domain activation loop (AL) or other areas. Point mutations within the juxtamembrane domain of FLT3 have also been detected and represent a rarer class of activating mutations. [7, 8] The crystal structure of FLT3 shows that the juxtamembrane domain makes contact with the activation loop and maintains FLT3 in an autoinhibited state. [9] Mutations in the juxtamembrane domain or in the activation loop destabilize the inhibitory conformation and lead to constitutive FLT3 activation. Transfection of plasmids containing either type of mutation into cytokine-dependent hematopoietic cell lines transforms cells to growth factor independence. [10] Activating mutations of FLT3 are also observed at lower frequencies in acute lymphoblastic leukemia (ALL), [11, 12] myelodysplastic syndrome (MDS) [13] and mixed-lineage leukemia (MLL) rearranged infant leukemias. [14, 15]
Kinase domain mutations are primarily restricted to the carboxy terminal portion of the catalytic domain, predominately at residues D835 and I836 on the activation loop. [6, 16, 17] The crystal structures of related tyrosine kinases indicate that the activation loop acts as a flexible gate to allow access of ATP and substrate to the nucleotide-binding site when the molecule assumes the “open” or active conformation. [9, 18-20] In the “closed” or autoinhibited conformation, the FLT3 activation loop swings inward and binds to D811, thus preventing ATP binding in this pocket. [9] Mutation of critical residues in the kinase domain destabilize the autoinhibited FLT3 structure, shifting the equilibrium towards the open conformation, leading to constitutive kinase activation in the absence of FLT3 ligand binding. While all FLT3 activating kinase domain mutations provide growth and survival advantages, there appear to be signaling differences compared to ITD mutations. [21, 22] Moreover, amongst the kinase domain mutations, there are wide differences in response to FLT3 tyrosine kinase inhibitors (TKI). [23, 24] All TKI described as FLT3 inhibitors target FLT3/ITD mutations, while some of these TKI show limited activity against wild-type FLT3, and most exhibit significantly reduced potency at inhibiting some of the AL and other point mutations. D835Y is the most frequently observed kinase domain mutation in AML patients, constituting ~50% of all FLT3 AL missense mutations. [25] Other substitution mutations detected less frequently at D835 include valine, histidine, glycine, and asparagine substitutions as well as aspartic acid codon deletion. [6, 16, 17, 26] The I836 codon also undergoes deletion and/or mutation to threonine, serine or leucine with or without an insertion. [6, 16] Several of the additional mutations dispersed on the activation loop or in the kinase domain have been documented in patients at D593∆ [27] , S840 [28] , N841 [29] and Y842 [30] .
In this study, BaF3 cells transfected with different FLT3 activating mutations reported in patients were tested against a panel of 13 compounds characterized as FLT3 inhibitors. Five of them are currently in clinical trials for treatment of acute myeloid leukemia including sorafenib [31] , quizartinib [32] , midostaurin [33] , crenolanib [34] and ponatinib [35] . Another FLT3 inhibitor, TTT-3002, demonstrated high potency in FLT3/ITD and FLT3/D835 mutated leukemia cell lines and patient samples in preclinical studies [36, 37] . A wide range of responses were noted to each mutation. The results show that most inhibitors are not effective against some activating mutations, or substantially higher drug concentrations are required to inhibit proliferation of cells bearing these particular mutations, concentrations unlikely to be reached in these patients. The data presented here indicate that a number of activating mutations render FLT3 insensitive to some TKI, and determination of the particular mutation expressed should be undertaken when deciding upon a FLT3 TKI regimen for AML patients with AL and other non-ITD point mutations.
Results
Differential sensitivity of FLT3 AL mutants to FLT3 TKI growth inhibition
The FLT3 AL mutations selected for use in this study were all originally isolated from AML patients and transfection of each into BaF3 cells led to IL-3 independent growth. Representative clones were obtained following limiting dilution and their responses to FLT3 TKI were measured by MTT (growth/survival) assay. Each of the 13 FLT3 TKI tested inhibited proliferation of BaF3/ITD mutant cells in a concentration-dependent manner, albeit with widely varying potencies (Table 1 and Figure 1). Lestaurtinib, midostaurin and TTT-3002 inhibited proliferation in all FLT3 AL mutants with generally similar or sometimes even greater potency compared to the FLT3/ITD cells. In contrast, 10 of the 13 TKI tested displayed a range of activity against FLT3 AL mutants. Multiple FLT3 TKI were active against the D835G, D835N, I836T and 840G+S mutants, with IC50 values within 10-fold of the values for inhibiting the FLT3/ITD mutant (midostaurin, lestaurtinib, linifanib, quizartinib, sutinib, sorafenib, TTT-3002 and KW2449). However, at least 7 of the 13 FLT3 TKI tested had little or no activity against the D593∆, D835Y, D835A, D835L+K, I836L+D and I836S mutants (AG1295, AGS324, linifanib, quizartinib, sunitinib, sorafenib and R406). For those 7 FLT3 TKI the IC50 was not reached over the concentration range tested.
Only 4 of the FLT3 TKI tested in this study inhibited growth of the most frequently seen clinical FLT3 AL mutant D835Y. Midostaurin, lestaurtinib, TTT-3002, and crenolanib inhibited growth of this mutant at concentrations that were at least within 20-fold the concentrations effective against the FLT3/ITD mutant, whereas the other inhibitors were largely ineffective at the FLT3/ITD inhibitory concentrations. Although ponatinib is a potent inhibitor of the FLT3/ITD cells with an IC50 below 1 nM, the D835Y mutation shifted the IC50 to 92 nM (Table 1). The IC50 values for the rest of the inhibitors evaluated against the D835Y mutation were beyond their respective testing ranges in this study. These findings are consistent with clinical data showing that treatment of FLT3/ITD positive AML patients with monotherapy with some FLT3 TKI has frequently given rise to blasts that express D835Y, rendering the cells insensitive to further therapy. [38] Midostaurin, lestaurtinib, TTT-3002, and crenolanib were also the only FLT3 TKI in this study that inhibited growth of the D593∆ mutant at IC50 levels close to those observed in the FLT3/ITD mutant cells.
The FLT3 TKI could be ranked based on the number of mutants they inhibited, with midostaurin, lestaurtinib, and TTT-3002 inhibiting growth of all mutants in the panel. Crenolanib and ponatinib inhibited all but 3 of the mutants (D835L+K, I836T, and I836I), although it should be noted that relatively high concentrations of ponatinib were required to inhibit proliferation in D835Y and D835A mutants. Sorafenib and KW2449 inhibited growth of all but 5 mutants (D835Y, D835L+K, I836L+D, D593∆ and D835A for sorafenib; D835Y, D835L+K, I836L+D, D593∆ and I836S for KW2449). Linifanib, quizartinib, and sunitinib each inhibited all but 6 of the mutants, whereas AGS324, AG1295, and R406 were ineffective against 7 of the panel members. These results indicate that treatment of AML patients using some FLT3 TKI would be more effective against a greater spectrum of FLT3 AL mutations than other FLT3 inhibitors.
Table 1: Proliferation IC50 (nM) of FLT3 TKI against AL mutants.
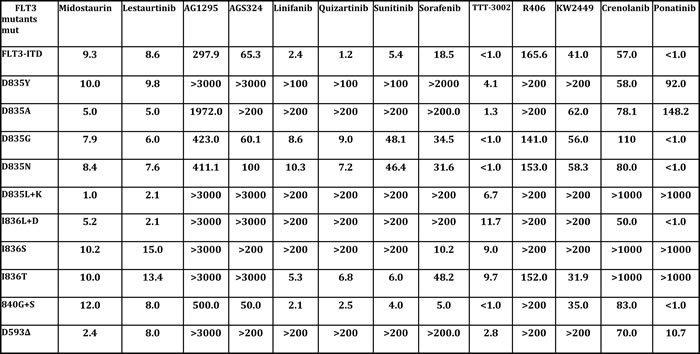
Proliferation half maximal inhibitory concentration (IC50) at 48 h by MTT assay. The cells were grown in the absence of IL3 and analyzed by treatment in increasing concentrations of FLT3 TKI for 48 hours, after which growth inhibition was assessed using the MTT assay. Each experiment was performed at least three times and representative results are shown.
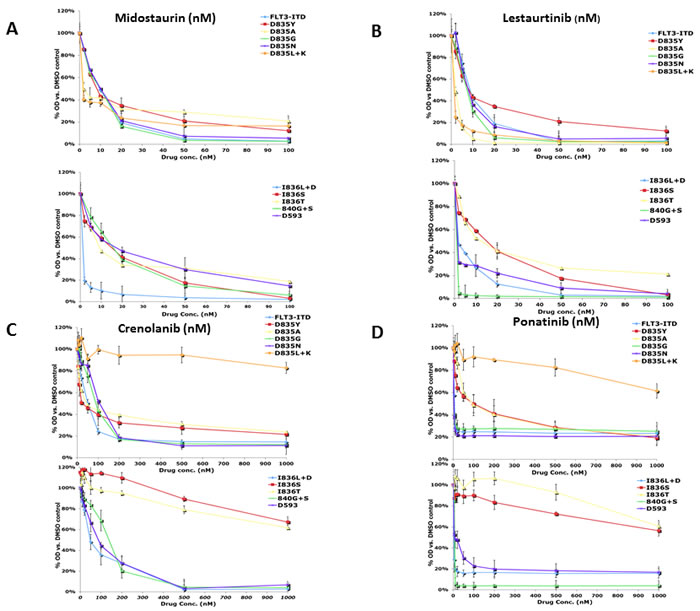
Figure 1: Growth of AL mutants in the presence of FLT3 TKI. The BaF3 AL mutants were grown in the absence of IL-3 and analyzed by treatment in increasing concentrations of FLT3 TKI for 48 hours, after which growth inhibition was assessed using the MTT assay. Means are representative of at least three independent experiments.
Uniform sensitivity of FLT3 AL mutants to lestaurtinib, midostaurin and TTT-3002
Of the 13 FLT3 TKI tested, only lestaurtinib, midostaurin and TTT-3002 inhibited proliferation in all FLT3 AL mutants tested. Lestaurtinib and midostaurin underwent phase 3 clinical trials for FLT3 mutant AML. To that end, we tested the ability of lestaurtinib and midostaurin to inhibit FLT3 autophosphorylation of each FLT3 AL mutant. Midostaurin (Figure 2a) and lestaurtinib (Figure 2b) both inhibited FLT3 autophosphorylation of all FLT3 AL mutants with IC50 values of 5 nM or lower. The Western blotting results examining FLT3 inhibition by lestaurtinib and midostaurin for each FLT3 AL mutant correlate well with the MTT data.
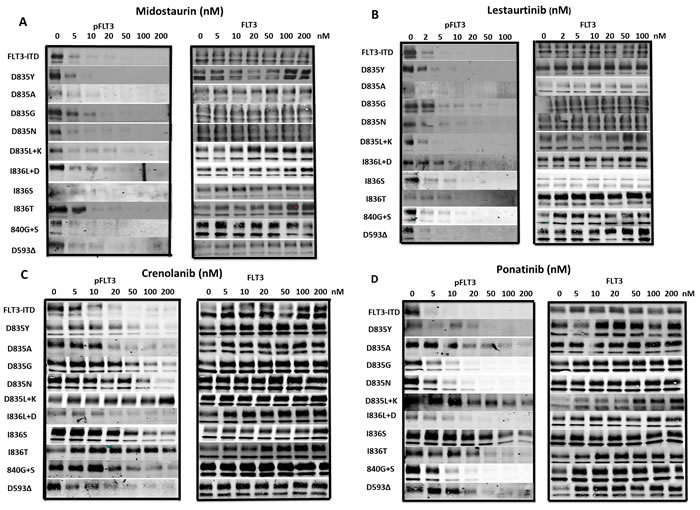
Figure 2: Inhibition of FLT3 AL mutant phosphorylation by FLT3 TKI. BaF3 cells expressing FLT3 AL mutations were treated with either a. midostaurin, b. lestaurtinib, c. crenolanib or d. ponatinib at the indicated concentrations for 1 h at 37°C followed by immunoprecipitation for FLT3, SDS-PAGE and WB for FLT3 and phospho-FLT3 (PFLT3). Each experiment was performed at least twice and representative results are shown.
Crenolanib and ponatinib have better activity against FLT3/D835 point mutants than the other FLT3 AL mutants
Crenolanib and ponatinib are both in clinical trials for the treatment of AML patients with FLT3 mutations. One study recently demonstrated that crenolanib displays activity against several of the FLT3 AL mutants as well as FLT3/ITD-TKD dual mutants. [39] We observed that crenolanib inhibited FLT3 autophosporylation of D835Y, D835A, D835G, D835N, I836L+D, 840G+S and D593∆ mutants with IC50s of 36, 20, 28, 30, 15, 104 and 12nM, respectively (Figure 1c). Ponatinib inhibited FLT3 autophosphorylation of D835Y, D835A, D835G, D835N, I836L+D, 840G+S and D593∆ mutants with IC50 values of 5 nM or less (Figure 1d). However, crenolanib and ponatinib were not nearly as effective at inhibiting phosphorylation of D835L+K, I836S and I836T FLT3 mutants, with significant autophosphorylation still observable even at concentrations of 200 nM (Figure 2c, 2d).
Effect of FLT3 TKI on wild-type FLT3 in the presence of FLT3 Ligand
Infant ALL, B-lineage ALL and AML samples with the highest levels of wild-type FLT3 expression often show constitutive FLT3 phosphorylation as a result of FL expression by autocrine or paracrine mechanisms. [40] These samples are often sensitive to FLT3 TKI, arguing that wild-type FLT3 signaling is important in these cells. Thus targeting of wild-type, non-mutant FLT3 may be important for this subset of ALL and AML patients. Thus it is important to study the effect of FLT3 TKI on wild-type FLT3 signaling as well. We determined the activity of FLT3 TKI against BaF3/wild-type FLT3 expressing cells that were stimulated with FL (Figure 3). Of the 12 FLT3 TKI tested, the majority of them inhibited FLT3 autophosphorylation of wild-type FLT3 with IC50 values of 20 nM or lower. Only Crenolanib, R406 and AG1295 were not nearly as effective at inhibiting wild-type FLT3 phosphorylation compared to FLT3/ITD ( IC50 values of 200 nM, 200 nM and 500 nM). Similar results were seen whether cells were incubated with FL first followed by FLT3 TKI treatment or FLT3 TKI followed by FL stimulation.

Figure 3: Inhibition of wild-type FLT3 signaling pathways by FLT3 TKI. BaF3/WT-FLT3 cells were treated with FLT3 TKI at the indicated concentrations for 1 h at 37°C then stimulated with 25ng/ml of human FLT3 ligand for 15 minutes, followed by immunoprecipitation for FLT3, SDS-PAGE and WB for FLT3 and phospho-FLT3 (PFLT3). Each experiment was performed at least twice and representative results are shown.
Effect of FLT3 TKI on ITD versus D835Y signaling pathways
The STAT5, PI-3 kinase/AKT and Ras/Map kinase pathways are activated by FLT3 and are important in cell survival and proliferation in cells that are dependent on FLT3 activity. However, there are also extrinsic mechanisms independent of FLT3, capable of maintaining signaling pathways downstream of FLT3 despite the presence of inhibitory FLT3 TKI levels. [41] In addition, off-target effects of some TKI that cause inhibition of downstream pathways might cause inhibition of growth despite lack of inhibition against a FLT3 mutant. To determine whether inhibition of FLT3 signaling pathways correlated with inhibition of FLT3 autophosphorylation, 3 FLT3 TKI representing different classes of inhibitors were tested against the FLT3/ITD and the FLT3 D835Y mutants. Treatement with lestaurtinib, sorafenib and AG1295 all inhibited FLT3 autophosphorylation as well as phosphorylation of STAT5, AKT and Map kinase in FLT3/ITD cells in a concentration-dependent manner (Figure 4). In FLT3 D835Y cells, lestaurtinib inhibited FLT3 autophosphorylation with an IC50 < 2 nM which resulted in termination of signaling through STAT5, AKT and MAP kinase pathways (Figure 5). In contrast, even the highest concentrations of sorafenib and AG1295 tested showed markedly reduced or absent inhibition of FLT3 autophosphorylation and a subsequent lack of inhibitory activity on phosphorylation of STAT5, AKT and MAP kinase. Thus, for the 3 FLT3 TKI tested against the FLT3/ITD and the FLT3 D835Y mutants, there was a good correlation between inhibition of FLT3 phosphorylation and inhibition of FLT3 dependent downstream signaling pathways.
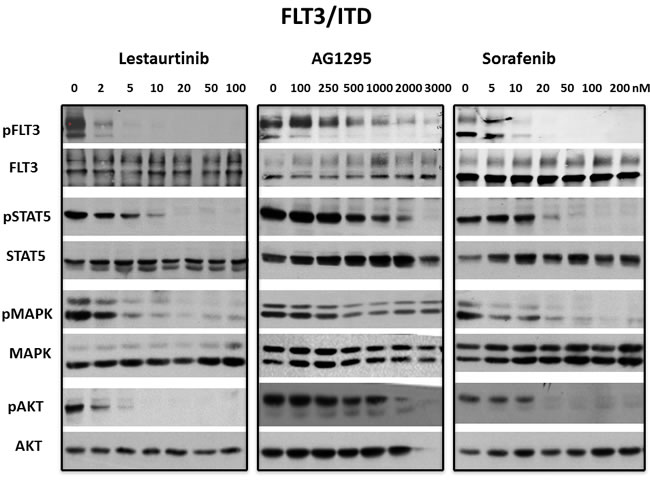
Figure 4: Inhibition of FLT3/ITD signaling pathways by FLT3 TKI. BaF3/ITD cells were treated with lestaurtinib, AG1295 or sorafenib at the indicated concentrations for 1 h. Inhibition of signaling pathways by WB was evaluated in whole cell lysates using antibodies described in Materials and Methods and visualizing bands using enhanced chemiluminescence. Each experiment was repeated at least three times and representative results are shown.
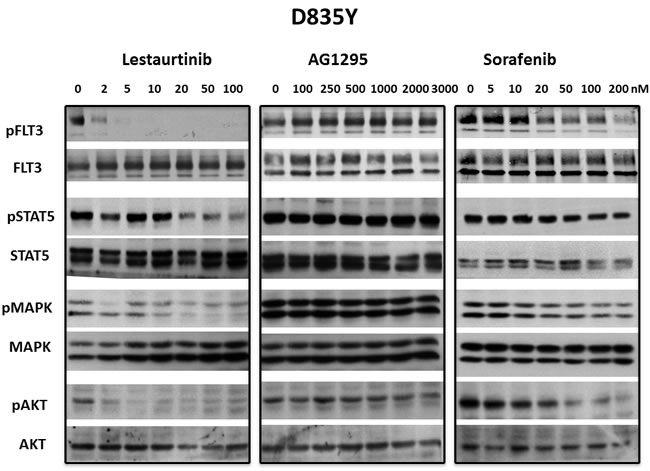
Figure 5: Inhibition of FLT3 D835Y signaling pathways by FLT3 TKI. BaF3 FLT3 AL mutant cells were treated with lestaurtinib, AG1295 or sorafenib at the indicated concentrations for 1 h. Inhibition of signaling pathways by WB was evaluated in whole cell lysates using antibodies described in Materials and Methods and visualizing bands using enhanced chemiluminescence. Each experiment was repeated at least three times and representative results are shown.
Effect of FLT3 TKI on engraftment levels of FLT3 D835Y mutant cells in BALB/c mice
Lestaurtinib and sorafenib both inhibit proliferation driven by signaling events in FLT3/ITD cells in vitro, but they have divergent effects on the FLT3 AL mutants. To assess their activity in vivo, BaF3 FLT3 D835Y cells were transplanted into syngeneic mice and were treated with lestaurtinib or sorafenib. Because the cells were stably transfected with luciferase, disease progression could be monitored by visualizing bioluminescence upon intraperitoneal injection of luciferin. Typically, these cells home to the bone marrow and spread to the spleen and peripheral blood before mice become moribund. By day 5 FLT3 D835Y cells were well engrafted in the mice. By day 9, 4 days after treatment was instituted, the sorafenib and vehicle treated mice demonstrated clear progression of leukemia. In contrast, leukemic burden was greatly reduced in mice treated with lestaurtinib, with only a small amount of D835Y cells detected in the bone marrow (Figure 6).
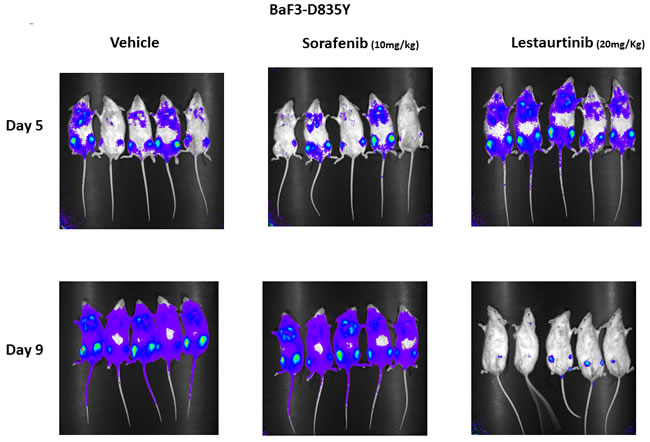
Figure 6: Effect of FLT3 TKI on progression of BaF3 D835Y cells engrafted in syngeneic BALB/c mice. BaF3 D835Y cells (2 × 106) transfected with a luciferase construct were transplanted into three cohorts of BALB/c mice via tail vein injection with 5 mice per group. On day 5 following transplantation, the level of engraftment was assessed by imaging mice for bioluminescence on an IVIS Spectrum imager. Starting on day 5, mice were then treated twice daily by vehicle, subcutaneous lestaurtinib (20 mg/kg) or once daily sorafenib (10 mg/kg) by oral gavage until day 9, at which point mice were again imaged. This experiment was repeated three times.
Discussion
Nearly half of acute myeloid leukemia patients treated with chemotherapy have a favorable outcome, but those who present with a FLT3/ITD mutation have a worse prognosis. [42-44] Preclinical and clinical evidence suggest that the addition of a FLT3 TKI to chemotherapy is synergistic and may lead to improved efficacy for those patients. [45] FLT3 AL mutations also constitutively activate FLT3 kinase activity and subsequent downstream signaling pathways that lead to transformation and cytokine independence, but do not appear to result in worse prognosis than non-FLT3 mutant AML patients. The addition of a FLT3 TKI to the therapy of FLT3 AL mutant patients might still further improve their outcome and appeared to do so in a recently reported trial of midostaurin. [46] Unfortunately, many FLT3 AL mutations fail to respond to many of the FLT3 TKI that have been selected for by their potency against FLT3 ITD mutations. [23, 24] Treatment of a FLT3 AL mutant patient with these FLT3 TKI would fail to inhibit FLT3 kinase activity and thus have no potential to contribute to the elimination of leukemic blasts. Thus, in this era of personalized medicine, it is important to know the exact FLT3 AL mutation each patient carries in order to be able to pick the most appropriate FLT3 TKI to use for that patient. This is also important during therapy for a patient with a FLT3 ITD mutation because the FLT3 AL mutations appear to arise frequently through selection in these patients under the pressure of FLT3 TKI therapy.
Most of the clinically available FLT3 TKI display reduced activity against at least some of the FLT3 AL mutants. In this study, only lestaurtinib, midostaurin and TTT-3002 appear to be universally effective against this type of mutation. Treatment with FLT3 TKIs other than lestaurtinib, midostaurin, or TTT-3002 therefore, could prove problematic if patients present with, or are selected for during treatment, one of the FLT3 AL mutations. With the increasing number of FLT3 TKI being developed clinically, it is important to determine the spectrum of FLT3 mutants that would respond to or be resistant to each TKI. [47, 48] For example, sorafenib and quizartinib have both demonstrated some efficacy in reducing peripheral blasts in FLT3 ITD mutant AML patients and bone marrow blasts in some cases, but the data from this study indicate that neither of these inhibitors would be effective against blasts harboring FLT3 mutations of D835Y, D835A, D835L+K or I836L+D. [49-53] In addition, the D835G and D835N mutations caused ~10-fold elevation in the IC50 for sunitinib that would likely preclude its use against these mutants.
We know that in infant ALL and in AML with the highest level of wild-type FLT3 expression, FLT3 is constitutively phosphorylated and the cells are sensitive to FLT3 TKI. Therefore, studying the effect of FLT3 TKI on wild-type FLT3 signaling is important. With the exception of one of the TKI (AG1295) tested in this study, all of the others showed a high level of activity against wild-type FLT3 signaling. Thus FLT3 TKIs might prove effective for upfront or relapsed therapy for these patients.
TKI are generally categorized as type I or type II inhibitors. [54] Type I inhibitors directly compete with ATP binding to the nucleotide-binding site and make contact with the tyrosine kinase hinge region. Crystal structures of kinases in complex with inhibitors indicate that type I TKI bind target when the phosphorylated kinase activation loop assumes either the active unfolded “open” conformation or the inactive closed conformation. [55] Type II inhibitors indirectly compete with ATP by binding a hydrophobic allosteric site adjacent to the nucleotide-binding site in addition to the nucleotide-binding site. These inhibitors only bind the target when the activation loop assumes the “closed” conformation. Dasatinib is an example of a type I inhibitor that binds BCR-ABL while imatinib and nilotinib are examples of type II inhibitors. [56] FLT3 mutations lead to constitutive activation by two different mechanisms: ITD mutations that relieve the negative regulatory influence of the juxtamembrane domain on the catalytic domain and the AL mutants that directly destabilize the activation loop. [9, 57] There are suggestions that FLT3 TKI also fall into the category of type I or type II inhibitors, but the biological data from this study argue that this categorization, while technically accurate, is likely an oversimplification. Midostaurin (and lestaurtinib) is thought to be a type I inhibitor whereas sorafenib and many other FLT3 TKI are thought to belong to the type II class. [58, 59] Since all FLT3 TKI inhibit ITD mutations but display variable activity against kinase domain mutants, the assertion that type II FLT3 TKI are ineffective against some AL mutations because the kinase is shifted toward the active conformation seems inadequate as it should then hold true for all FLT3 AL mutations. Instead, it is more probable that the activation loop of specific FLT3 AL mutants adopts a conformation that prohibits the DFG residues from forming hydrogen bonds that are characteristic of binding with type II inhibitors, thereby reducing drug affinity. The phenylalanine in the DFG region of activated kinases has been shown to move more than 10 Å from its position in the inhibited conformation. [54] Almost certainly, different substitutions on the activation loop will have varying effects on the position of the DFG residues with subsequently varying effects on the ability of type II FLT3 TKI to tightly bind these mutants. An alternative explanation as to why type II inhibitors might not be effective against FLT3 AL mutants could be that some FLT3 AL mutations don’t fully transition to the inactive conformation seen in the autoinhibited form where the activation loop is completely folded between the N and C kinase lobes.
The number of FLT3 activating kinase domain mutations continues to grow and with that the need to define the spectrum of activity of each FLT3 TKI against these mutations. In this report, we demonstrated that TTT-3002, lestaurtinib and midostaurin are fairly effective against the known FLT3 AL mutants reported to date. Despite the development of subsequent generations of FLT3 TKI, many would not be effective in patients that express a FLT3 AL mutation. Thus, there is still a need to identify FLT3 TKI that can target multiple FLT3 activating mutations, both FLT3 ITD and FLT3 AL types, in order to improve upfront AML patient responses and to reduce the incidence of selection of FLT3 AL resistance mutations.
Materials and Methods
Reagents and antibodies
All compounds were dissolved in dimethyl sulfoxide (DMSO) at stock concentrations of 10 mM and stored at -800C. Lestaurtinib, quizartinib, midostaurin,, ponatinib, sunitinib and sorafenib were purchased from LCLabs (Ontario, Canada). Linifanib, crenolanib and R406 were purchased from Selleckchem (Houston, Texas, USA). KW2449 was from Kyowa Hakko Kirin Co., Ltd. (Tokyo, Japan). AG1295 and AGS324 were provided by Aviv Gazit (The Hebrew University, Israel). TTT-3002 was a generous gift of TauTaTis, Inc. (San Diego, CA). The final DMSO concentration was maintained at < 0.2% in cell suspension for each drug concentration and control group in all assays. Recombinant human interleukin-3 (IL-3) and human FLT3 ligand (FL) were purchased from Pepro Tech, Inc. (Rocky Hill, NJ, USA). The FLT3 S-18 antibody (#SC-480) was from Santa Cruz Biotechnology (Santa Cruz, CA, USA), 4G10 phosphotyrosine mouse monoclonal antibody and recombinant protein A-agarose were from Upstate Biotechnology (Lake Placid, NY, USA), and CD135-phycoerythrin (PE) conjugated antibody (#558996) was from BD Pharmingen (San Jose, CA, USA). AlexaFluor 680 goat anti-mouse (# A21058) and Daylight 800 goat anti-rabbit antibodies (#611-145-122) were purchased from Invitrogen Corporation (Grand Island, NY, USA) and Rockland Immunochemicals Inc. (Gilbertsville, PA, USA) respectively. PhosphoMAP kinase (#9101), phosphoSTAT5 (#9351), phosphoAKT (#9271), MAP kinase (#9102), STAT5 (#9363) and AKT (#9272) antibodies were from Cell Signaling Technologies, Inc. (Beverly, MA, USA). Goat anti-mouse (#NA931) and goat anti-rabbit (#NA934) horseradish peroxidase antibodies and the enhanced chemiluminescence kit were from Amersham Biosciences (Arlington Heights, IL, USA).
DNA constructs and cells
BaF3 cells were grown cultured at 370C in 5% CO2 in RPMI medium (Invitrogen Corporation) with 10% fetal bovine serum (Gemini Bio-Products, Woodland, CA) containing penicillin/streptomycin (Invitrogen Corporation) and supplemented with 1 ng/ml recombinant human IL-3. FLT3 point mutations in BaF3 cells were generated by site-directed mutagenesis in the pBabe Neo vector containing wild-type FLT3 cDNA using the QuickChange Site-Directed Mutagenesis kit following the manufacturer’s recommendations (Stratagene, La Jolla, CA, USA). Primers for each point mutation were designed using Primer 3 software. All primers were ordered through Invitrogen Corporation. Primer sequences are listed in Supplemental Table 1.
All mutations were confirmed by sequencing. Transfection was performed using the Nucleofector II from Amaxa Biosystems (Walkersville, MD, USA), after which cells were selected in 1 mg/ml G418 in the presence of IL-3 and analyzed for FLT3 expression using CD135-PE antibody on a Becton-Dickinson FACSCalibur flow cytometer (Becton-Dickinson, San Jose, CA, USA) using CellQuest software. Clones were subsequently obtained by limiting dilution and tested for their ability to grow without IL-3. BaF3/ITD cells were established from a patient sample as previously described. [60]
Growth inhibition assay
Based on Trypan Blue exclusion, 2 × 104 viable cells were seeded in quadruplicate in a 96-well microtiter plate in the presence or absence of inhibitor for 48 h. The effect of drug activity on cell viability was measured using the 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay according to the manufacturer’s instructions (Roche Applied Science, Indianapolis, IN, USA). Following treatment, cells were incubated for 4 h in MTT followed by dissolution in solubilization buffer overnight. Absorbance was measured at 570 nm on a Bio-Rad plate reader (Hercules, CA, USA). The concentration of drug that inhibited absorbance by 50% (IC50) was calculated using CalcuSyn software. Results shown are presented as means and are representative of at least three independent experiments.
Immunoprecipitation and western blotting
Activated FLT3 expression was measured by performing immunoprecipitation, SDS-PAGE and Western blotting as previously described. [60] Specifically, 107 cells were washed in medium and treated with inhibitor for 1 h followed by stimulation with 25 ng/ml of FLT3 ligand for 15 minutes where indicated. Cells were washed twice with ice-cold PBS and lysed in 300 µl of NP-40 lysis buffer (150 mM NaCl, 20 mM TrisHCl (pH7.4), 10% glycerol, 1% NP-40, 10 mM EDTA, 100 mM NaF) with 2 mM Na3VO4 and protease inhibitors (Complete Tablets, Roche, Mannheim, Germany) at 4°C for 30 m. Cell lysate was harvested by centrifugation at 13,000 × g for 30 m at 4°C. FLT3 S-18 antibody was incubated with 500 µg lysate overnight at 4°C followed by immunoprecipitation with protein A-agarose beads for 2 h. Beads were washed once in Tris buffered saline with 1% Tween-20 (TBST) and twice in TBS. Samples were boiled in sodium dodecyl sulfate (SDS) sample buffer prior to separating in a 8% SDS-polyacrylamide gel (SDS-PAGE). Proteins were transferred to a polyvinyl diflouride (PVDF) membrane (Millipore, Bedford, MA, USA) and detected by 4G10 phosphotyrosine antibody and anti-FLT3 S-18 antibody. FLT3 was visualized using the Odyssey Infra Red Imaging System from Li-Cor (Lincoln, NE, USA) using AlexaFluor 680 goat anti-mouse and AlexaFluor 800 goat anti-rabbit antibodies. Phosphorylation of the FLT3 downstream signaling mediators, STAT5, AKT and MAP kinase was investigated by loading 50 µg of whole cell lysate and probing with the indicated antibodies. The membrane was washed and incubated in goat anti-rabbit secondary antibody conjugated to horseradish peroxidase followed by enhanced chemiluminescence (ECL). The membrane was subsequently stripped in 200 mM glycine buffer, pH 2.5 with 0.1% Tween-20 and reprobed for STAT5, AKT and MAP kinase expression using the respective antibodies.
Engraftment in BALB/c mice
BaF3 D835Y cells were transfected with the L3GFP plasmid (a gift from Dr. Linzhao Cheng of Johns Hopkins University) containing genes for luciferase and green fluorescent protein (GFP). Cells were sorted for GFP and CD135 expression on a FACS AriaII cytometer using FACSDiva software. For engraftment, 2 ×106 cells were injected via tail vein into BALB/c mice (Jackson Laboratories, Bar Harbor, ME, USA) (day 0). Starting five days later, mice were treated with either 20 mg/kg of lestaurtinib twice daily by subcutaneous injection as previously described or with the lestaurtinib vehicle to serve as a control or 10 mg/kg sorafenib suspended in 30%(w/v) Cremophor EL, 30% (w/v) PEG 400, 10% ethanol, and 10% glucose (all Sigma-Aldrich), once daily by oral gavage. [36, 37] These drug doses have previously been demonstrated to be effective against FLT3/ITD in mice. Mice were imaged by intraperitoneal injection of luciferin (3 mg) and visualizing on an IVIS Spectrum imager (Caliper LifeSciences, Hopkinton, MA, USA) using Living Image software for analysis on day 5 following inoculation to monitor engraftment and on day 9 to assess drug effect. All animal procedures were conducted in accordance with the policy of the Johns Hopkins University School of Medicine Animal Care and Use Committee.
Acknowledgments
We are grateful to Dr. Linzhao Cheng (Johns Hopkins University) for providing us with the L3GFP vector used to visualize FLT3/ITD engraftment and to members of the lab for numerous thoughtful discussions. This work was supported by grants from the NCI (CA006973 and CA90668), Alex’s Lemonade Stand, and Giant Food Pediatric Cancer Research Fund. D.S. is also supported by the Kyle Haydock Professorship.
Conflicts of Interest
The authors declare no competing financial interests.
Authorship
B.N. and A.B.W. designed experiments, performed research, analyzed data and wrote the manuscript; H.M. revised the manuscript; L.L. performed research; P.B. analyzed data; M.L. analyzed data; D.S. designed experiments, supervised the project, analyzed data and wrote the manuscript.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Small D, Levenstein M, Kim E, Carow C, Amin S, Rockwell P, Witte L, Borruw C, Ratajczak MZ, Gewirtz AM, Civin C. STK-1, the human homolog of Flk-2/Flt-3, is selectively expressed in CD34+ human bone marrow cells and is involved in the proliferation of early progenitor/stem cells. Proc Natl Acad Sci U S A 1994;91: 459-63.
2. Rosnet O, Schiff C, Pebusque MJ, Marchetto S, Tonnelle C, Toiron Y, Birg F. Birnbaum D, Human FLT3/FLK2 gene: cDNA cloning and expression in hematopoietic cells. Blood 1993;82: 1110-9.
3. Lavagna-Sevenier C, Marchetto S, Birnbaum D, Rosnet O. FLT3 signaling in hematopoietic cells involves CBL, SHC and an unknown P115 as prominent tyrosine-phosphorylated substrates. Leukemia 1998;12: 301-10.
4. Zhang S, Broxmeyer HE. Flt3 ligand induces tyrosine phosphorylation of gab1 and gab2 and their association with shp-2, grb2, and PI3 kinase. Biochem Biophys Res Commun 2000;277: 195-9.
5. Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, Sonoda Y, Fujimoto T, Misawa S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996;10: 1911-8.
6. Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, Asou N, Kuriyama K, Yagasaki F, Shimazaki C, Akiyama H, Saito K, Nishimura M, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 2001;97: 2434-9.
7. Stirewalt DL, Meshinchi S, Kussick SJ, Sheets KM, Pogosova-Agadjanyan E, Willman CL, Radich JP. Novel FLT3 point mutations within exon 14 found in patients with acute myeloid leukaemia. Br J Haematol 2004;124: 481-4.
8. Reindl C, Bagrintseva K, Vempati S, Schnittger S, Ellwart JW, Wenig K, Hopfner KP, Hiddemann W, Spiekermann K. Point mutations in the juxtamembrane domain of FLT3 define a new class of activating mutations in AML. Blood 2006;107: 3700-7.
9. Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F, Lippke J, Saxena K. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell 2004;13: 169-78.
10. Tse KF, Mukherjee G, Small D. Constitutive activation of FLT3 stimulates multiple intracellular signal transducers and results in transformation. Leukemia 2000;14: 1766-76.
11. Nakao M, Janssen JW, Erz D, Seriu T, Bartram CR. Tandem duplication of the FLT3 gene in acute lymphoblastic leukemia: a marker for the monitoring of minimal residual disease. Leukemia 2000;14: 522-4.
12. Xu F, Taki T, Yang HW, Hanada R, Hongo T, Ohnishi H, Kobayashi M, Bessho F, Yanagisawa M, Hayashi Y. Tandem duplication of the FLT3 gene is found in acute lymphoblastic leukaemia as well as acute myeloid leukaemia but not in myelodysplastic syndrome or juvenile chronic myelogenous leukaemia in children. Br J Haematol 1999;105: 155-62.
13. Yokota S, Kiyoi H, Nakao M, Iwai T, Misawa S, Okuda T, Sonoda Y, Abe T, Kahsima K, Matsuo Y, Naoe T. Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines. Leukemia 1997;11: 1605-9.
14. Brown P, Hunger SP, Smith FO, Carroll WL, Reaman GH. Novel targeted drug therapies for the treatment of childhood acute leukemia. Expert Rev Hematol 2009;2: 145.
15. Carroll WL, Raetz EA. Clinical and laboratory biology of childhood acute lymphoblastic leukemia. J Pediatr 2012;160: 10-8.
16. Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U, Wermke M, Bornhäuser M, Ritter M, Neubauer A, Ehninger G, Illmer T. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002;99: 4326-35.
17. Abu-Duhier FM, Goodeve AC, Wilson GA, Care RS, Peake IR, Reilly JT. Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br J Haematol 2001;113: 983-8.
18. Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell 2002;109: 275-82.
19. Hubbard SR. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. Embo J 1997;16: 5572-81.
20. Mol CD, Lim KB, Sridhar V, Zou H, Chien EY, Sang BC, Nowakowski J, Kassel DB, Cronin CN, McRee DE. Structure of a c-kit product complex reveals the basis for kinase transactivation. J Biol Chem 2003;278: 31461-4.
21. Grundler R, Miething C, Thiede C, Peschel C, Duyster J. FLT3-ITD and tyrosine kinase domain mutants induce 2 distinct phenotypes in a murine bone marrow transplantation model. Blood 2005;105: 4792-9.
22. Choudhary C, Schwäble J, Brandts C, Tickenbrock L, Sargin B, Kindler T, Fischer T, Berdel WE, Müller-Tidow C, Serve H. AML-associated Flt3 kinase domain mutations show signal transduction differences compared with Flt3 ITD mutations. Blood 2005;106: 265-73.
23. Grundler R, Thiede C, Miething C, Steudel C, Peschel C, Duyster J. Sensitivity toward tyrosine kinase inhibitors varies between different activating mutations of the FLT3 receptor. Blood 2003;102: 646-51.
24. Clark JJ, Cools J, Curley DP, Yu JC, Lokker NA, Giese NA, Gilliland DG. Variable sensitivity of FLT3 activation loop mutations to the small molecule tyrosine kinase inhibitor MLN518. Blood 2004;104: 2867-72.
25. Whitman SP, Ruppert AS, Radmacher MD, Mrózek K, Paschka P, Langer C, Baldus CD, Wen J, Racke F, Powell BL, Kolitz JE, Larson RA, Caligiuri MA et al. FLT3 D835/I836 mutations are associated with poor disease-free survival and a distinct gene-expression signature among younger adults with de novo cytogenetically normal acute myeloid leukemia lacking FLT3 internal tandem duplications. Blood 2008;111: 1552-9.
26. Fröhling S, Schlenk RF, Breitruck J, Benner A, Kreitmeier S, Tobis K, Döhner H, Döhner K; AML Study Group Ulm. Acute myeloid leukemia. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood 2002;100: 4372-80.
27. Armstrong SA, Mabon ME, Silverman LB, Li A, Gribben JG, Fox EA, Sallan SE, Korsmeyer SJ. FLT3 mutations in childhood acute lymphoblastic leukemia. Blood 2004;103: 3544-6.
28. Spiekermann K, Bagrintseva K, Schoch C, Haferlach T, Hiddemann W, Schnittger S. A new and recurrent activating length mutation in exon 20 of the FLT3 gene in acute myeloid leukemia. Blood 2002;100: 3423-5.
29. Jiang J, Paez JG, Lee JC, Bo R, Stone RM, DeAngelo DJ, Galinsky I, Wolpin BM, Jonasova A, Herman P, Fox EA, Boggon TJ, Eck MJ, et al. Identifying and characterizing a novel activating mutation of the FLT3 tyrosine kinase in AML. Blood 2004;104: 1855-8.
30. Kindler T, Breitenbuecher F, Kasper S, Estey E, Giles F, Feldman E, Ehninger G, Schiller G, Klimek V, Nimer SD, Gratwohl A, Choudhary CR, Mueller-Tidow C, et al. Identification of a novel activating mutation (Y842C) within the activation loop of FLT3 in patients with acute myeloid leukemia (AML). Blood 2005;105: 335-40.
31. Borthakur G, Kantarjian H, Ravandi F, Zhang W, Konopleva M, Wright JJ, Faderl S, Verstovsek S, Mathews S, Andreeff M, Cortes JE. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica 2010;96: 62-8.
32. Cortes JE, Pearl A, Dombret H, Kayser S, Steffen B, Rousselot P, Martinelli G, Estey E, Burnett A, Gammon G, Trone D, Leo E and Levis M. Final results of a phase 2 open-label, monotherapy efficacy and safety study of Quizartinib (AC220) in patients with FLT3-ITD positive or negative relapse/refractory Acute Myeloid Leukemia. after second-line chemotherapy or hematopoetic stem cell transplantation. Blood (ASH) 2012.
33. Fischer T, Stone RM, Deangelo DJ, Galinsky I, Estey E, Lanza C, Fox E, Ehninger G, Feldman EJ, Schiller GJ, Klimek VM, Nimer SD, Gilliland DG, et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol 2010;28: 4339-45.
34. Zimmerman EI, Turner DC, Buaboonnam J, Hu S, Orwick S, Roberts MS, Janke LJ, Ramachandran A, Stewart CF, Inaba H, Baker S. Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood;122: 3607-15.
35. Shah NP, Talpaz M, Deininger MW, Mauro MJ, Flinn IW, Bixby D, Lustgarten S, Gozgit JM, Clackson T, Turner CD, Haluska FG, Kantarjian H, Cortes JE. Ponatinib in patients with refractory acute myeloid leukaemia: findings from a phase 1 study. Br J Haematol 2013;162: 548-52.
36. Ma HS, Nguyen B, Duffield AS, Li L, Galanis A, Williams AB, Brown PA, Levis MJ, Leahy DJ, Small D. FLT3 kinase inhibitor TTT-3002 overcomes both activating and drug resistance mutations in FLT3 in acute myeloid leukemia. Cancer Res 2014;74: 5206-17.
37. Ma H, Nguyen B, Li L, Greenblatt S, Williams A, Zhao M, Levis M, Rudek M, Duffield A, Small D. TTT-3002 is a novel FLT3 tyrosine kinase inhibitor with activity against FLT3-associated leukemias in vitro and in vivo. Blood 2014;123: 1525-34.
38. Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, Perl AE, Travers KJ, Wang S, Hunt JP, Zarrinkar PP, Schadt EE, Kasarskis A,et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 2012;485: 260-3.
39. Smith CC, Lasater EA, Lin KC, Wang Q, McCreery MQ, Stewart WK, Damon LE, Perl AE, Jeschke GR, Sugita M, Carroll M, Kogan SC, Kuriyan J,et al. Crenolanib is a selective type I pan-FLT3 inhibitor. Proc Natl Acad Sci U S A 2014;111: 5319-24.
40. Brown P, Levis M, Shurtleff S, Campana D, Downing J, Small D. FLT3 inhibition selectively kills childhood acute lymphoblastic leukemia cells with high levels of FLT3 expression. Blood 2005;105: 812-20.
41. Piloto O, Wright M, Brown P, Kim KT, Levis M, Small D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood 2007;109: 1643-52.
42. Kiyoi H, Naoe T, Nakano Y, Yokota S, Minami S, Miyawaki S, Asou N, Kuriyama K, Jinnai I, Shimazaki C, Akiyama H, Saito K, Oh H, et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood 1999;93: 3074-80.
43. Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, Walker H, Wheatley K, Bowen DT, Burnett AK, Goldstone AH, Linch DC. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood 2001;98: 1752-9.
44. Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, Löffler H, Sauerland CM, Serve H, Büchner T, Haferlach T, Hiddemann W. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 2002;100: 59-66.
45. Levis M, Pham R, Smith BD, Small D. In Vitro studies of a FLT3 inhibitor combined with chemotherapy: sequence of administration is important to achieve synergistic cytotoxic effects. Blood 2004;104: 1145-50.
46. Stone RM, Mandrekar S, Sanford BL, Geyer S, Bloomfield CD, Dohner K, Thiede C, Marcucci G, Lo-Coco F, Klisovic RB, Wei A, Sierra J, Sanz MA et al. The Multi-Kinase Inhibitor Midostaurin (M) Prolongs Survival Compared with Placebo (P) in Combination with Daunorubicin (D)/Cytarabine (C) Induction (ind), High-Dose C consolidation (consol), and As Maintenance (maint) Therapy in Newly Diagnosed Acute Myeloid Leukemia (AML) Patients (pts) Age 18-60 with FLT3 Mutations (muts): An International Prospective Randomized (rand) P-Controlled Double-Blind Trial (CALGB 10603/RATIFY [Alliance]) ASH 2015.
47. el-Shami K, Stone RM, Smith BD. FLT3 inhibitors in acute myeloid leukemia. Expert Rev Hematol 2008;1: 153-60.
48. Pemmaraju N, Kantarjian H, Ravandi F, Cortes J. FLT3 inhibitors in the treatment of acute myeloid leukemia: the start of an era? Cancer;117: 3293-304.
49. O’Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, Wong LM, Hong W, Lee LB, Town A, Smolich BD, Manning WC, Murray LJ,et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 2003;101: 3597-605.
50. Lee SH, Paietta E, Racevskis J, Wiernik PH. Complete resolution of leukemia cutis with sorafenib in an acute myeloid leukemia patient with FLT3-ITD mutation. Am J Hematol 2009;84: 701-2.
51. Nepomuceno R, Gunawardane R, Rooks A, Armstrong R. Detection of Phosphorylated and Total Flt3 and STAT5 in Whole Blood: Modulation by AC220 From Phase I and II Trials in AML Blood (ASH) 2011.
52. Perl A, Jeschke G, Smith C, Mangan J, M. Luger S, Carroll M. Phospho-Specific Flow Cytometry of Fixed Whole Blood Demonstrates In Vivo FLT3 Inhibition in Circulating Leukemic Blasts During AC220 Therapy and Accurately Detects the Development of Therapeutic Resistance. Blood (ASH) 2011.
53. Cortes JE, Perl A, Smith C, Kovascovics T, Dombret H, Dohner H, Steffen B, Pigneux A, Rousselot P, Krauter J, Martenelli G, Estey E, Burnett A, et al. A Phase II Open-Label, AC220 Monotherapy Efficacy Study In Patients with Refractory/Relapsed Flt3-Itd Positive Acute Myeloid Leukemia : Updated Interim Results. Blood (ASH) 2011.
54. Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol 2006;2: 358-64.
55. Traxler P, Furet P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol Ther 1999;82: 195-206.
56. Johnson LN. Protein kinase inhibitors: contributions from structure to clinical compounds. Q Rev Biophys 2009;42: 1-40.
57. Torrent M, Rickert K, Pan BS, Sepp-Lorenzino L. Analysis of the activating mutations within the activation loop of leukemia targets Flt-3 and c-Kit based on protein homology modeling. J Mol Graph Model 2004;23: 153-65.
58. Weisberg E, Roesel J, Furet P, Bold G, Imbach P, Flörsheimer A, Caravatti G, Jiang J, Manley P, Ray A, Griffin JD. Antileukemic Effects of Novel First- and Second-Generation FLT3 Inhibitors: Structure-Affinity Comparison. Genes Cancer 2010;1: 1021-32. doi: 10.1177/1947601910396505.
59. Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R; Cancer Genome Project. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004;116: 855-67.
60. Tse KF, Allebach J, Levis M, Smith BD, Bohmer FD, Small D. Inhibition of the transforming activity of FLT3 internal tandem duplication mutants from AML patients by a tyrosine kinase inhibitor. Leukemia 2002;16: 2027-36.