
INTRODUCTION
With the explosive growth of biological sequences in the post-genomic age, we are facing a lot of binary classification problems. For DNA/RNA sequences, these problems are about how to identify the recombination spots [1–4], nucleosome positioning [5–9], promoters [10], microRNA precursors [11–13], enhancers [14, 15], translation initiation sites [16, 17], various PTRM (postpost-replication modification) sites in DNA [18] and PTCM (post-transcriptiom modification) sites in RNA [19, 20], RNA pseudouridine sites [21], DNA origin of replication [22, 23], adenosine to inosine editing sites in RNA [24], and many more other topics as mentioned in a recent review article [25].
For protein/peptide sequences, they are about how to identify various PTM (Posttranslational Modification) sites [26–42], anticancer peptides [43, 44], interactions between drugs and target proteins [45–49], PPI (protein-protein interaction) [50]. PPBS (proire-protein binding sites [51, 52], as well as a long list of references cited in a recent comprehensive review [53].
It is quite laborious even if using computational approches to deal with these problems since the development of each computational predictor needs to undergo the following five steps [54]: (1) benchmark dataset preparation, (2) optimise sample formulation, (3) optimize operation engine, (4) conduct cross-validations, and (5) establish a web-server. Each of the five procedures is time-consuming and tedious, particularly in how to select the optimal parameters [55–60] for the samples concerned and for the operation engine adopted.
To speed up such processes, we are to propose a Python package called Pse-Analysis, which is based on the framework of LIBSVM [61] and which can automatically generate the predictor desired by users. The users only need to input their benchmark dataset and the query biological sequences, followed by getting their desired results from the output of the Pse-Analysis system. All the tedious things in the aforementioned steps (2)–(5) can be totally skipped and leave them to be fulfilled by the computer.
RESULTS AND DISCUSSION
A powerful Python package, called Pse-Analysis, has been developed, and its web-server established at http://bioinformatics.hitsz.edu.cn/Pse-Analysis/. It is formed by two important parts: one is “train.py”, and the other is “predict.py” (Figure 1).
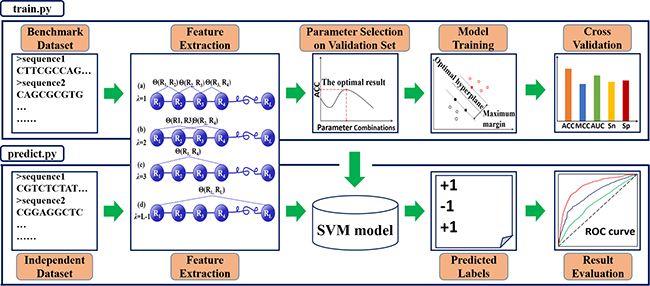
Figure 1: The flowchart of Pse-Analysis Python package. The “train.py” script is for training the predictive model based on the benchmark dataset submitted by the user. It contains four procedures; i.e., feature extraction, parameter selection, model training, and cross validation. The “predict.py” is for using the trained model to predict the query samples and evaluate their prediction quality by a set of widely used metrics Acc, MCC, Sn, Sp [25], and AUC [68].
The “train.py” is designed for training a Support Vector Machine (SVM) model. It includes four steps: (1) feature extraction, (2) parameter selection, (3) model training, and (4) cross validation.
The “predict.py” is to generate the output. Note: the meaning of the “output” here is not limited in the predicted results for the original query biological sequence data submitted along with the benchmark dataset, but also include an optimal predictor. Users can directly apply it on various relevant problems, substantially saving a lot of time to repeat tedious for developing an effective predictor.
For instance, it is a very important task to effectively predict nucleosome positioning in genomes. To deal with the problem, Guo et al. [7] had praiseworthily developed a predictor called iNuc-PseKNC by going thru all the five procedures described in the Introduction section. Now, with the Pse-Analysis package, what we need to do is just to input the benchmark dataset used by Guo et al. [7] into the package, and Pse-Analysis will automatically do all the remaining jobs: optimising sample formulation; optimising operation engine; conducting cross-validations; and forming a web-server that is fully equivalent to the iNuc-PseKNC of [7].
The computational speed in optimizing many different parameters is a bottleneck for the efficiency of the Pse-Analysis platform. In this regard, the multiprocessing technique has been applied to significantly speed up the computational processes. It has been shown when dealing with the above case that the computing time for the parameter optimization process can be reduced by 6 folds when using 10 cores instead of a single core, as shown in Figure 2.
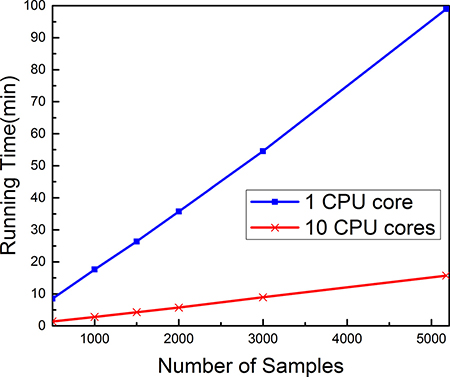
Figure 2: The computational cost of Pse-Analysis can be significantly reduced by using multiprocessing technique. The blue curve reflects the computational time time for the parameter optimization process when using Pse-Analysis of one CPU core to process the five subsets for nucleosome positioning prediction of Caenorhabditis elegans [7], while the red curve reflects the corresponding computational time when using Pse-Analysis of ten CPU cores to do the same.
As pointed out in a comprehensive review paper [25], the general form of PseKNC (pseudo K-tuple nucleotide composition) can cover all the existing feature vectors for DNA/RNA sequences. And the general form of PseACC (pseudo amino acid composition) can cover all the existing feature vectors for protein/peptide sequences [54, 56]. Particularly, the very powerful web-server Pse-in-One [60] developed recently not only can cover all the existing feature vectors for DNA/RNA and protein/peptide sequences, but also can cover those defined by the users themselves. Accordingly, the pseudo components in the Pse-Analysis package have virtually covered all the feature vectors for DNA/RNA or protein/peptide sequences.
MATERIALS AND METHODS
Feature extraction
In the Pse-Analysis, various state-of-the-art algorithms are employed, including pseudo k-tuple nucleotide composition (PseKNC) [25, 57–59, 62] and pseudo amino acid composition (PseAAC) [56, 60, 63–67] for extracting the features of DNA, RNA, and protein sequences, respectively. The details of these algorithms have been clearly elaborated in the aforementioned papers, and hence there is no need to repeat here.
Parameter selection
The aforementioned algorithms contain some uncertain parameters, such as k, λ, and w. All these parameters are automatically determined by train.py in processing the benchmark dataset submitted by users. The concrete process is to optimize the following five commonly used success scores: (1) accuracy (Acc), (2) Mathew’s Correlation Coefficient (MCC), (3) sensitivity (Sn), (4) specificity (Sp), and (5) area under ROC curve (AUC). As for their rigorous definitions and intuitive formulations, see [21, 23, 24, 35, 38, 60]. Furthermore, the corresponding ROC (receiver operating characteristic) [68] curve is also provided and saved in a PNG file. Finally, by optimizing these scores with respect to all possible parameters, the corresponding best model will be generated.
Model training
The model is trained with LIBSVM [61] using the RBF kernel. The trained model thus obtained and all its optimized parameters are saved in a separate file, which will be used as the input for “predict.py”.
Cross validation
Built-in the Pse-Analysis package is also a set of validation operators, which can be used to automatically validate the model from sub-sampling (or K-fold cross-validation) test, and jackknife (or leave-one-out) test, the three most used cross-validation approaches [69].
Manual of Pse-Analysis
To maximize users’ convenience, the manual of how to use Pse-Analysis is provided, which can be directly downloaded at http://bioinformatics.hitsz.edu.cn/Pse-Analysis/static/download/Pse-Analysis_manual.pdf.
CONCLUSIONS
Now we are living in a century or era to pursue the goal to minimize various tedious things and leave them to be done by robots or computers, such as in developing autonomous cars or self-driving cars. The present study represents one step forward to such a goal in genome and proteome analyses. It has not escaped our notice that the idea and approach can also be used to many other areas so as to substantially speed up their development accordingly.
ACKNOWLEDGMENTS AND FUNDING
The authors wish to thank the five anonymous reviewers, whose constructive comments are very helpful for strengthening the presentation of this paper. This work was supported by National Natural Science Foundation of China (No. 61672184, 61300112, and 61573118), the Natural Science Foundation of Guangdong Province (2014A030313695), Guangdong Natural Science Funds for Distinguished Young Scholars (2016A030306008), and Scientific Research Foundation in Shenzhen (Grant No. JCYJ20150626110425228).
CONFLICTS OF INTEREST
None.
REFERENCES
1. Chen W, Feng PM, Lin H, Chou KC. iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition Nucleic Acids Res. 2013; 41:e68.
2. Qiu WR, Xiao X. iRSpot-TNCPseAAC: Identify recombination spots with trinucleotide composition and pseudo amino acid components. Int J Mol Sci. 2014; 15:1746–1766.
3. Kabir M, Hayat M. iRSpot-GAEnsC: identifing recombination spots via ensemble classifier and extending the concept of Chou’s PseAAC to formulate DNA samples. Molecular Genetics and Genomics. 2016; 291:285–296.
4. Liu B, Wang S, Long R. iRSpot-EL: identify recombination spots with an ensemble learning approach. Bioinformatics. 2016; doi: 10.1093/bioinformatics/btw539.
5. Yi XF, He ZS, Kong XY. Nucleosome positioning based on the sequence word composition. Protein & Peptide Letters. 2012; 19:79–90.
6. Chen W, Lin H, Feng PM, Ding C. iNuc-PhysChem: A Sequence-Based Predictor for Identifying Nucleosomes via Physicochemical Properties. PLoS ONE. 2012; 7:e47843.
7. Guo SH, Deng EZ, Xu LQ, Ding H, Lin H. iNuc-PseKNC: a sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics. 2014; 30:1522–1529.
8. Tahir M, Hayat M. iNuc-STNC: a sequence-based predictor for identification of nucleosome positioning in genomes by extending the concept of SAAC and Chou’s PseAAC. Mol Biosyst. 2016; 12:2587–2593.
9. Chen W, Feng P, Ding H. Using deformation energy to analyze nucleosome positioning in genomes. Genomics. 2016; 107:69–75.
10. Lin H, Deng EZ, Ding H, Chen W, Chou KC. iPro54-PseKNC: a sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res. 2014; 42:12961–12972.
11. Liu B, Fang L, Wang S, Wang X. Identification of microRNA precursor with the degenerate K-tuple or Kmer strategy. Journal of Theoretical Biology. 2015; 385:153–159.
12. Liu B, Fang L, Liu F, Wang X. Identification of real microRNA precursors with a pseudo structure status composition approach. PLoS ONE. 2015; 10:e0121501.
13. Liu B, Fang L, Liu F, Wang X. iMiRNA-PseDPC: microRNA precursor identification with a pseudo distance-pair composition approach. J Biomol Struct Dyn. 2016; 34:223–235.
14. Lee D, Karchin R, Beer MA. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res. 2011; 21:2167–2180.
15. Liu B, Fang L, Long R, Lan X. iEnhancer-2L: a two-layer predictor for identifying enhancers and their strength by pseudo k-tuple nucleotide composition Bioinformatics. 2016; 32:362–369.
16. Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010; 11:113–127.
17. Chen W, Feng PM, Deng EZ. iTIS-PseTNC: a sequence-based predictor for identifying translation initiation site in human genes using pseudo trinucleotide composition. Anal Biochem. 2014; 462:76–83.
18. Liu Z, Xiao X, Qiu WR. iDNA-Methyl: Identifying DNA methylation sites via pseudo trinucleotide composition. Analytical Biochemistry (also, Data in Brief, 2015, 4:87–89). 2015; 474:69–77.
19. Chen W, Feng P, Ding H, Lin H. iRNA-Methyl: Identifying N6-methyladenosine sites using pseudo nucleotide composition. Analytical Biochemistry (also, Data in Brief, 2015, 5: 376–378). 2015; 490:26–33.
20. Liu Z, Xiao X, Yu DJ, Jia J, Qiu WR. pRNAm-PC: Predicting N-methyladenosine sites in RNA sequences via physical-chemical properties. Anal Biochem. 2016; 497:60–67.
21. Chen W, Tang H, Ye J, Lin H. iRNA-PseU: Identifying RNA pseudouridine sites Molecular Therapy-Nucleic Acids 2016; 5:e332.
22. Xiao X, Ye HX, Liu Z, Jia JH. iROS-gPseKNC: predicting replication origin sites in DNA by incorporating dinucleotide position-specific propensity into general pseudo nucleotide composition. Oncotarget. 2016; 7:34180–34189. doi: 10.18632/oncotarget.9057.
23. Zhang CJ, Tang H, Li WC, Lin H. iOri-Human: identify human origin of replication by incorporating dinucleotide physicochemical properties into pseudo nucleotide composition. Oncotarget. 2016; 7:69783–69793. doi: 10.18632/oncotarget.11975.
24. Chen W, Feng P, Yang H, Ding H. iRNA-AI: identifying the adenosine to inosine editing sites in RNA sequences. Oncotarget. 2017; 8:4208–4217. doi: 10.18632/oncotarget.13758.
25. Chen W, Lin H, Chou KC. Pseudo nucleotide composition or PseKNC: an effective formulation for analyzing genomic sequences. Mol BioSyst. 2015; 11:2620–2634.
26. Xu Y, Ding J, Wu LY. iSNO-PseAAC: Predict cysteine S-nitrosylation sites in proteins by incorporating position specific amino acid propensity into pseudo amino acid composition PLoS ONE. 2013; 8:e55844.
27. Xu Y, Shao XJ, Wu LY, Deng NY. iSNO-AAPair: incorporating amino acid pairwise coupling into PseAAC for predicting cysteine S-nitrosylation sites in proteins. PeerJ. 2013; 1:e171.
28. Qiu WR, Xiao X, Lin WZ. iMethyl-PseAAC: Identification of Protein Methylation Sites via a Pseudo Amino Acid Composition Approach. Biomed Res Int. 2014; 2014:947416.
29. Xu Y, Wen X, Shao XJ. iHyd-PseAAC: Predicting hydroxyproline and hydroxylysine in proteins by incorporating dipeptide position-specific propensity into pseudo amino acid composition. Int J Mol Sci. 2014; 15:7594–7610.
30. Jia C, Lin X, Wang Z. Prediction of Protein S-Nitrosylation Sites Based on Adapted Normal Distribution Bi-Profile Bayes and Chou’s Pseudo Amino Acid Composition. Int J Mol Sci. 2014; 15:10410–10423.
31. Zhang J, Zhao X, Sun P, Ma Z. PSNO: Predicting Cysteine S-Nitrosylation Sites by Incorporating Various Sequence-Derived Features into the General Form of Chou’s PseAAC. Int J Mol Sci. 2014; 15:11204–11219.
32. Xu Y, Wen X, Wen LS, Wu LY. iNitro-Tyr: Prediction of nitrotyrosine sites in proteins with general pseudo amino acid composition. PLoS ONE. 2014; 9:e105018.
33. Qiu WR, Xiao X, Lin WZ. iUbiq-Lys: Prediction of lysine ubiquitination sites in proteins by extracting sequence evolution information via a grey system model. Journal of Biomolecular Structure and Dynamics. 2015; 33:1731–1742.
34. Jia J, Liu Z, Xiao X, Liu B. iSuc-PseOpt: Identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset. Anal Biochem. 2016; 497:48–56.
35. Jia J, Liu Z, Xiao X. pSuc-Lys: Predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach. J Theor Biol. 2016; 394:223–230.
36. Qiu WR, Sun BQ, Xiao X. iPhos-PseEvo: Identifying human phosphorylated proteins by incorporating evolutionary information into general PseAAC via grey system theory. Molecular Informatics. 2016; doi: 10.1002/minf.201600010.
37. Jia J, Liu Z, Xiao X. iCar-PseCp: identify carbonylation sites in proteins by Monto Carlo sampling and incorporating sequence coupled effects into general PseAAC. Oncotarget. 2016; 7:34558–34570. doi: 10.18632/oncotarget.9148.
38. Jia J, Zhang L, Liu Z, Xiao X. pSumo-CD: Predicting sumoylation sites in proteins with covariance discriminant algorithm by incorporating sequence-coupled effects into general PseAAC. Bioinformatics. 2016; 32:3133–3141.
39. Qiu WR, Sun BQ, Xiao X, Xu ZC. iHyd-PseCp: Identify hydroxyproline and hydroxylysine in proteins by incorporating sequence-coupled effects into general PseAAC. Oncotarget. 2016; 7:44310–44321. doi: 10.18632/oncotarget.10027.
40. Qiu WR, Sun BQ, Xiao X. iPTM-mLys: identifying multiple lysine PTM sites and their different types. Bioinformatics. 2016; 32:3116–3123.
41. Qiu WR, Xiao X, Xu ZH. iPhos-PseEn: identifying phosphorylation sites in proteins by fusing different pseudo components into an ensemble classifier. Oncotarget. 2016; 7:51270–51283. doi: 10.18632/oncotarget.9987.
42. Xu Y. Recent progress in predicting posttranslational modification sites in proteins. Curr Top Med Chem. 2016; 16:591–603.
43. Hajisharifi Z, Piryaiee M, Mohammad Beigi M, Behbahani M, Mohabatkar H. Predicting anticancer peptides with Chou’s pseudo amino acid composition and investigating their mutagenicity via Ames test. J Theor Biol. 2014; 341:34–40.
44. Chen W, Ding H, Feng P, Lin H. iACP: a sequence-based tool for identifying anticancer peptides. Oncotarget. 2016; 7:16895–16909. doi: 10.18632/oncotarget.7815.
45. Min JL, Xiao X. iEzy-Drug: A web server for identifying the interaction between enzymes and drugs in cellular networking. BioMed Research International. 2013; 2013:701317.
46. Xiao X, Min JL, Wang P. iGPCR-Drug: A web server for predicting interaction between GPCRs and drugs in cellular networking. PLoS ONE. 2013; 8:e72234.
47. Xiao X, Min JL, Wang P. iCDI-PseFpt: Identify the channel-drug interaction in cellular networking with PseAAC and molecular fingerprints. J Theor Biol. 2013; 337C:71–79.
48. Fan YN, Xiao X, Min JL. iNR-Drug: Predicting the interaction of drugs with nuclear receptors in cellular networking. Intenational Journal of Molecular Sciences. 2014; 15:4915–4937.
49. Xiao X, Min JL, Lin WZ, Liu Z. iDrug-Target: predicting the interactions between drug compounds and target proteins in cellular networking via the benchmark dataset optimization approach. J Biomol Struct Dyn. 2015; 33:2221–2233.
50. Jia J, Liu Z, Xiao X. iPPI-Esml: an ensemble classifier for identifying the interactions of proteins by incorporating their physicochemical properties and wavelet transforms into PseAAC. J Theor Biol. 2015; 377:47–56.
51. Jia J, Liu Z, Xiao X, Liu B. Identification of protein-protein binding sites by incorporating the physicochemical properties and stationary wavelet transforms into pseudo amino acid composition (iPPBS-PseAAC). J Biomol Struct Dyn. 2016; 34:1946–1961.
52. Jia J, Liu Z, Xiao X. iPPBS-Opt: A Sequence-Based Ensemble Classifier for Identifying Protein-Protein Binding Sites by Optimizing Imbalanced Training Datasets. Molecules. 2016; 21:95.
53. Chou KC. Impacts of bioinformatics to medicinal chemistry. Medicinal Chemistry. 2015; 11:218–234.
54. Chou KC. Some remarks on protein attribute prediction and pseudo amino acid composition (50th Anniversary Year Review). J Theor Biol. 2011; 273:236–247.
55. Chou KC, Shen HB. Review: Recent progresses in protein subcellular location prediction. Anal Biochem. 2007; 370:1–16.
56. Chou KC. Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology. Current Proteomics. 2009; 6:262–274.
57. Chen W, Lei TY, Jin DC. PseKNC: a flexible web-server for generating pseudo K-tuple nucleotide composition. Anal Biochem. 2014; 456:53–60.
58. Liu B, Liu F, Fang L, Wang X. repDNA: a Python package to generate various modes of feature vectors for DNA sequences by incorporating user-defined physicochemical properties and sequence-order effects. Bioinformatics. 2015; 31:1307–1309.
59. Liu B, Liu F, Fang L. repRNA: a web server for generating various feature vectors of RNA sequences. Molecular Genetics and Genomics. 2016; 291:473–481.
60. Liu B, Liu F, Wang X, Chen J, Fang L, Chou KC. Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences Nucleic Acids Res. 2015; 43:W65–W71.
61. Chang CC, Lin CJ. LIBSVM: A Library for Support Vector Machines. ACM Trans Intell Syst Technol. 2011; 2:1–27.
62. Chen W, Zhang X, Brooker J, Lin H. PseKNC-General: a cross-platform package for generating various modes of pseudo nucleotide compositions. Bioinformatics. 2015; 31:119–120.
63. Chou KC. Prediction of protein cellular attributes using pseudo amino acid composition. PROTEINS: Structure, Function, and Genetics (Erratum: ibid, 2001, Vol44, 60). 2001; 43:246–255.
64. Chou KC. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics. 2005; 21:10–19.
65. Shen HB. PseAAC: a flexible web-server for generating various kinds of protein pseudo amino acid composition. Anal Biochem. 2008; 373:386–388.
66. Cao DS, Xu QS, Liang YZ. propy: a tool to generate various modes of Chou’s PseAAC. Bioinformatics. 2013; 29:960–962.
67. Du P, Gu S, Jiao Y. PseAAC-General: Fast building various modes of general form of Chou’s pseudo amino acid composition for large-scale protein datasets. International Journal of Molecular Sciences. 2014; 15:3495–3506.
68. Fawcett JA. An Introduction to ROC Analysis. Pattern Recognition Letters. 2005; 27:861–874.
69. Chou KC, Zhang CT. Review: Prediction of protein structural classes. Crit Rev Biochem Mol Biol. 1995; 30:275–349.