
INTRODUCTION
Therapy-related myeloid neoplasms include myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) occurring as a late effect of chemo- and/or radiotherapy for a primary malignancy or an autoimmune disease and have been included in the WHO classification of acute leukemia [1–3]. Although the majority of t-MN are of myeloid lineage, therapy-related acute lymphoblastic leukemia (t-ALL) has been also reported [4–5].
t-MN are characterized by high incidence of complex karyotypes, frequent abnormalities of chromosome 7 and/or 5 (monosomies and/or deletions) and TP53 mutations in 10-30% of patients [1, 6–8]. On the other hand, somatic mutations recently identified in patients with de novo AML and MDS, such as those of epigenetic regulators, spliceosome machinery and SETBP1, are rare, with the exception of SRSF2 [9–11]. t-ALL are frequently associated with exposure to alkylating agents and/or topoisomerase II inhibitors [5–12]. Balanced translocations involving the gene KMT2A at 11q23, with a high prevalence of the t(4;11)(q21;q23) translocation have been commonly reported [5–12].
Cytotoxic therapy may induce chromosomal alterations and genetic mutations in hematopoietic progenitors, leading to a high incidence of complex karyotypes. TP53 mutations in the founding clone are major contributors to genetic instability and have also been associated to the occurrence of cytogenetic abnormalities and poor response to chemotherapy that are typical of t-MN. A different scenario has been recently depicted by Wong et al, who showed that the number of somatic single nucleotide variants (SNV) in t-MN was similar to that of de novo AML and MDS, suggesting that cytotoxic therapy does not induce genome-wide DNA damage [13]. On the other hand, using a modified next generation sequencing (NGS) protocol that allows the detection of very low TP53 variant allele frequencies, the authors of this study found the same TP53 mutation identified at the time of t-MN diagnosis in bone marrow (BM) samples collected prior to any chemotherapy. In this line, somatic mutations in genes associated to hematological malignancies have been found in the peripheral blood of elderly otherwise healthy individuals, suggesting that mutations can naturally occur in hematopoietic cells [14–15].
The incidence of t-MN has been increasing in the past years [16]. Although this increase can be attributed to the numerical expansion and the prolonged survival of treated patients, the identification of mechanisms explaining the underlying individual susceptibility to t-MN development could be useful to limit the risk of this complication.
The aim of our study was to investigate the role of common somatic mutations and other gene alterations during clonal evolution of 14 therapy-related leukemia patients (13 t-MN and 1 t- ALL) for whom we had collected BM samples at the time of primary diagnosis of a hematological neoplasm and/or during its follow-up. At the time of t-MN diagnosis, we studied mutations in genes belonging to epigenetic regulators (DNMT3A, IDH1, IDH2 and ASXL1), spliceosome enzymes (U2AF1, SF3B1 and SRSF2), TP53, SETBP1, NRAS and KRAS. Mutations identified in the t-MN were then tracked backwards in previous samples using high sensitivity techniques, as NGS or pyrosequencing.
RESULTS
Screening of genetic changes in therapy-related neoplasms
Our first aim was the genetic characterization of 13 t-MN and one t-ALL patient to identify molecular markers of the disease. Mutational screening of spliceosome machinery (U2AF1 S34 and R35, SF3B1 exons 13–16 and SRSF2 exon 1), epigenetic enzyme (IDH1 R132, IDH2 R140 and R172, DNMT3A R882 and ASXL1 exon 12), SETBP1 (SKI homologous domain), N-RAS (exons 2-3) and K-RAS (exons 2-3) was performed using Sanger sequencing, whereas TP53 (exons 4-9) mutations were studied by standard NGS technology. We identified 8 mutations (IDH1 R132H, ASXL1 Y591*, ASXL1 S689*, ASXL1 R693*, SRSF2 P95H, SF3B1 K700E, SETBP1 G870R and TP53 Y220C) in seven of thirteen t-MN patients (54%), whereas the t-ALL patient was KMT2A-AFF1-positive. No other recurrent somatic mutations in these genes were detected in the remaining 6 patients (Table 1).
Table 1: Patient characteristics
UPN |
Age at t-MN diagnosis (yrs) |
Gender |
Primary Malignancy |
Treatment of primary malignancy |
BM-blasts (%) |
Diagnosis |
Latency (months) |
Karyotype |
Molecular marker |
|---|---|---|---|---|---|---|---|---|---|
1 |
73 |
F |
NHL |
ProMACE-Cytabom + RT |
12 |
t-MDS |
100 |
46,XX [25] |
ASXL1 Y591* |
2 |
62 |
F |
APL |
AIDA 2000 [17] |
14 |
t-MDS |
30 |
TP53 Y220C |
|
3 |
63 |
M |
NHL |
R-CHOP, R-MICMA, autoSCT; bortezomib/lenalidomide |
4 |
t-MDS |
83 |
47,XY,+21 [12] |
ASXL1 S689* |
4 |
62 |
F |
NHL |
CHOP, MICMA, autoSCT, fludarabin |
8.5 |
t-MDS |
124 |
ASXL1 R693* |
|
5 |
30 |
M |
NHL |
CODOX-M/IVAC, |
10 |
t-MDS |
54 |
46,XY,-7 [10] |
absent |
6 |
63 |
F |
NK-AML and Breast Ca |
AML-12 [18], Carboplatin, taxotere, herceptin + RT |
10 |
t-MDS |
83 |
SF3B1 |
|
7 |
74 |
F |
NHL |
CHOP, R-FC, chlorambucil |
19 |
t-MDS |
66 |
46,XX [25] |
absent |
8 |
60 |
F |
APL |
AIDA + AutoSCT |
6 |
t-MDS |
100 |
absent |
|
9 |
81 |
M |
NHL |
Chlorambucil + RT |
13 |
t-MDS |
100 |
Not available |
IDH1 R132H |
10 |
74 |
M |
NHL |
R-FND, R-MICMA |
4.5 |
t-MDS |
74 |
46,XY [19]/44,X,-7, del(1)(p35), del(5)(q13), del(11)(q14) [6] |
absent |
11 |
37 |
F |
HL |
BEACOPP escalated |
18 |
t-MDS |
83 |
absent |
|
12 |
50 |
F |
HL |
BEACOPP escalated |
43 |
t-AML |
18 |
46,XX [25] |
absent |
13 |
43 |
M |
ALL |
GMALL 05/93 [19] |
7 |
t-MDS |
32 |
45,XY,-7 [6] |
SETBP1 G870R |
14 |
40 |
F |
APL |
AIDA 2000 [17] |
80 |
t-ALL |
18 |
46,XX, |
KMT2A/AFF1 |
Legend: NK: normal karyotype; RT: radiotherapy; ProMACE-CytaBOM: cyclophosphamide, doxorubicin, etoposide, bleomycin, vincristine, methotrexate and prednisone; AIDA: ATRA, idarubicine, mitoxantrone; R: Rituximab; CHOP: cyclophosphamide, adriblastin, vincristine, prednisone; MICMA: mitoxantrone, carboplatin, cytarabine, methylprednisolone; CODOX-M/IVAC: cyclophosphamide, doxorubicin, vincristine, methotrexate, etoposide, ifosfamide and cytarabine; FC: fludarabin, cyclophosphamide; FND: fludarabin, mitoxantrone, desametazone; AutoSCT: autologous stem cell transplantation; BEACOPP: bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone.
The apparently low frequency of TP53 mutations identified in the present patient cohort (1 in 14 patients) may not be considered representative of t-MN in general, since it is the result of the particularly restrictive patient selection, which included only patients with a previous history of hematological malignancies, and available bone marrow samples prior to t-MN development.
Validation and quantification of variants
Pyrosequencing analyses, specifically designed to quantify the mutations confirmed the data obtained from Sanger sequencing in all 8 cases. Mutations identified in epigenetic regulators, spliceosome enzymes and SETBP1 were present at a variant allele frequency (VAF) of 20 to 42% at the time of t-MN diagnosis (Figure 1), whereas the TP53 mutation was detectable at a lower frequency (6.75%, Figure 2). Interestingly, UPN9 carried two mutations (IDH1 R132H and SRSF2 P95H), at a similar VAF (38% and 35%, respectively), suggesting that the two mutations were present in the same leukemic clone. The mutations were somatically acquired in the myeloid compartment, as shown by their presence also in PB-granulocytes, whereas they were not detectable in the CD3+ T-lymphocyte population.
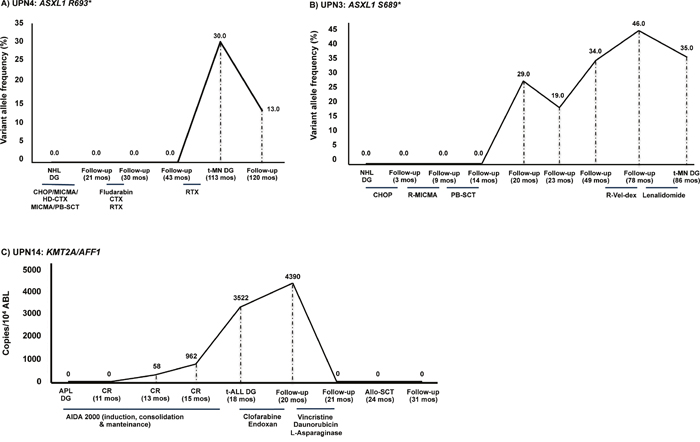
Figure 1: Genetic changes present in t-MN were not detectable at primary cancer diagnosis. A. In UPN4, the ASXL1 R693* mutation detected in the t-MN sample was undetectable at the time of NHL diagnosis (113 months prior to t-MN onset). B. In UPN3, the ASXL1 S689* mutation was undetectable at the time of primary cancer diagnosis (NHL) but was detectable for the first time during NHL follow-up (at 20 months) and increased until the time of t-MN diagnosis (at 86 months). Of note, the mutation was first detected after high-dose therapy and PB-SCT. C. Similarly, the KMT2A-AFF1 fusion identified in UPN 14 at t-ALL diagnosis was, as expected, undetectable in the primary APL diagnostic sample, and was detected at low levels by Q-RT-PCR thirteen months after achievement of complete molecular remission of APL, using the AIDA 2000 protocol [17]. Notably, a constant increase in the transcript copy number was evident from first identification (58 copies/104 ABL) to t-ALL onset (3522 copies/104 ABL). The fusion transcript became undetectable only after re-induction treatment according to the LAL0904 protocol [27] and resulted to date undetectable (54+ months) after ASCT. The variant allele frequency (VAF) is indicated. Legend. NHL DG: non-Hodgkin lymphoma diagnosis; t-MN DG: therapy-related myeloid neoplasm diagnosis; APL DG: acute promyelocitic leukemia diagnosis; CR: complete remission; t-ALL DG: therapy-related acute lymphoblastic leukemia diagnosis; Allo-SCT: allogeneic stem cell transplantation; CHOP: cyclophosphamide, adriblastin, vincristine, prednisone; MICMA: mitoxantrone, carboplatin, cytarabine, methylprednisolone; PB-SCT: peripheral blood stem cell transplantation; RTX: radiotherapy; CTX: cyclophosphamide; HD-CTX: high-dose cyclophosphamide; R-MICMA: mitoxantrone, carboplatin, cytosine arabinoside, and methylprednisolone; R-Vel-dex: lenalidomide, bortezomib, dexametasone; AIDA: ATRA, idarubicine.
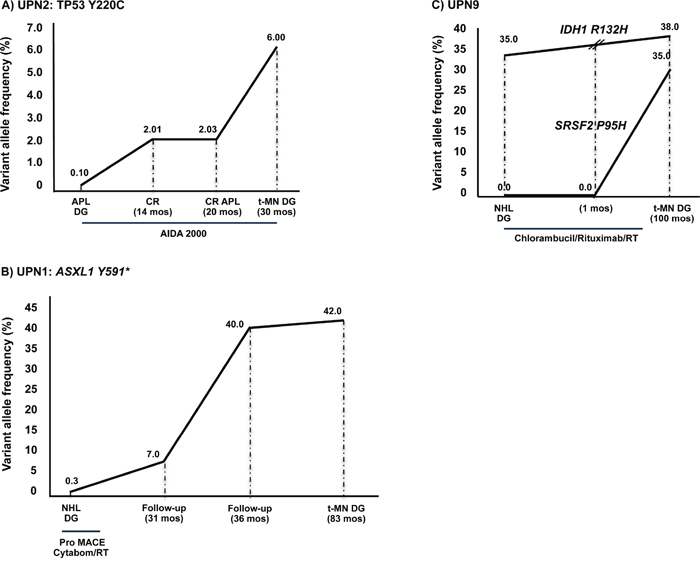
Figure 2: Somatic mutations were present prior to any cytotoxic treatment. A. A TP53 Y220C mutation was identified in UPN2 in all the available BM-MNC specimens, from APL diagnosis to complete remission, and expanded in the t-MN clone. B. Similarly, in UPN1 the ASXL1 Y591* mutation was identified in all the available BM-MNC specimens, from NHL diagnosis to t-MN. Notably, this mutation was detected at very low levels in the BM NHL specimen (0.3%), significantly increased at 31 months follow-up (7%) and reached the highest VAF at t-MN diagnosis (42%). C. In UPN9, the IDH1 R132H mutation was originally present at a high allele frequency at NHL diagnosis, 9 years before t-MN onset, when the patient was 72 years old, indicating that this mutation could have occurred as a pre-leukemic event. The SRSF2 P95H mutation was acquired later, at the time of t-MN diagnosis. Legend: APL DG: acute promyelocitic leukemia diagnosis; CR: complete remission; NHL DG: non-Hodgkin lymphoma diagnosis; t-MN DG: therapy-related myeloid neoplasm diagnosis; AIDA: ATRA, idarubicine; RT: radiotherapy; ProMACE-CytaBOM: cyclophosphamide, doxorubicin, etoposide, bleomycin, vincristine, methotrexate and prednisone.
Quantitative evaluation of the KMT2A-AFF1 transcript in UPN 14 revealed strong positivity for the chimeric transcript at the time of t-ALL diagnosis (3500 copies/104 ABL).
Disease markers and clonal evolution
Using ultra-deep NGS, we then tracked backwards by NGS the t-MN clonal markers in BM samples collected at the time of the primary hematological malignancy, and identified at least two different patterns.
In the first scenario somatic mutations characterizing the t-MN clone were not present at primary cancer diagnosis and appeared after cytotoxic treatment. In UPN 3, UPN4, UPN6 and UPN13, the ASXL1 S689*, ASXL1 R693*, SF3B1 K700E and SETBP1 G870R mutations, studied using ultra-deep NGS, were undetectable at the time of primary cancer (NHL in UPN3 and UPN4, normal karyotype AML in UPN6 and B-ALL with a complex karyotype in UPN13) and were detected after 20, 113, 83 and 32 months from start of primary tumor treatment. In UPN4 (Figure 1A), UPN6 and UPN13 the identified mutations were detectable for the first time at t-MN diagnosis, suggesting a dominant role of these mutations in t-MN evolution. In UPN3, the ASXL1 S689* mutation was detectable for the first time at NHL follow-up (VAF: 29%, at 20 months), and increased until the time of t-MN diagnosis (VAF: 35%, 86 months). Of note, the ASXL1 S689* was first detected following peripheral blood stem cell transplantation (PB-SCT, Figure 1B). In UPN 6, who had two primary malignancies (AML and BC), the SF3B1 mutation may have been induced by the chemotherapy used to treat AML or by the combined radio-chemotherapy approach used to treat BC. In this case we have no data to precisely unravel the genomic effects of the different treatments.
Similarly, the KMT2A-AFF1 fusion identified in UPN14 at t-ALL diagnosis, as expected, was undetectable in the primary APL diagnostic sample, and was detected at low levels by Q-RT-PCR 13 months after achievement of complete remission of APL, treated according to the AIDA protocol (Figure 1C, 17). Notably, a constant increase in the transcript copy number was evident from first identification (58 copies/104 ABL) to t-ALL onset (3522 copies/104 ABL). The patient did not achieve molecular response after induction treatment for ALL using clofarabine and cyclophosphamide (4390 copies/104 ABL), whereas re-induction treatment according to the LAL0904 protocol (3 courses of Vincristine and Daunorubicin, 20) resulted in complete hematological and molecular remission. UPN14 subsequently underwent allogeneic stem cell transplantation (ASCT) from an HLA-identical related donor with T-cell depletion and is to-date in complete hematological and molecular remission 54+ months after ASCT.
A different pattern was observed in UPN1, UPN2 and UPN9, where mutations were detectable in the primary BM sample, prior to any cytotoxic treatment. In particular, the specific TP53 Y220C mutation was identified in UPN2 in all available BM-MNC specimens, from APL diagnosis through complete remission phases, and finally in t-MN. However, the TP53 Y220C mutation was undetectable in mesenchymal stromal cells (MSC) cultured from the BM at t-MN diagnosis, indicating that the mutation had been acquired by hematopoietic cells only. A high-throughput NGS analysis revealed a constant increase in the mutated allele frequency from APL to t-MN onset (Figure 2A).
Similarly, the ASXL1 Y591* mutation was identified in UPN1 in all the available BM-MNC specimens, from NHL diagnosis to t-MN. Notably, ASXL1 Y591* mutation was detected at very low level in the NHL BM-specimen (0.3%), significantly increased at 31 month follow-up (7%) and reached the highest VAF at t-MN diagnosis (42%, Figure 2B).
UPN9 was characterized by a particular pattern, with the IDH1 R132H mutation present at NHL diagnosis, whereas the SRSF2 P95H mutation appeared at t-MN onset (Figure 2C). In particular, the IDH1 R132H mutation, originally present at a high allele frequency (35%) 9 years before t-MN onset, when the patient was 72 years old, indicates that this mutation could have occurred as a pre-leukemic event. The later acquisition of a SRSF2 P95H mutation suggests its role in t-MN pathogenesis in a susceptible individual.
Altogether, these data suggest that different patterns of therapy-related leukemogenesis may occur in different patients.
DISCUSSION
Changes in cytotoxic treatment for different malignancies have modified the incidence of t-MN [16]. Since only a small proportion of cancer patients develop a t-MN, individual susceptibility to this disease is one of the topics of major interest. In the past years, several authors have suggested that germ-line variants may play a major role in t-MN predisposition. Candidates are polymorphisms of genes implicated in detoxification, DNA repair and apoptosis pathways [21–23], or single nucleotide variants (SNV) affecting cancer predisposition genes, such as Fanconi anemia genes [7, 24]. Although next generation sequencing approaches have allowed the acquisition of detailed information, a direct and univocal mechanism to explain t-MN susceptibility has not been identified yet.
Recently, somatic mutations in critical genes, including DNMT3A, ASXL1 and TP53 have been identified in the PB of healthy elderly individuals, indicating that mutated clones may spontaneously emerge as a result of the aging process [14–15]. In this scenario, therapy-related leukemia would not be a direct consequence of cytotoxic treatment, but the result of clonal selection due to cytotoxic treatment administered to predisposed individuals.
In the present investigation, we studied the time-course of the appearance of somatic mutations in genes known to be involved in hematological malignancies during therapy-related leukemogenesis.
Our results highlight at least two different models. In a first model, mutations were acquired probably as a consequence of cytotoxic treatment, where ASXL1 S689* (UPN3), ASXL1 R693* (UPN4), SF3B1 K700E (UPN6) and SETBP1 G870R (UPN13) mutations were only detectable at the time of t-MN diagnosis, and were apparently absent in the BM-MNC harvested at time of primary tumor diagnosis and during follow-up. Although we cannot exclude the presence of minor clones (VAF <0.1%) in UPN3, UPN4, UPN6 and UPN13, our results suggest that the ASXL1 S689*, ASXL1 R693*, SF3B1 K700E and SETBP1 G870R mutations may be directly induced by chemo- and/or radiotherapy. The acquisition of these mutations at high VAF may play a pivotal role in the development of t-MN.
Similarly, in UPN14, the KMT2A-AFF1 transcript was not amplified at APL diagnosis and was detected for the first time 14 months after APL treatment start, during maintenance treatment that was being given according to the AIDA 2000 protocol [17]. The KMT2A/AFF1 copy number progressively increased later on, until t-ALL diagnosis. The standard AIDA protocol includes consolidation with mitoxantrone, a topoisomerase-II inhibitor, known to be involved in DNA-damage specifically targeting the KMT2A gene [25]. t-MN following Topo-II inhibitors are also characterized by short latency, as in this case, where the KMT2A/AFF1-positive clone became detectable 13 months after treatment start. The high curability of APL highlights the importance of avoiding genotoxic and mutagenic drugs in this disease, where chemotherapy-free regimens including arsenic-trioxide have shown high curative potential at least in non-high risk patients [26].
In the second scenario, somatic mutations were present in the marrow harvested at the time of primary cancer diagnosis. This is exemplified by UPN2, who carried a TP53 Y220C mutation at a frequency of 0.1% at primary APL diagnosis, progressively increasing in serial follow-up samples, and until t-MN onset. A low, but significant number (6%), of TP53 mutated cells may suffice to guide t-MN development. This same pattern has been previously identified by Wong et al. in four of seven t-MN patients, suggesting that cytotoxic therapy does not directly induce TP53 mutations, but selects for hematopoietic progenitor cells carrying spontaneous TP53 mutations, which are more resistant to chemotherapy than TP53 wild-type cells [13]. The presence of TP53 mutations could contribute to the typical genetic instability of t-MN, associated with complex karyotypes and poor response to chemotherapy.
The same scenario identified in UPN2 could be hypothesized in UPN1, where the ASXL1 Y591* mutation was detected at low frequency (0.3%) at primary NHL diagnosis and progressively increased in serial follow-up samples, reaching a very high VAF at the time of t-MN diagnosis (42%).
In UPN9, we found two mutations in the t-MN sample (IDH1 R132H and SRSF2 P95H), but only the IDH1 R132H was present at the time of NHL diagnosis, probably acting as predisposing factor for the acquisition of the second mutation. The similar VAF (35-38%) of the two mutations suggests their presence in the same cell clone, although we cannot exclude that two independent clones were simultaneously present. According to the literature, IDH1 mutations are rare or absent in NHL, therefore it seems unlikely that these were present in the NHL clone in our case [27]. The high VAF of the IDH1 mutation in the initial BM sample (35%) which remained virtually unchanged at t-MN diagnosis (38%) may indicate independence of this clone from the pressure of cytotoxic therapy with chlorambucil. Since this drug has only rarely been associated with t-MN, it remains unclear whether this was a true therapy-related MDS or a second neoplasm occurring in a susceptible patient [28]. In this line, Lindsley et al, have recently shown that mutations of splicing and epigenetic enzymes may distinguish AML subtypes which are “secondary” to MDS, from truly therapy-related forms [9].
In conclusion, our data show that secondary leukemogenesis is a heterogeneous process at least in cases with a previous hematological malignancy. Somatic mutations in critical genes may precede and favor leukemic development or may be induced by the cytotoxic treatment. Additional studies in extended patient cohorts with a longitudinal follow-up, together with further advances in NGS technology and digital PCR, are needed to define the contribution of minor clones to therapy-related leukemogenesis. This will also help to identify patients at major risk at the time of primary cancer diagnosis, where more leukemogenic treatments could be avoided and a more stringent follow-up monitoring should be recommended.
MATERIALS AND METHODS
Study population
Our study population included 14 patients (13 t-MN and one t-ALL), diagnosed at the Department of Hematology of the Università Cattolica del Sacro Cuore and at Tor Vergata University between November 2003 and March 2014. Cases were selected based on availability of DNA from secondary leukemia diagnosis and at least one preceding phase (e.g. marrows collected during follow-up of the primary tumor). There were 5 males and 9 females, with a median age at t-MN diagnosis of 58 years (range 30–81). According to the proportion of blasts, there were 12 MDS, 1 AML and 1 B-lineage ALL. The primary malignancies were all hematological and included non-Hodgkin lymphoma (NHL) in 7 patients, Hodgkin lymphoma (HL) in 2 patients, acute promyelocytic leukemia (APL) in 3 patients, normal-karyotype AML followed by breast cancer (BC) in 1 patient and B-ALL in 1 patient. Detailed patient characteristics are reported in Table 1. All peripheral blood (PB) and BM samples were obtained after informed consent. The study has been approved by the Institutional Ethical Committees of the Università Cattolica del Sacro Cuore and Tor Vergata University.
Mutational analysis
Mononuclear cells (MNCs) were separated from patients’ BM at the time of the primary cancer diagnosis, during its follow-up and at t-MN diagnosis by Ficoll gradient centrifugation using Lympholyte-H (Cedarlane, Ontario, Canada). As previously reported [29], BM-mesenchymal stem cells (MSC) were expanded in UPN2 using Mesencult medium (Stem Cell Technologies, Voden Medical Instruments, Milan, Italy) in plastic-adherent cultures up to the second passage. Granulocyte, CD3+ and CD3- cells were isolated at t-MN diagnosis from the peripheral blood of UPN9 by Ficoll gradient centrifugation followed by MACS cell separation (Miltenyi biotec, Italy).
Genomic DNA was extracted using the QIAamp DNA Mini Kit (Qiagen Srl., Milan, Italy), following the manufacturer’s instructions, whereas total RNA was extracted using standard procedures and reverse-transcribed using random hexamers as primers [30]. The following mutations were studied at the time of t-MN diagnosis by Sanger sequencing of genomic DNA: IDH1 R132, IDH2 R140 and R172, DNMT3A R882, ASXL1 exon 12, U2AF1 S34 and R35, SF3B1 exons 13–16, SRSF2 exon 1, SETBP1 SKI homologous domain, NRAS exons 2-3 and KRAS exons 2-3 as previously described (ABI PRISM 3100; Applied Biosystems/Life Technologies, Milan, Italy, 10-11, 31). All mutations were confirmed in independent experiments and quantified by pyrosequencing using specifically designed oligonucleotides targeting the mutated region (shown in Supplementary Table 1). Reagents (PyroMark Gold Q96, QiagenSrl, Milan, Italy), instrumentation and analysis software used for pyrosequencing analysis were as recommended by the manufacturers (PyroMark Q96 ID, DiatechPharmacogenetics, Jesi, Italy, PyroMark Assay Design and PyroMark Q24 version 2.0.6).
TP53 mutations were studied at the time of t-MN diagnosis by standard NGS, since mutations of this gene have been detected at low VAF by several authors at t-MN diagnosis. Briefly, genomic DNA was amplified using Fast Start High Fidelity Polymerase (Roche, Monza, Italy) and fusion primers (Roche, Monza, Italy) containing M13 adapter sequences and the sequence-specific primers (Supplementary Table 1) spanning TP53 exons 4-9 (RefSeq NM_000546.5, representing the longer transcript encoding the longest protein isoform). Amplicon library A and B sequencing adapters and multiplex identifier (MID) tags were then added to both tails of amplicons by a second amplification step. PCR products were visualized on agarose gel, purified using AMPure XP DNA-binding paramagnetic beads (Agencourt Bioscience Corp., Beckman Coulter S.p.A, Milan, Italy), and quantified using the picogreen dye (Life Technologies, Carlsbad, California) and the Victor X2 fluorometer (Perkin Elmer, Waltham, Massachusetts). Samples were then pooled at equimolar ratios to prepare for Roche/454 pyrosequencing. The obtained amplicon library was added to the emulsion PCR at a ratio of 0.8 molecules per bead and subjected to deep sequencing on the Genome Sequencer Junior instrument (Roche-454 Life Sciences, Branford, Connecticut). The obtained sequencing reads were mapped to the TP53 human reference sequence (RefSeq NC_000017.11) and analyzed by the Amplicon Variant Analyzer (AVA) software version 3.0 (Roche-454 Life Sciences) to establish the mutant allele frequency.
The mutations identified in the t-MN sample were then studied in marrow samples collected at the time of primary diagnosis and/or during follow-up using high-throughput NGS (Sistemas Genòmicos S.L., Valencia, Spain). Briefly, genomic DNA was amplified by PCR using specific homemade designed primers linked to specific flags (Supplementary Table 1). Amplicons resequencing was performed using the Illumina MiSeq sequencing platform and NexteraXT PCR enrichment (Illumina, San Diego, CA, USA). Paired-end’ reads of 250 nt were generated. Then, targeted regions were enriched using the Nextera XT DNA kit (Illumina, San Diego, CA, USA). For the amplicon data analysis, three different plots were generated for each sample’s read group: per base sequence quality, per base sequence content and per sequence quality scores. To remove the background noise and to assure that only very confident high quality reads were used to perform read alignment, Nextera adapters were removed using FASTQ-MCF software. Reads were then collapsed using UPARSE [32] to merge the paired-end reads into a single better quality read (no mismatches allowed). Then PRINSEQ [33] was used to remove low quality reads in both 5’ and 3’ ends. Collapsed reads were aligned separately against the human reference genome version GRCh38. Read alignment was performed using BWA and ‘in-house’ scripts (Sistemas Genòmicos S.L., Valencia, Spain). From the BAM formatted file obtained after reads mapping, low quality reads were removed. In addition, the overall sample coverage and the efficiency of the combination of the selected strategy (PCR enrichment system plus NGS platform) were evaluated. Filtering processes were performed using Picard-tools (http://picard.sourceforge.net/) and SAM tools [34]. Coverage metrics and the evaluation of the target enrichment system were performed using custom scripts (Sistemas Genómicos S.L., Valencia, Spain). For each of the corresponding specific amplicon regions average coverage was above 120000x for all the samples in the experiment (detailed read depth in Supplementary Table 2). To avoid bias, for the quantification of studied mutations all collapsed mapped reads were used to establish the count and frequencies in % (alternative allele count/total depth x100).
Detection and quantification of the KMT2A-AFF1, transcript
Qualitative screening of the t(4;11)(q21;q23) translocation resulting into the KMT2A-AFF1 fusion transcript was performed in UPN14 at the time of diagnosis of a therapy-related B-ALL, according to van Dongen, et al. [35]. The quantitative analysis for KMT2A-AFF1 was performed according to Gabert J, et al. [36] at the time of the primary acute promyelocytic leukemia and at various time points during its follow-up.
ACKNOWLEDGMENTS
The authors would like to acknowledge research funding from FIRB (RBAP11TF7Z) and Associazione Italiana Ricerca sul Cancro (A.I.R.C, Grant N. 16952).
CONFLICTS OF INTEREST
The authors declared no conflicts of interest.
REFERENCES
1. Pedersen-Bjergaard J, Andersen MK, Andersen MT, Christiansen DH. Genetics of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2008; 22:240-8. doi: 10.1038/sj.leu.2405078.
2. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, Harris NL, Le Beau MM, Hellström-Lindberg E, Tefferi A, Bloomfield CD. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937-51. doi: 10.1182/blood-2009-03-209262.
3. Fianchi L, Pagano L, Piciocchi A, Candoni A, Gaidano G, Breccia M, Criscuolo M, Specchia G, Maria Pogliani E, Maurillo L, Aloe-Spiriti MA, Mecucci C, Niscola P, et al. Characteristics and outcome of therapy-related myeloid neoplasms: Report from the Italian network on secondary leukemias. Am J Hematol. 2015;90:E80-5. doi: 10.1002/ajh.23966.
4. Pagano L, Pulsoni A, Tosti ME, Annino L, Mele A, Camera A, Martino B, Guglielmi C, Cerri R, Di Bona E, Invernizzi R, Castagnola C, Bassan R, et al. Acute lymphoblastic leukaemia occurring as second malignancy: report of the GIMEMA archive of adult acute leukaemia. Gruppo Italiano Malattie Ematologiche Maligne dell'Adulto. Br J Haematol. 1999;106:1037-40.
5. Pedersen-Bjergaard J. Acute lymphoid leukemia with t(4;11)(q21;q23) following chemo-therapy with cytostatic agents targeting at DNA-topoisomerase II. Leuk Res. 1992;16:733-5.
6. Shih AH, Chung SS, Dolezal EK, Zhang SJ, Abdel-Wahab OI, Park CY, Nimer SD, Levine RL, Klimek VM. Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica. 2013;98:908-12.
7. Voso MT, Fabiani E, Zang Z, Fianchi L, Falconi G, Padella A, Martini M, Li Zhang S, Santangelo R, Larocca LM, Criscuolo M, La Brocca A, Cutcutache I, et al. Fanconi anemia gene variants in therapy-related myeloid neoplasms. Blood Cancer J. 2015; 5:e323. doi: 10.1038/bcj.2015.44.
8. Stoddart A, McNerney ME, Bartom E, Bergerson R, Young DJ, Qian Z, Wang J, Fernald AA, Davis EM, Larson RA, White KP, Le Beau MM. Genetic pathways leading to therapy-related myeloid neoplasms. Mediterr J Hematol Infect Dis. 2011;3:e2011019. doi: 10.4084/MJHID.2011.019.
9. Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, Pigneux A, Wetzler M, Stuart RK, Erba HP, Damon LE, Powell BL, Lindeman N, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125:1367-76. doi: 10.1182/blood-2014-11-610543.
10. Voso MT, Fabiani E, Fianchi L, Falconi G, Criscuolo M, Santangelo R, Chiusolo P, Betti S, D'Alo' F, Hohaus S, De Stefano V, Leone G. Mutations of epigenetic regulators and of the spliceosome machinery in therapy-related myeloid neoplasms and in acute leukemias evolved from chronic myeloproliferative diseases. Leukemia. 2013;27:982-5. doi: 10.1038/leu.2012.267.
11. Fabiani E, Falconi G, Fianchi L, Criscuolo M, Leone G, Voso MT. SETBP1 mutations in 106 patients with therapy-related myeloid neoplasms. Haematologica. 2014;99:e152-3. doi: 10.3324/haematol.2014.108159.
12. Ishizawa S, Slovak ML, Popplewell L, Bedell V, Wrede JE, Carter NH, Snyder DS, Arber DA. High frequency of pro-B acute lymphoblastic leukemia in adults with secondary leukemia with 11q23 abnormalities. Leukemia. 2003;17:1091-5.
13. Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, Lamprecht TL, Shen D, Hundal J, Fulton RS, Heath S, Baty JD, Klco JM, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518:552-5. doi: 10.1038/nature13968.
14. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488-98. doi: 10.1056/NEJMoa1408617.
15. Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, Purcell SM, Svantesson O, Landén M, et al. Clonal hematopoiesis and blood cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477-87. doi: 10.1056/NEJMoa1409405.
16. Morton LM, Dores GM, Tucker MA, Kim CJ, Onel K, Gilbert ES, Fraumeni JF Jr, Curtis RE. Evolving risk of therapy-related acute myeloid leukemia following cancer chemotherapy among adults in the United States, 1975-2008. Blood. 2013;121:2996-3004. doi: 10.1182/blood-2012-08-448068.
17. Lo-Coco F, Avvisati G, Vignetti M, Breccia M, Gallo E, Rambaldi A, Paoloni F, Fioritoni G, Ferrara F, Specchia G, Cimino G, Diverio D, Borlenghi E, et al. Front-line treatment of acute promyelocytic leukemia with AIDA induction followed by risk-adapted consolidation for adults younger than 61 years: results of the AIDA-2000 trial of the GIMEMA Group. Blood. 2010;116:3171-9. doi: 10.1182/blood-2010-03-276196.
18. Willemze R, Suciu S, Meloni G, Labar B, Marie JP, Halkes CJ, Muus P, Mistrik M, Amadori S, Specchia G, Fabbiano F, Nobile F, Sborgia M, et al. High-dose cytarabine in induction treatment improves the outcome of adult patients younger than age 46 years with acute myeloid leukemia: results of the EORTC-GIMEMA AML-12 trial. J Clin Oncol. 2014;32:219-28. doi: 10.1200/JCO.2013.51.8571.
19. Hoelzer D, Gökbuget N, Digel W, Faak T, Kneba M, Reutzel R, Romejko-Jarosinska J, Zwolinski J, Walewski J. Outcome of adult patients with T-lymphoblastic lymphoma treated according to protocols for acute lymphoblastic leukemia. Blood. 2002;99:4379-85.
20. Chiaretti S, Vitale A, Cazzaniga G, Orlando SM, Silvestri D, Fazi P, Valsecchi MG, Elia L, Testi AM, Mancini F, Conter V, te Kronnie G, Ferrara F, et al. Clinico-biological features of 5202 patients with acute lymphoblastic leukemia enrolled in the Italian AIEOP and GIMEMA protocols and stratified in age cohorts. Haematologica. 2013;98:1702-10. doi: 10.3324/haematol.2012.080432.
21. Felix CA, Walker AH, Lange BJ, Williams TM, Winick NJ, Cheung NK, Lovett BD, Nowell PC, Blair IA, Rebbeck TR. Association of CYP3A4 genotype with treatment-related leukemia. Proc Natl Acad Sci U S A. 1998;95:13176-81.
22. Allan JM, Wild CP, Rollinson S, Willett EV, Moorman AV, Dovey GJ, Roddam PL, Roman E, Cartwright RA, Morgan GJ. Polymorphism in glutathione S-transferase P1 is associated with susceptibility to chemotherapy-induced leukemia. Proc Natl Acad Sci U S A. 2001;98:11592-7.
23. Fabiani E, Fianchi L, Falconi G, Boncompagni R, Boncompagni R, Criscuolo M, Guidi F, La Brocca A, Hohaus S, Leone G, Voso MT. The BCL2L10 Leu21Arg variant and risk of therapy-related myeloid neoplasms and de novo myelodysplastic syndromes. Leuk Lymphoma. 2014;55:1538-43. doi: 10.3109/10428194.2013.845885.
24. Churpek JE, Marquez R, Neistadt B, Claussen K, Lee MK, Churpek MM, Huo D, Weiner H, Bannerjee M, Godley LA, Le Beau MM, Pritchard CC, Walsh T, et al. Inherited mutations in cancer susceptibility genes are common among survivors of breast cancer who develop therapy-related leukemia. Cancer. 2015; 122:304-11. doi: 10.1002/cncr.29615.
25. Cowell IG, Austin CA. Mechanism of generation of therapy related leukemia in response to anti-topoisomerase II agents. Int J Environ Res Public Health. 2012;9:2075-2091. doi: 10.3390/ijerph9062075.
26. Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, Ferrara F, Fazi P, Cicconi L, Di Bona E, Specchia G, Sica S, Divona M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111-21. doi: 10.1056/NEJMoa1300874.
27. Zou Y, Zeng Y, Zhang DF, Zou SH, Cheng YF, Yao YG. IDH1 and IDH2 mutations are frequent in Chinese patients with acute myeloid leukemia but rare in other types of hematological disorders. Biochem Biophys Res Commun. 2010;402:378-383. doi: 10.1016/j.bbrc.2010.10.038.
28. Gale RP, Bennett JM, Hoffman FO. Therapy-related AML: a slip of the lip can sink a ship. Leuk Res. 2014;38:418-20. doi: 10.1016/j.leukres.2013.
29. Falconi G, Fabiani E, Fianchi L, Criscuolo M, Raffaelli CS, Bellesi S, Hohaus S, Voso MT, D'Alò F, Leone G. Impairment of PI3K/AKT and WNT/β-catenin pathways in bone marrow mesenchymal stem cells isolated from patients with myelodysplastic syndromes. Exp Hematol. 2016;44:75-83. doi: 10.1016/j.exphem.2015.
30. Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidiniumthiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156-9.
31. Chou WC, Huang HH, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Ko BS, Wu SJ, Huang SY, Hsu SC, Chen YC, Huang YN, Chang YC, Lee FY, Liu MC, Liu CW, Tseng MH, Huang CF, Tien HF. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood. 2010 Nov 18;116:4086-94. doi: 10.1182/blood-2010-05-283291.
32. Edgar RC. UPARSE: highly accurate CLUSTER sequences from microbial amplicon reads. Nat Methods. 2013;10:996-8. doi: 10.1038/nmeth.2604.
33. Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863-4. doi: 10.1093/bioinformatics/btr026.
34. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078-9. doi: 10.1093/bioinformatics/btp352.
35. vanDongen JJ, Macintyre EA, Gabert JA, Delabesse E, Rossi V, Saglio G, Gottardi E, Rambaldi A, Dotti G, Griesinger F, Parreira A, Gameiro P, Diáz MG, et al. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 Concerted Action: investigation of minimal residual disease in acute leukemia. Leukemia. 1999;13:1901-28.
36. Gabert J, Beillard E, van der Velden VH, Bi W, Grimwade D, Pallisgaard N, Barbany G, Cazzaniga G, Cayuela JM, Cavé H, Pane F, Aerts JL, De Micheli D, et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia – A Europe Against Cancer Program. Leukemia. 2003;17:2318-57.