
Introduction
Systemic Anaplastic Large Cell Lymphomas (ALCLs) refer to a group of malignancies of mature T lymphocytes characterized by large lymphoid cells (“hallmark cells”) and strong expression of CD30 [1]. The CD30 antigen has historically been instrumental in defining ALCL as a distinct category; however, its expression is not restricted to this pathology. CD30 is also found in activated non-neoplastic lymphoid cells [2, 3], in a subset of Peripheral T-cell Lymphoma - Not Otherwise Specified (PCTL-NOS) [4], in Hodgkin’s lymphoma (HL) [1], and solid neoplasms [5]. The discovery of recurrent chromosomal translocations involving the anaplastic lymphoma kinase (ALK) gene in approximately 50% of ALCL patients [6] led to the delineation of ALK positive and ALK negative as two distinct subtypes [7, 8]. Of note, ALK activation is necessary and sufficient for promoting ALCL tumorigenesis and its inhibition is key for the therapeutic treatment of ALK positive ALCL [9-14]. Therefore, ALK positive ALCL was identified as a distinct disease. Conversely, ALK negative ALCL was defined as a provisional entity, lacking distinctive features.
In recent years, great advances have been made in dissecting the heterogeneity of ALK negative ALCLs and in providing new diagnostic and treatment options for these patients [15-20]. Consequently, the new revision of the World Health Organization (WHO) classification has promoted ALK negative ALCL to a definite entity that includes distinct cytogenetic subsets with prognostic implications [21].
This review will focus on advances in understanding the biology and pathogenesis of ALK negative ALCL, evaluating the clinical relevance of these findings.
ALCLs features
Systemic ALCL comprises approximately 3% of all adult NHLs and 10% to 20% of childhood lymphomas. Both subtypes are characterized by male predominance (60%). Most patients present advanced stage disease (III to IV stage) often with B symptoms. ALK positive ALCL mostly affects young patients (10-19 years), whereas ALK negative ALCL occurs in older patients (peak of incidence in the sixth decade of life) [22]. Systemic ALCL frequently presents as a nodal disease, however extranodal involvement is seen in approximately 20% of cases, especially in skin, soft tissues, liver, and bone marrow.
ALK positive ALCL displays a better outcome compared to the ALK negative, with a 5-year overall survival rate of about 80% in ALK positive and 50% in ALK negative [6]. Nevertheless, when ALCL patients are stratified according to age and/or stage ALK positive and ALK negative individuals result in similar prognosis [23, 24].
ALCLs are characterized by high morphological heterogeneity, ranging from small-cell neoplasms to cases where very large and anaplastic cells predominate. However, almost all cases share a common feature, which is the presence of so called ‘hallmark cells’ characterized by abundant cytoplasm and large horseshoe-shaped nuclei. Neoplastic cells grow cohesively in a sheet-like pattern and preferentially involve lymph node sinuses [25].
Irrespective of ALCL subtype, strong expression of CD30 can be detected on the cell membrane and Golgi region. ALK positive ALCL cases show more often positivity for EMA (epithelial membrane antigen; 83% vs 43%; P < 0.001). Cytotoxic protein expression (TIA1, granzyme B, or perforin) is slightly more pronounced in ALK positive compared to ALK negative, but the difference is not statistically significant. ALK negative is most frequently CD3 positive compared to ALK positive ALCL [22, 23].
Another peculiar feature of ALCLs is the significant repression of the T-cell expression program. Even though nearly all ALCL cases (74-90%) show clonal TCR gene rearrangements, both ALK positive and ALK negative ALCLs lack T-cell receptors (TCRs) and related signaling molecules such as CD3, ZAP70, LAT and SLP76 [26, 27]. Paradoxically, ALCL cells display morphology, migration efficiency, and cytoskeletal rearrangements consistent with those of activated T-cells. In ALK positive ALCLs, it has been demonstrated that oncogene-deregulated tyrosine kinase activity controls T cell identity by transcriptional regulation and epigenetic silencing of key signaling molecules [28, 29].
Very recently Hassler et al. provided insights into the pathogenesis of ALCL through genome-wide DNA methylation profiling. The study found that ALK positive and ALK negative ALCL share common DNA methylation changes for genes involved in T cell differentiation and immune response [30].
A growing amount of literature has reported a link between breast implant and ALK negative ALCL designated as Breast Implant-Associated ALCL (BIA-ALCL or I-ALCL) [21]. Neoplastic cells are characterized by anaplastic features such as cytotoxic T-cell phenotype, CD30 and EMA co-expression, and ALK negativity [31-33]. Two distinct clinicopathological subtypes have been identified according to tumor localization: in situ BIA-ALCL (anaplastic cells confined to the fibrous capsule) and infiltrative I-ALCL (pleomorphic cells infiltrating adjacent tissue). In situ BIA-ALCLs have an indolent clinical course and generally remain free of disease after implant removal. On the contrary, infiltrative BIA-ALCLs have a more aggressive clinical course that might require systemic chemotherapy [34]. Chronic inflammation, implant immunogenicity, and sub-clinical infections have been implicated as driving mechanisms of BIA-ALCL tumorigenesis [32].
Therapeutic options
Optimal therapy for ALK negative ALCL patients has not yet been identified due to the rarity of the disease and the lack of randomized trials.
CHOP (Cyclophosphamide, doxorubicin, vincristine, and prednisone), or CHOP-like regimen, is currently the standard of care in the initial management of ALCL patients [22].
After induction chemotherapy with CHOP, ALK negative ALCL patients often receive a high-dose chemotherapy followed by consolidative autologous stem cell transplantation [19, 21, 23, 35]. The outcome of ALK negative ALCL is consistently worse using CHOP-like regimens than in ALK positive ALCL and no improved survival rate could be achieved using dose-intensive chemotherapies [36, 37]. The poor outcome of ALK negative patients likely reflects the clinical and genetic heterogeneity of the disease and suggests that more specific therapeutic strategies should be explored.
In the last decade there have been a limited number of trials evaluating novel therapies specific for systemic ALK negative ALCL. Among these, CD30-directed therapies with Brentuximab Vedotin (BV) received great attention and displayed promising results [38]. BV is composed of an anti-CD30 antibody conjugated to the anti-microtubule agent monomethyl auristatin E (MMAE). Based on the positive responses to BV in relapsed/refractory ALCL (ORR: 86%; CR: 57%), the drug was approved in 2012 for relapsed/refractory ALCL following one line of therapy [39]. A subsequent study including 32 ALCLs patients (6 ALK positive and 26 ALK negative) demonstrated that BV treatment in combination with CHOP or CHP (CHOP without vincristine) exhibits substantial antitumor activity with a manageable safety profile (ORR: 100%; CR: 88%) [40]. The use of BV in combination with chemotherapy as front-line treatment is now being investigated in the ECHELON-2 phase III trial (NCT01777152).
Transcriptional profiles
Gene expression profiling (GEP) is a recognized tool to identify differentially expressed genes between two or more groups. This analysis has been widely applied to identify novel diagnostic and prognostic biomarkers for the peripheral T-cell lymphoma patients’ stratification [41-44].
Thompson et al. first demonstrated the ability of GEP to correctly distinguish between ALK positive and ALK negative ALCL based on the differential expression of genes encoding signal transduction molecules (SYK, LYN, CDC37), transcription factors (including HOXC6 and HOXA3), and cell cycle regulators (CCND3 and CDKN2D) [45].
A subsequent study performed on 32 systemic ALCL samples and 5 cell lines, identified ALK, BCL6, PTPN12, CEBPB, and SERPINA1 as the most discriminating genes between ALK positive and ALK negative ALCL. Moreover, a molecular signature of ALK negative included the overexpression of CCR7, CNTFR, IL22, and IL21 genes [46]. However, these studies have analyzed a small number of patients and lacked of objective quality control criteria.
A GEP analysis of T-cell non-Hodgkin’s lymphoma samples, including angioimmunoblastic lymphomas (AILT), ALK positive ALCL, ALK negative ALCL, PTCL-NOS and normal T-cells, identified a genomic classifier for the recognition of ALCL patients [16]. Specifically, a set of 14 genes was able to distinguish ALK negative ALCL from PTCL-NOS and AILT. This study showed that ALCL patients share a cluster of transcripts, which allow their stratification and distinction from other T-cell lymphomas, and suggests that all ALCL may have a common cell of origin.
Piccaluga et al. developed a GEP-based molecular classifier that improved classification and prognostication among ALK negative ALCL, AITL, and PTCL-NOS patients [47]. This classifier displayed very high accuracy both in frozen and FFPE samples, however its clinical application remains limited due to the large number of genes required for ALK negative distinction.
A meta-analysis of several expression data sets [16, 48-50] identified and validated a 3-gene model (TNFRSF8, BATF3, and TMOD1) able to separate ALK negative ALCL from PTCL-NOS with a 97% accuracy [18]. The application of RT-qPCR protocols to FFPE tissues allowed the translation of GEP studies to routine clinical settings and the correct stratification of T-NHL.
To explore boundaries between PTCL-NOS and ALK negative ALCL, Bisig et al. analyzed the immunophenotype of different T-NHL subtypes [51]. The study found a substantial overlap between CD30-positive PTCL-NOS and ALK negative ALCL signatures. Specifically, CD30-positive PTCL-NOS were significantly enriched in ALK-negative related genes. The authors introduced a new hypothesis stating a biological continuum across CD30 positive PTCLs in contrast with other studies that demonstrated that PTCL-NOS and ALK negative ALCL are separated entities [16, 18, 23, 41]. The discordant observations were probably due to the high heterogeneity of these pathologies, different criteria for the samples’ characterization and the relatively small number of patients.
A more recent GEP analysis revealed that ALK negative ALCL were enriched for MYC and IRF4 target gene signature in comparison with PTCL-NOS [44]. The same study pointed out other differences between ALCL subtypes, such as the overexpression of the PI3K pathway- in ALK negative cases and the overrepresentation of HIF1A, IL10 and HRAS/KRAS-induced genes in the ALK positive patients. MYC inhibition has been demonstrated to be critical for ALCL survival and may represent a therapeutic target for ALCL therapy [52, 53].
ALK negative ALCL characterization was also improved by microRNA expression profiling. MicroRNAs (miRNAs) are small non-coding RNA molecules that play a crucial role in regulating gene expression at post-transcriptional level in a sequence-specific manner [54, 55]. miRNAs can act as oncogenes or tumor suppressors according to their target mRNAs. Recent works demonstrated the diagnostic and prognostic value of miRNA profiling for ALK negative patients. Liu et al proposed an 11-miRNA signature including 4 upregulated (miR-210, miR-197, miR-191, and miR-512-3p) and 7 downregulated miRNA (miR-451, miR- 146a, miR-22, miR-455-3p, miR-455-5p, miR-143, and miR-494), which distinguished ALK negative patients from PTCL-NOS with a 90% probability [56]. Merkel et al highlighted miRNA signatures associated with ALCL subtypes. The study described miR-17-92 cluster and miR-155 highly expressed in ALK positive and ALK negative ALCL patients, respectively [57]. Accordingly, Spaccarotella et al demonstrated that miR 17-92 cluster promotes proliferation and survival of ALK-positive anaplastic large cell lymphoma [58]. Moreover, miR-155 silencing results in increased levels of cleaved caspase-3 and SOCS1, which leads to STAT3 signaling suppression and tumor growth reduction in murine models of ALK negative ALCL [59]. These data suggested that miR-155 could act as a tumor driver in ALK negative ALCL. However, mir-155 is consistently over-expressed in the majority of T-NHL samples, indicating that its levels could not be used as a marker for differential diagnosis [18]. Another study identified a five miRNAs signature able to discriminate PTCL-NOS from ALK negative ALCL with high accuracy. This signature was validated in FFPE samples and was suggested to be predictive for the distinction between CD30 positive PTCL-NOS and ALK negative ALCLs [60]. Small RNA sequencing was recently used to investigate the differential expression of miRNA between ALCL subgroups. Steinhilber et al. identified a 56-miRNA signature distinguishing ALK positive, ALK negative and normal T-cells. This signature shows overlapping results with 26 miRNA identified by Merkel et al. and Liu et al. [61].
GEP has had a clear impact on elucidating ALK negative ALCL biology, defining the borders with other PTCL subtypes and providing new genomic classifiers for the correct stratification of patients. However, applying gene-expression profiling analysis is currently impractical and not yet standardized in routine clinical settings. Alternative strategies should be considered to translate the knowledge gained from GEP studies to the clinical arena. A three gene classifier able to discerne ALK negative ALCL showed potential clinical utility [18]. Recently, Nanostring nCounter technology has been developed to quantify a high number of RNA transcripts derived from formalin-fixed paraffin-embedded tissues [62, 63]. This methodology provides results concordant to conventional GEP with high reproducibility [64]. It is expected that the application of RT-qPCR or Nanostring protocols to FFPE tissues in clinical settings will allow the development of precise molecular diagnostic tools able to reduce errors and ambiguity in the stratification of T-NHL.
Somatic copy number alterations
Comparative genomic hybridization (CGH) and single nucleotide polymorphism (SNP) arrays have thoroughly portrayed the profile of chromosomal imbalances of ALCLs. One of the first CGH studies in ALCLs and PTCL-NOS identified recurrent chromosomal gains of 1q (1q41-qter) in 46%, and losses of 6q (6q21) and 13q (13q21-q22) in 31% and 23% ALK negative ALCL patients, respectively [65]. The authors demonstrated that, despite a considerable overlap between the genetic features of ALK negative ALCL and PTCL-NOS (such as loss of 6q and 13q), the profile of chromosomal imbalances segregate PTCL-NOS from ALK negative ALCL.
Salaverria et al. performed CGH analysis in a large series of ALK positive and ALK negative ALCL [66]. Chromosomal imbalances were detected in 58% of ALK positive and 65% of ALK negative ALCL. ALK positive ALCL cases displayed recurrent 17p and 17q24-qter gains and 4q13-q21, and 11q14 losses, gains of 1q and 6p21 were more frequently observed in ALK negative ALCL, whereas gains of chromosome 7 and 6q and 13q losses were seen in both types of ALCL tumors. The authors demonstrated that ALK positive and ALK negative ALCL harbor different genetic aberrations, confirming that they correspond to separated genetic entities.
More recently, the genomic profile of ALCL was analyzed with a different approach. Genome-wide DNA profiling of ALCL using high-density, single nucleotide polymorphism (SNP) arrays identified concomitant losses at 17p13 and at 6q21, encompassing the TP53 and PRDM1/BLIMP1, in up to one quarter of ALCL cases. Loss of TP53 and/or PRDM1 was present in 52% ALK negative ALCL, and in 29% of all ALCL cases. In particular, PRDM1 displayed a tumor suppressive role in the ALCL model [67]. BLIMP1 is a critical factor for B and T cell differentiation, and its onco-suppressive role has been documented in different models including diffuse large B cell lymphomas and natural killer cell lymphoma [68].
Copy number alteration studies have provided the landscape of chromosomal aberrations in ALCLs. However, these findings did not find their application in the routine clinical setting.
Mutations
Using classical DNA Sanger sequencing, PRF1 monoallelic germline mutations were frequently found in patients with childhood ALCL (27% of cases). Current opinion is that PRF1 mutations are not oncogenic per se but they could represent a predisposition factor for the disease by partially impairing the cytotoxic machinery [69, 70].
Next generation sequencing (NGS) technologies have emerged over the past decade providing new insights into the biology of ALCL. Different NGS based approaches ranging from amplicon-based (targeted), whole exome or whole genome sequencing were applied to identify new translocations and somatic mutations in ALK negative ALCL [17, 71, 72].
Whole exome sequencing was used to investigate the frequency of somatic mutation and associated to copy number variation analysis in ALK negative ALCL [71]. Among the plethora of mutations including PRDM1, TP53, TET2, FAS and STIM2 genes, JAK1 and STAT3 genes were the most recurrently mutated accounting for 18% of systemic ALK negative ALCL. The authors demonstrated that JAK1/STAT3 mutations lead to STAT3 activation and transformation. Interestingly, constitutive STAT3 phosphorylation was observed in a significant proportion of JAK1/STAT3 non-mutated cases, suggesting alternative mechanisms of pathway activation.
JAK/STAT3 signaling is frequently deregulated in hematopoietic and solid tumors [73]. In ALK positive ALCL, the oncogenic effect of ALK chimeras is mostly mediated by STAT3 [74-78]. The discovery of STAT3 activation in ALK negative ALCLs suggests that the STAT3-mediated oncogenic mechanism may be shared by all ALCLs, independently of ALK status. As a result of these findings Crescenzo et al. demonstrated that JAK/STAT3 inhibition impaired tumor growth in a preclinical ALK-negative ALCL-patient derived tumorgraft model, providing new potential therapeutic targets for ALK negative treatment [71]. Activating STAT3 mutations have been observed in other T cell and B cell disorders [79, 80], suggesting that STAT3 might be considered as a therapeutic target in several malignancies. Among JAK/STAT3 pathway inhibitors, Ruxolitinib displays promising results in different pathological models and has been approved by the FDA to treat myeloproliferative disorders [81]. The identification of ALCL patients that could benefit from this therapy can be promptly achieved by immunohistochemical staining for activated STAT3.
Chromosomal Translocations
Next generation sequencing performed on mate-paired libraries, identified numerous rearrangements in ALK negative ALCL. The first translocation described involves DUSP22-IRF4 locus on 6p25.3 and the FRA7H fragile site on 7q32.3 [17]. The presence of a DUSP22 rearrangement was associated with down-regulation of DUSP22 expression and upregulation of mir29A levels. DUSP22 is a dual-specificity phosphatase involved in JNK activation [82, 83], suppression of IL-6-induced STAT3 activation [84] and TCR signaling down-regulation in reactive T cells through ERK2 inactivation [85]. DUSP22 has a tumor suppressor function in B-cell lymphomas [86], T-lymphoblastic leukemias [87] and ALK positive ALCLs [88]. FRAH7H site contains a miRNA gene cluster that includes miR-29b. Accordingly, ALCL with 7q32.3 rearrangements show miR-29b over expression. The role of miR-29b is still controversial, however its up-regulation suggests a role as tumor promoter in ALCL, AML [89], bladder cancer [90], and breast cancer [91]. At present, the biological significance of DUSP22 rearrangements has still to be demonstrated.
With the same approach thirteen recurrent rearrangements were identified in PTCLs, five of these are p53-related genes, including TP53, TP63, CDKN2A, WWOX, and ANKRD11. The authors focused their attention on inv(3)(q26q28) that leads to the expression of a fusion transcript TBL1XR1/TP63 with structural homology to oncogenic deltaNp63, a p63 isoform lacking transactivation domain [72]. DeltaNp63 acts as a dominant negative by inhibiting the p53 pathway. Its oncogenic role has been demonstrated in several models including breast [92, 93], lung [94], and head and neck cancers [95]. TP63 rearrangements were exclusively found in PTCL-NOS (9.4%), ALK negative ALCL (12.5%), and primary cutaneous ALCL (10.5%). Of note, the large majority of TP63-positive PTCL-NOS show CD30 expression higher than 80%. Moreover, TP63 was associated to inferior overall survival [72].
Subsequent analyses were aimed to test DUSP22 and TP63 rearrangements as biomarkers for diagnosis and risk stratification of ALK negative ALCL patients. A multi-institutional study on 105 ALCL patients (32 ALK positive and 73 ALK negative) revealed that DUSP22 and TP63 rearrangements are present in 30% and 8% of ALK-negative ALCL patients, respectively. These rearrangements were mutually exclusive and specifically expressed in ALK negative ALCL. On morphologic grounds DUSP22-rearranged ALCL display significant differences from other ALK negative ALCL, typically showing sheet-like growth with doughnut cells and few large pleomorphic cells. Tumor cells are CD30 positive, ALK, TIA-1 and granzyme B negative. TP-63 rearranged cases show more heterogenic features with hallmark cells always present. However, the small number of TP63-rearranged cases limited the identification of general features [19, 96].
Patients with DUSP22 rearrangement had better outcomes, similar to ALK positive ALCL (five years overall survival: 90% DUSP22 and 85% ALK positive ALCL). Patients with TP63-rearrangements had overall survival rates significantly worse than those with ALK positive ALCL (five years overall survival: 17%). Therefore, DUSP22 and TP63 rearrangements have important prognostic relevance and may serve as predictive biomarkers [19].
The study by Crescenzo et al. further depicted the heterogeneity of ALK negative ALCL by the recognition of numerous sporadic fusion transcripts [71]. As a common feature, chimeric proteins recurrently involved tyrosine kinases (i.e. ROS1 or TYK2) triggering the activation of JAK/STAT3 pathway. Interestingly, these gene fusions were mutually exclusive with JAK1/STAT3 mutations, suggesting convergent pathogenetic mechanisms and therapeutic targets for ALK negative ALCL (Figure 1).
Whole genome sequencing analyses have shed light on genetic and biological heterogeneity among ALK negative ALCL, supporting the idea that this entity is composed by different subgroups. DUSP22 and TP63 translocations defined three different subgroups (DUPSP22-positive, TP63-positive and triple negative ALCL) with clear prognostic implications. The predictive value of these rearrangements can be successfully translated to routine clinical setting by performing fluorescence in situ hybridization assays, in order to deliver the most appropriated therapeutic protocol to ALK negative patients. Early autologous Stem Cell Transplantation (SCT) is indicated for ALK negative patients but not for ALK positive because of their favorable outcomes following chemotherapy. The discovery that DUSP22 positive patients show similar outcomes to ALK positive may play a role in the decision to employ early SCT [97].
ROS and TYK translocations assume clinical value in the light of their ability to activate JAK/STAT pathway which represent an attractive therapeutic target for ALK negative ALCL, as discussed above. Immunofluorescence analysis for routine clinical detection of these translocations in primary samples remains to be confirmed on a larger cohort of ALCL patients.
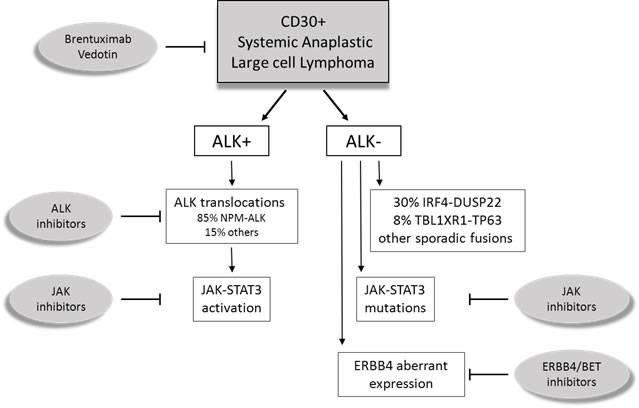
Figure 1: Schematic representation of systemic Anaplastic Large Cell Lymphomas (ALCL). ALK positive (ALK+) ALCL is a well-defined entity, characterized by ALK translocations. ALK negative (ALK-) ALCL were promoted to a definite entity that includes different cytogenetic subsets with prognostic and pharmacological significance.
Aberrant Transcripts
Using integrated bioinformatics approaches Scarfò et al. recently identified a novel diagnostic subclass of ALK negative ALCL coexpressing ERBB4 and COL29A1 and featuring a specific gene signature [20]. ERBB4 encodes for a member of the tyrosine kinase receptor superfamily which includes ERBB1 (EGFR) and ERBB2 (HER2), known to be deregulated in several solid tumors [98]. ERBB4 was found to be mutated and potentially oncogenic in several cancer types, such as melanoma and lung adenocarcinoma [99-103]. Moreover, it has been demonstrated that ERBB4 mediates acquired resistance to ERBB2 inhibitors in breast cancer cells [104, 105]. Very recently, Boddicker et al. described a novel translocation involving ERBB4 in one PTCL-NOS patient. This rearrangement contains IKZF2 gene (exons 1-2) fused with ERBB4 (exons 2-28) [106].
Scarfò et al. found ERBB4 expression in ~25% ALK negative ALCL, but not in PTCL-NOS nor in ALK positive ALCL. Interestingly, ERBB4 ectopic expression in ALK negative ALCL patients resulted from two different truncated transcripts: I20ΔERBB4 and I12ΔERBB4. The study suggests that ERBB4 aberrant expression is not due to a genomic alteration, rather it is driven by reactivation of normally dormant long terminal repeat elements (LTRs) located in ERBB4 introns [20]. Examples of ancient LTR promoters awakening have been previously reported in Hodgkin lymphoma (HL). Lamprecht and colleagues demonstrated that the activation of an endogenous LTR leads to the expression of colony-stimulating factor 1 receptor (CSF1R), which results oncogenic and correlates with a poor outcome of HL patients [107]. More recently, Babaian et al. reported the activation of the LOR1a LTR with consequent ectopic overexpression of IRF5 [108]. Notably, ERBB4 positive ALCL frequently displayed Hodgkin-like features, usually rare among conventional ALCL. Considering the shared awakening of ancient LTR promoters in HL and ERBB4 positive ALCL, it will be interesting to analyze ERBB4 expression in Hodgkin Lymphoma samples.
The study by Scarfò et al. indicated that ERBB4-truncated forms show oncogenic potentials. Nevertheless, the pharmacologic inhibition of ERBB4 only partially controls ALCL cell growth and disease progression in a preclinical model, indicating the need for combination therapies in relapsed or refractory ERBB4-positive ALCL patients.
The identification of a subset of ERBB4 expressing ALK negative ALCL confirms the commonly accepted hypothesis that ALK negative includes multiple subgroups driven by different aberrations. ERBB4 represents a new diagnostic marker and a potential therapeutic target for this novel subclass. Clinical diagnosis of ERBB4 positive patients can be established using droplet digital PCR analysis for ERBB4 detection and/or immunohistochemical staining for MMP9, a protein highly correlated with ERBB4 expression. The diagnostic value of this finding is reinforced by observing that ERBB4 expression is mutually exclusive with other rearrangements such as: TP63, DUSP22 and ROS or TYK translocations. However these data have to be confirmed in a larger panel of patients.
LTR de-repression raises the perspective of clinical use of epigenetic drugs in tumors driven by transposable elements (TE) and refractory to current standard therapeutic regimens. Histone methylation and acetylation are among the most relevant modifications that guide chromatin remodeling and epigenetic control [109]. Targeting genes that regulate these modifications can represent valid therapeutic strategies to repress TE-driven oncogenic transcription. Indeed, the use of demethylases (KDM) and bromodomains (BET) inhibitors to block aberrant transcription could be an attractive option [110]. In particular, numerous studies have highlighted BET inhibitors as a novel category of anti-cancer agents, with preclinical and clinical evidence both in solid and hematological malignancies [111-113].
Conclusions
ALK negative ALCL is a genetically and biologically heterogeneous neoplasm previously considered a provisional entity because of the lack of specific biomarker. Many efforts have been made over the past few decades to identify precise, reproducible and clinically applicable biomarkers that have led to the recognition of ALK negative ALCL as a distinct clinicopathologic entity. The discovery of key driver mutations and therapeutic targets has been slowed down by the intrinsic molecular heterogeneity and the relative rarity of this disease. However, recent advances in next generation sequencing and bioinformatics approaches allowed the recognition of different subgroups of ALK negative ALCL with prognostic or pharmacological relevance. Integrating these findings with each other will be critical to understand the molecular heterogeneity of ALK negative ALCL and to select therapeutic strategies specifically tailored for each subgroup of ALK negative patients.
Acknowledgments
This work has been supported by: Investigator Grant IG-13358 and Special Program Molecular Clinical Oncology 5 x 1000 No. 10007, Associazione Italiana per la Ricerca sul Cancro (AIRC), Milan, Italy; Grant TO_Call2_2012_0061, Compagnia di San Paolo, Turin, Italy; Grant 2014_1105, Fondazione CRT, Turin, Italy.
Conflicts of interest
None declared.
References
1. Stein H, Mason DY, Gerdes J, O’Connor N, Wainscoat J, Pallesen G, Gatter K, Falini B, Delsol G, Lemke H, Schwarting R, Lennert K. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985; 66: 848-58.
2. Werner B, Massone C, Kerl H, Cerroni L. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008; 35: 1100-7.
3. Chiarle R, Podda A, Prolla G, Gong J, Thorbecke GJ, Inghirami G. CD30 in normal and neoplastic cells. Clin Immunol. 1999; 90: 157-64.
4. Went P, Agostinelli C, Gallamini A, Piccaluga PP, Ascani S, Sabattini E, Bacci F, Falini B, Motta T, Paulli M, Artusi T, Piccioli M, Zinzani PL, et al. Marker expression in peripheral T-cell lymphoma: a proposed clinical-pathologic prognostic score. J Clin Oncol. 2006; 24: 2472-9.
5. Falini B, Flenghi L, Fedeli L, Broe MK, Bonino C, Stein H, Durkop H, Bigerna B, Barbabietola G, Venturi S, et al. In vivo targeting of Hodgkin and Reed-Sternberg cells of Hodgkin’s disease with monoclonal antibody Ber-H2 (CD30): immunohistological evidence. Br J Haematol. 1992; 82: 38-45.
6. Vose J, Armitage J, Weisenburger D. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008; 26: 4124-30.
7. Stein H, Foss HD, Durkop H, Marafioti T, Delsol G, Pulford K, Pileri S, Falini B. CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood. 2000; 96: 3681-95.
8. Jaffe ES. Anaplastic large cell lymphoma: the shifting sands of diagnostic hematopathology. Mod Pathol. 2001; 14: 219-28.
9. Coluccia AM, Gunby RH, Tartari CJ, Scapozza L, Gambacorti-Passerini C, Passoni L. Anaplastic lymphoma kinase and its signalling molecules as novel targets in lymphoma therapy. Expert Opin Ther Targets. 2005; 9: 515-32.
10. Ergin M, Denning MF, Izban KF, Amin HM, Martinez RL, Saeed S, Alkan S. Inhibition of tyrosine kinase activity induces caspase-dependent apoptosis in anaplastic large cell lymphoma with NPM-ALK (p80) fusion protein. Exp Hematol. 2001; 29: 1082-90.
11. Piva R, Chiarle R, Manazza AD, Taulli R, Simmons W, Ambrogio C, D’Escamard V, Pellegrino E, Ponzetto C, Palestro G, Inghirami G. Ablation of oncogenic ALK is a viable therapeutic approach for anaplastic large-cell lymphomas. Blood. 2006; 107: 689-97.
12. Piva R, Pellegrino E, Mattioli M, Agnelli L, Lombardi L, Boccalatte F, Costa G, Ruggeri BA, Cheng M, Chiarle R, Palestro G, Neri A, Inghirami G. Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCL2A1 as critical target genes. J Clin Invest. 2006; 116: 3171-82.
13. Turturro F, Arnold MD, Frist AY, Pulford K. Model of inhibition of the NPM-ALK kinase activity by herbimycin A. Clin Cancer Res. 2002; 8: 240-5.
14. Wan W, Albom MS, Lu L, Quail MR, Becknell NC, Weinberg LR, Reddy DR, Holskin BP, Angeles TS, Underiner TL, Meyer SL, Hudkins RL, Dorsey BD, et al. Anaplastic lymphoma kinase activity is essential for the proliferation and survival of anaplastic large-cell lymphoma cells. Blood. 2006; 107: 1617-23.
15. Feldman AL, Law M, Remstein ED, Macon WR, Erickson LA, Grogg KL, Kurtin PJ, Dogan A. Recurrent translocations involving the IRF4 oncogene locus in peripheral T-cell lymphomas. Leukemia. 2009; 23: 574-80.
16. Piva R, Agnelli L, Pellegrino E, Todoerti K, Grosso V, Tamagno I, Fornari A, Martinoglio B, Medico E, Zamo A, Facchetti F, Ponzoni M, Geissinger E, et al. Gene expression profiling uncovers molecular classifiers for the recognition of anaplastic large-cell lymphoma within peripheral T-cell neoplasms. J Clin Oncol. 2010; 28: 1583-90.
17. Feldman AL, Dogan A, Smith DI, Law ME, Ansell SM, Johnson SH, Porcher JC, Ozsan N, Wieben ED, Eckloff BW, Vasmatzis G. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood. 2011; 117: 915-9.
18. Agnelli L, Mereu E, Pellegrino E, Limongi T, Kwee I, Bergaggio E, Ponzoni M, Zamo A, Iqbal J, Piccaluga PP, Neri A, Chan WC, Pileri S, et al. Identification of a 3-gene model as a powerful diagnostic tool for the recognition of ALK-negative anaplastic large-cell lymphoma. Blood. 2012; 120: 1274-81.
19. Parrilla Castellar ER, Jaffe ES, Said JW, Swerdlow SH, Ketterling RP, Knudson RA, Sidhu JS, Hsi ED, Karikehalli S, Jiang L, Vasmatzis G, Gibson SE, Ondrejka S, et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood. 2014; 124: 1473-80.
20. Scarfo I, Pellegrino E, Mereu E, Kwee I, Agnelli L, Bergaggio E, Garaffo G, Vitale N, Caputo M, Machiorlatti R, Circosta P, Abate F, Barreca A, et al. Identification of a new subclass of ALK-negative ALCL expressing aberrant levels of ERBB4 transcripts. Blood. 2016; 127: 221-32.
21. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, Jaffe ES. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016; 127: 2375-90.
22. Hapgood G, Savage KJ. The biology and management of systemic anaplastic large cell lymphoma. Blood. 2015.
23. Savage KJ, Harris NL, Vose JM, Ullrich F, Jaffe ES, Connors JM, Rimsza L, Pileri SA, Chhanabhai M, Gascoyne RD, Armitage JO, Weisenburger DD. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood. 2008; 111: 5496-504.
24. Sibon D, Fournier M, Briere J, Lamant L, Haioun C, Coiffier B, Bologna S, Morel P, Gabarre J, Hermine O, Sonet A, Gisselbrecht C, Delsol G, et al. Long-term outcome of adults with systemic anaplastic large-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte trials. J Clin Oncol. 2012; 30: 3939-46.
25. Benharroch D, Meguerian-Bedoyan Z, Lamant L, Amin C, Brugieres L, Terrier-Lacombe MJ, Haralambieva E, Pulford K, Pileri S, Morris SW, Mason DY, Delsol G. ALK-positive lymphoma: a single disease with a broad spectrum of morphology. Blood. 1998; 91: 2076-84.
26. Bonzheim I, Geissinger E, Roth S, Zettl A, Marx A, Rosenwald A, Muller-Hermelink HK, Rudiger T. Anaplastic large cell lymphomas lack the expression of T-cell receptor molecules or molecules of proximal T-cell receptor signaling. Blood. 2004; 104: 3358-60.
27. Foss HD, Anagnostopoulos I, Araujo I, Assaf C, Demel G, Kummer JA, Hummel M, Stein H. Anaplastic large-cell lymphomas of T-cell and null-cell phenotype express cytotoxic molecules. Blood. 1996; 88: 4005-11.
28. Turner SD, Yeung D, Hadfield K, Cook SJ, Alexander DR. The NPM-ALK tyrosine kinase mimics TCR signalling pathways, inducing NFAT and AP-1 by RAS-dependent mechanisms. Cell Signal. 2007; 19: 740-7.
29. Ambrogio C, Martinengo C, Voena C, Tondat F, Riera L, di Celle PF, Inghirami G, Chiarle R. NPM-ALK oncogenic tyrosine kinase controls T-cell identity by transcriptional regulation and epigenetic silencing in lymphoma cells. Cancer Res. 2009; 69: 8611-9.
30. Hassler MR, Pulverer W, Lakshminarasimhan R, Redl E, Hacker J, Garland GD, Merkel O, Schiefer AI, Simonitsch-Klupp I, Kenner L, Weisenberger DJ, Weinhaeusel A, Turner SD, et al. Insights into the Pathogenesis of Anaplastic Large-Cell Lymphoma through Genome-wide DNA Methylation Profiling. Cell Rep. 2016; 17: 596-608.
31. Miranda RN, Aladily TN, Prince HM, Kanagal-Shamanna R, de Jong D, Fayad LE, Amin MB, Haideri N, Bhagat G, Brooks GS, Shifrin DA, O’Malley DP, Cheah CY, et al. Breast implant-associated anaplastic large-cell lymphoma: long-term follow-up of 60 patients. J Clin Oncol. 2014; 32: 114-20.
32. Ye X SK, Rozen WM, Conyers R, Wright P, Kenner L, Turner SD,, IS W. Anaplastic large cell lymphoma (ALCL) and breast implants: breaking down the evidence. Mutat Res Rev Mutat Res. 2014; 762: 123-32.
33. Brody GS, Deapen D, Taylor CR, Pinter-Brown L, House-Lightner SR, Andersen JS, Carlson G, Lechner MG, Epstein AL. Anaplastic large cell lymphoma occurring in women with breast implants: analysis of 173 cases. Plast Reconstr Surg. 2015; 135: 695-705.
34. Laurent C, Delas A, Gaulard P, Haioun C, Moreau A, Xerri L, Traverse-Glehen A, Rousset T, Quintin-Roue I, Petrella T, Emile JF, Amara N, Rochaix P, et al. Breast implant-associated anaplastic large cell lymphoma: two distinct clinicopathological variants with different outcomes. Ann Oncol. 2016; 27: 306-14.
35. Swerdlow S, Campo E, Harris N, eds. ea. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues.IARC Press; 2008. IARC Press, Lyon, France. 2008.
36. Escalon MP, Liu NS, Yang Y, Hess M, Walker PL, Smith TL, Dang NH. Prognostic factors and treatment of patients with T-cell non-Hodgkin lymphoma: the M. D. Anderson Cancer Center experience. Cancer. 2005; 103: 2091-8.
37. Abramson JS, Feldman T, Kroll-Desrosiers AR, Muffly LS, Winer E, Flowers CR, Lansigan F, Nabhan C, Nastoupil LJ, Nath R, Goy A, Castillo JJ, Jagadeesh D, et al. Peripheral T-cell lymphomas in a large US multicenter cohort: prognostication in the modern era including impact of frontline therapy. Ann Oncol. 2014; 25: 2211-7.
38. Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL, Forero-Torres A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2010; 363: 1812-21.
39. Pro B, Advani R, Brice P, Bartlett NL, Rosenblatt JD, Illidge T, Matous J, Ramchandren R, Fanale M, Connors JM, Yang Y, Sievers EL, Kennedy DA, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol. 2012; 30: 2190-6.
40. Fanale MA, Horwitz SM, Forero-Torres A, Bartlett NL, Advani RH, Pro B, Chen RW, Davies A, Illidge T, Huebner D, Kennedy DA, Shustov AR. Brentuximab vedotin in the front-line treatment of patients with CD30+ peripheral T-cell lymphomas: results of a phase I study. J Clin Oncol. 2014; 32: 3137-43.
41. Piccaluga PP, Agostinelli C, Califano A, Rossi M, Basso K, Zupo S, Went P, Klein U, Zinzani PL, Baccarani M, Dalla Favera R, Pileri SA. Gene expression analysis of peripheral T cell lymphoma, unspecified, reveals distinct profiles and new potential therapeutic targets. J Clin Invest. 2007; 117: 823-34.
42. Piccaluga PP, Agostinelli C, Tripodo C, Gazzola A, Bacci F, Sabattini E, Pileri SA. Peripheral T-cell lymphoma classification: the matter of cellular derivation. Expert Rev Hematol. 2011; 4: 415-25.
43. Iqbal J, Wilcox R, Naushad H, Rohr J, Heavican TB, Wang C, Bouska A, Fu K, Chan WC, Vose JM. Genomic signatures in T-cell lymphoma: How can these improve precision in diagnosis and inform prognosis? Blood Rev. 2016; 30: 89-100.
44. Iqbal J, Wright G, Wang C, Rosenwald A, Gascoyne RD, Weisenburger DD, Greiner TC, Smith L, Guo S, Wilcox RA, Teh BT, Lim ST, Tan SY, et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014; 123: 2915-23.
45. Thompson MA, Stumph J, Henrickson SE, Rosenwald A, Wang Q, Olson S, Brandt SJ, Roberts J, Zhang X, Shyr Y, Kinney MC. Differential gene expression in anaplastic lymphoma kinase-positive and anaplastic lymphoma kinase-negative anaplastic large cell lymphomas. Hum Pathol. 2005; 36: 494-504.
46. Lamant L, de Reynies A, Duplantier MM, Rickman DS, Sabourdy F, Giuriato S, Brugieres L, Gaulard P, Espinos E, Delsol G. Gene-expression profiling of systemic anaplastic large-cell lymphoma reveals differences based on ALK status and two distinct morphologic ALK+ subtypes. Blood. 2007; 109: 2156-64.
47. Piccaluga PP, Fuligni F, De Leo A, Bertuzzi C, Rossi M, Bacci F, Sabattini E, Agostinelli C, Gazzola A, Laginestra MA, Mannu C, Sapienza MR, Hartmann S, et al. Molecular profiling improves classification and prognostication of nodal peripheral T-cell lymphomas: results of a phase III diagnostic accuracy study. J Clin Oncol. 2013; 31: 3019-25.
48. Piccaluga PP, Agostinelli C, Califano A, Carbone A, Fantoni L, Ferrari S, Gazzola A, Gloghini A, Righi S, Rossi M, Tagliafico E, Zinzani PL, Zupo S, et al. Gene expression analysis of angioimmunoblastic lymphoma indicates derivation from T follicular helper cells and vascular endothelial growth factor deregulation. Cancer Res. 2007; 67: 10703-10.
49. Eckerle S, Brune V, Doring C, Tiacci E, Bohle V, Sundstrom C, Kodet R, Paulli M, Falini B, Klapper W, Chaubert AB, Willenbrock K, Metzler D, et al. Gene expression profiling of isolated tumour cells from anaplastic large cell lymphomas: insights into its cellular origin, pathogenesis and relation to Hodgkin lymphoma. Leukemia. 2009; 23: 2129-38.
50. Iqbal J, Weisenburger DD, Greiner TC, Vose JM, McKeithan T, Kucuk C, Geng H, Deffenbacher K, Smith L, Dybkaer K, Nakamura S, Seto M, Delabie J, et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood. 2010; 115: 1026-36.
51. Bisig B, de Reynies A, Bonnet C, Sujobert P, Rickman DS, Marafioti T, Delsol G, Lamant L, Gaulard P, de Leval L. CD30-positive peripheral T-cell lymphomas share molecular and phenotypic features. Haematologica. 2013; 98: 1250-8.
52. Boi M, Todaro M, Vurchio V, Cvitkovic E, Riveiro E, Bertoni F, Inghirami G. OTX015, a bromodomain and extraterminal inhibitor, represents a novel agent for ALK positive anaplastic large cell lymphoma. Mol Cancer Ther. 2013; 12: A219.
53. Weilemann A, Grau M, Erdmann T, Merkel O, Sobhiafshar U, Anagnostopoulos I, Hummel M, Siegert A, Hayford C, Madle H, Wollert-Wulf B, Fichtner I, Dorken B, et al. Essential role of IRF4 and MYC signaling for survival of anaplastic large cell lymphoma. Blood. 2015; 125: 124-32.
54. Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012; 13: 271-82.
55. Wilczynska A, Bushell M. The complexity of miRNA-mediated repression. Cell Death Differ. 2015; 22: 22-33.
56. Liu C, Iqbal J, Teruya-Feldstein J, Shen Y, Dabrowska MJ, Dybkaer K, Lim MS, Piva R, Barreca A, Pellegrino E, Spaccarotella E, Lachel CM, Kucuk C, et al. MicroRNA expression profiling identifies molecular signatures associated with anaplastic large cell lymphoma. Blood. 2013.
57. Merkel O, Hamacher F, Laimer D, Sifft E, Trajanoski Z, Scheideler M, Egger G, Hassler MR, Thallinger C, Schmatz A, Turner SD, Greil R, Kenner L. Identification of differential and functionally active miRNAs in both anaplastic lymphoma kinase (ALK)+ and ALK- anaplastic large-cell lymphoma. Proc Natl Acad Sci U S A. 2010; 107: 16228-33.
58. Spaccarotella E, Pellegrino E, Ferracin M, Ferreri C, Cuccuru G, Liu C, Iqbal J, Cantarella D, Taulli R, Provero P, Di Cunto F, Medico E, Negrini M, et al. STAT3-mediated activation of microRNA cluster 17~92 promotes proliferation and survival of ALK-positive anaplastic large cell lymphoma. Haematologica. 2014; 99: 116-24.
59. Merkel O, Hamacher F, Griessl R, Grabner L, Schiefer AI, Prutsch N, Baer C, Egger G, Schlederer M, Krenn PW, Hartmann TN, Simonitsch-Klupp I, Plass C, et al. Oncogenic role of miR-155 in anaplastic large cell lymphoma lacking the t(2;5) translocation. J Pathol. 2015; 236: 445-56.
60. Laginestra MA, Piccaluga PP, Fuligni F, Rossi M, Agostinelli C, Righi S, Sapienza MR, Motta G, Gazzola A, Mannu C, Sabattini E, Bacci F, Tabanelli V, et al. Pathogenetic and diagnostic significance of microRNA deregulation in peripheral T-cell lymphoma not otherwise specified. Blood Cancer J. 2015; 4: 259.
61. Steinhilber J, Bonin M, Walter M, Fend F, Bonzheim I, Quintanilla-Martinez L. Next-generation sequencing identifies deregulation of microRNAs involved in both innate and adaptive immune response in ALK+ ALCL. PLoS One. 2015; 10: e0117780.
62. Chen X, Deane NG, Lewis KB, Li J, Zhu J, Washington MK, Beauchamp RD. Comparison of Nanostring nCounter(R) Data on FFPE Colon Cancer Samples and Affymetrix Microarray Data on Matched Frozen Tissues. PLoS One. 2016; 11: e0153784.
63. Veldman-Jones MH, Brant R, Rooney C, Geh C, Emery H, Harbron CG, Wappett M, Sharpe A, Dymond M, Barrett JC, Harrington EA, Marshall G. Evaluating Robustness and Sensitivity of the NanoString Technologies nCounter Platform to Enable Multiplexed Gene Expression Analysis of Clinical Samples. Cancer Res. 2015; 75: 2587-93.
64. Scott DW, Wright GW, Williams PM, Lih CJ, Walsh W, Jaffe ES, Rosenwald A, Campo E, Chan WC, Connors JM, Smeland EB, Mottok A, Braziel RM, et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood. 2014; 123: 1214-7.
65. Zettl A, Rudiger T, Konrad MA, Chott A, Simonitsch-Klupp I, Sonnen R, Muller-Hermelink HK, Ott G. Genomic profiling of peripheral T-cell lymphoma, unspecified, and anaplastic large T-cell lymphoma delineates novel recurrent chromosomal alterations. Am J Pathol. 2004; 164: 1837-48.
66. Salaverria I, Bea S, Lopez-Guillermo A, Lespinet V, Pinyol M, Burkhardt B, Lamant L, Zettl A, Horsman D, Gascoyne R, Ott G, Siebert R, Delsol G, et al. Genomic profiling reveals different genetic aberrations in systemic ALK-positive and ALK-negative anaplastic large cell lymphomas. Br J Haematol. 2008; 140: 516-26.
67. Boi M, Rinaldi A, Kwee I, Bonetti P, Todaro M, Tabbo F, Piva R, Rancoita PM, Matolcsy A, Timar B, Tousseyn T, Rodriguez-Pinilla SM, Piris MA, et al. PRDM1/BLIMP1 is commonly inactivated in anaplastic large T-cell lymphoma. Blood. 2013; 122: 2683-93.
68. Boi M, Zucca E, Inghirami G, Bertoni F. Advances in understanding the pathogenesis of systemic anaplastic large cell lymphomas. Br J Haematol. 2015; 168: 771-83.
69. Cannella S, Santoro A, Bruno G, Pillon M, Mussolin L, Mangili G, Rosolen A, Arico M. Germline mutations of the perforin gene are a frequent occurrence in childhood anaplastic large cell lymphoma. Cancer. 2007; 109: 2566-71.
70. Ciambotti B, Mussolin L, d’Amore ES, Pillon M, Sieni E, Coniglio ML, Ros MD, Cetica V, Arico M, Rosolen A. Monoallelic mutations of the perforin gene may represent a predisposing factor to childhood anaplastic large cell lymphoma. J Pediatr Hematol Oncol. 2014; 36: e359-65.
71. Crescenzo R, Abate F, Lasorsa E, Tabbo F, Gaudiano M, Chiesa N, Di Giacomo F, Spaccarotella E, Barbarossa L, Ercole E, Todaro M, Boi M, Acquaviva A, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015; 27: 516-32.
72. Vasmatzis G, Johnson SH, Knudson RA, Ketterling RP, Braggio E, Fonseca R, Viswanatha DS, Law ME, Kip NS, Ozsan N, Grebe SK, Frederick LA, Eckloff BW, et al. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas. Blood. 2012; 120: 2280-9.
73. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014; 14: 736-46.
74. Zamo A, Chiarle R, Piva R, Howes J, Fan Y, Chilosi M, Levy DE, Inghirami G. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene. 2002; 21: 1038-47.
75. Zhang Q, Raghunath PN, Xue L, Majewski M, Carpentieri DF, Odum N, Morris S, Skorski T, Wasik MA. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J Immunol. 2002; 168: 466-74.
76. Ruchatz H, Coluccia AM, Stano P, Marchesi E, Gambacorti-Passerini C. Constitutive activation of Jak2 contributes to proliferation and resistance to apoptosis in NPM/ALK-transformed cells. Exp Hematol. 2003; 31: 309-15.
77. Amin HM, Medeiros LJ, Ma Y, Feretzaki M, Das P, Leventaki V, Rassidakis GZ, O’Connor SL, McDonnell TJ, Lai R. Inhibition of JAK3 induces apoptosis and decreases anaplastic lymphoma kinase activity in anaplastic large cell lymphoma. Oncogene. 2003; 22: 5399-407.
78. Chiarle R, Simmons WJ, Cai H, Dhall G, Zamo A, Raz R, Karras JG, Levy DE, Inghirami G. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med. 2005; 11: 623-9.
79. Ohgami RS, Ma L, Monabati A, Zehnder JL, Arber DA. STAT3 mutations are present in aggressive B-cell lymphomas including a subset of diffuse large B-cell lymphomas with CD30 expression. Haematologica. 2014; 99: e105-7.
80. Couronne L, Scourzic L, Pilati C, Della Valle V, Duffourd Y, Solary E, Vainchenker W, Merlio JP, Beylot-Barry M, Damm F, Stern MH, Gaulard P, Lamant L, et al. STAT3 mutations identified in human hematologic neoplasms induce myeloid malignancies in a mouse bone marrow transplantation model. Haematologica. 2013; 98: 1748-52.
81. Roskoski R, Jr. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol Res. 2016; 111: 784-803.
82. Huang CY, Tan TH. DUSPs, to MAP kinases and beyond. Cell Biosci. 2012; 2: 24.
83. Chen AJ, Zhou G, Juan T, Colicos SM, Cannon JP, Cabriera-Hansen M, Meyer CF, Jurecic R, Copeland NG, Gilbert DJ, Jenkins NA, Fletcher F, Tan TH, et al. The dual specificity JKAP specifically activates the c-Jun N-terminal kinase pathway. J Biol Chem. 2002; 277: 36592-601.
84. Sekine Y, Tsuji S, Ikeda O, Sato N, Aoki N, Aoyama K, Sugiyama K, Matsuda T. Regulation of STAT3-mediated signaling by LMW-DSP2. Oncogene. 2006; 25: 5801-6.
85. Li H, Hsu HC, Wu Q, Yang P, Li J, Luo B, Oukka M, Steele CH, 3rd, Cua DJ, Grizzle WE, Mountz JD. IL-23 promotes TCR-mediated negative selection of thymocytes through the upregulation of IL-23 receptor and RORgammat. Nat Commun. 2014; 5: 4259.
86. Chen L, Juszczynski P, Takeyama K, Aguiar RC, Shipp MA. Protein tyrosine phosphatase receptor-type O truncated (PTPROt) regulates SYK phosphorylation, proximal B-cell-receptor signaling, and cellular proliferation. Blood. 2006; 108: 3428-33.
87. Kleppe M, Lahortiga I, El Chaar T, De Keersmaecker K, Mentens N, Graux C, Van Roosbroeck K, Ferrando AA, Langerak AW, Meijerink JP, Sigaux F, Haferlach T, Wlodarska I, et al. Deletion of the protein tyrosine phosphatase gene PTPN2 in T-cell acute lymphoblastic leukemia. Nat Genet. 2010; 42: 530-5.
88. Han Y, Amin HM, Frantz C, Franko B, Lee J, Lin Q, Lai R. Restoration of shp1 expression by 5-AZA-2’-deoxycytidine is associated with downregulation of JAK3/STAT3 signaling in ALK-positive anaplastic large cell lymphoma. Leukemia. 2006; 20: 1602-9.
89. Garzon R, Heaphy CE, Havelange V, Fabbri M, Volinia S, Tsao T, Zanesi N, Kornblau SM, Marcucci G, Calin GA, Andreeff M, Croce CM. MicroRNA 29b functions in acute myeloid leukemia. Blood. 2009; 114: 5331-41.
90. Xu F, Zhang Q, Cheng W, Zhang Z, Wang J, Ge J. Effect of miR-29b-1* and miR-29c knockdown on cell growth of the bladder cancer cell line T24. J Int Med Res. 2013; 41: 1803-10.
91. Wang C, Bian Z, Wei D, Zhang JG. miR-29b regulates migration of human breast cancer cells. Mol Cell Biochem. 2011; 352: 197-207.
92. Amin R, Morita-Fujimura Y, Tawarayama H, Semba K, Chiba N, Fukumoto M, Ikawa S. DeltaNp63alpha induces quiescence and downregulates the BRCA1 pathway in estrogen receptor-positive luminal breast cancer cell line MCF7 but not in other breast cancer cell lines. Mol Oncol. 2016; 10: 575-93.
93. Dang TT, Westcott JM, Maine EA, Kanchwala M, Xing C, Pearson GW. DeltaNp63alpha induces the expression of FAT2 and Slug to promote tumor invasion. Oncotarget. 2016; 7: 28592-611. doi: 10.18632/oncotarget.8696.
94. Massion PP, Taflan PM, Jamshedur Rahman SM, Yildiz P, Shyr Y, Edgerton ME, Westfall MD, Roberts JR, Pietenpol JA, Carbone DP, Gonzalez AL. Significance of p63 amplification and overexpression in lung cancer development and prognosis. Cancer Res. 2003; 63: 7113-21.
95. Kakuki T, Kurose M, Takano K, Kondoh A, Obata K, Nomura K, Miyata R, Kaneko Y, Konno T, Takahashi S, Hatakeyama T, Kohno T, Himi T, et al. Dysregulation of junctional adhesion molecule-A via p63/GATA-3 in head and neck squamous cell carcinoma. Oncotarget. 2016; 7: 33887-900. doi: 10.18632/oncotarget.8432.
96. King R, Dao LN, McPhail ED, Jaffe ES, Said J, Swerdlow SH, Sattler CA, Ketterling RP, Sidhu JS, Hsi ED, Karikehalli S, Jiang L, Gibson SE, Ondrejka SL, Nicolae A, Macon WR, Dasari S, Parrilla Castellar E, Feldman AL. Morphologic Features of ALK-negative Anaplastic Large Cell Lymphomas With DUSP22 Rearrangements. Am J Surg Pathol. 2016; 40 36-43.
97. Boddicker RL, Feldman AL. Progress in the identification of subgroups in ALK-negative anaplastic large-cell lymphoma. Biomark Med. 2015; 9: 719-22.
98. Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2014; 25: 282-303.
99. Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008; 455: 1069-75.
100. Prickett TD, Agrawal NS, Wei X, Yates KE, Lin JC, Wunderlich JR, Cronin JC, Cruz P, Rosenberg SA, Samuels Y. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet. 2009; 41: 1127-32.
101. Veikkolainen V, Vaparanta K, Halkilahti K, Iljin K, Sundvall M, Elenius K. Function of ERBB4 is determined by alternative splicing. Cell Cycle. 2011; 10: 2647-57.
102. Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer. 2012; 12: 553-63.
103. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013; 502: 333-9.
104. Kiuchi T, Ortiz-Zapater E, Monypenny J, Matthews DR, Nguyen LK, Barbeau J, Coban O, Lawler K, Burford B, Rolfe DJ, de Rinaldis E, Dafou D, Simpson MA, et al. The ErbB4 CYT2 variant protects EGFR from ligand-induced degradation to enhance cancer cell motility. Sci Signal. 2014; 7: ra78.
105. Canfield K, Li J, Wilkins OM, Morrison MM, Ung M, Wells W, Williams CR, Liby KT, Vullhorst D, Buonanno A, Hu H, Schiff R, Cook RS, et al. Receptor tyrosine kinase ERBB4 mediates acquired resistance to ERBB2 inhibitors in breast cancer cells. Cell Cycle. 2015; 14: 648-55.
106. Boddicker RL, Razidlo GL, Dasari S, Zeng Y, Hu G, Knudson RA, Greipp PT, Davila JI, Johnson SH, Porcher JC, Smadbeck JB, Eckloff BW, Billadeau DD, et al. Integrated mate-pair and RNA sequencing identifies novel, targetable gene fusions in peripheral T-cell lymphoma. Blood. 2016; 128: 1234-45.
107. Lamprecht B, Walter K, Kreher S, Kumar R, Hummel M, Lenze D, Kochert K, Bouhlel MA, Richter J, Soler E, Stadhouders R, Johrens K, Wurster KD, et al. Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat Med. 2010; 16: 571-9, 1p following 579.
108. Babaian A, Romanish MT, Gagnier L, Kuo LY, Karimi MM, Steidl C, Mager DL. Onco-exaptation of an endogenous retroviral LTR drives IRF5 expression in Hodgkin lymphoma. Oncogene. 2016; 35: 2542-6.
109. Brien GL, Valerio DG, Armstrong SA. Exploiting the Epigenome to Control Cancer-Promoting Gene-Expression Programs. Cancer Cell. 2016; 29: 464-76.
110. Maes T, Carceller E, Salas J, Ortega A, Buesa C. Advances in the development of histone lysine demethylase inhibitors. Curr Opin Pharmacol. 2015; 23: 52-60.
111. Chaidos A, Caputo V, Karadimitris A. Inhibition of bromodomain and extra-terminal proteins (BET) as a potential therapeutic approach in haematological malignancies: emerging preclinical and clinical evidence. Ther Adv Hematol. 2015; 6: 128-41.
112. Jung M, Gelato KA, Fernandez-Montalvan A, Siegel S, Haendler B. Targeting BET bromodomains for cancer treatment. Epigenomics. 2015; 7: 487-501.
113. Boi M, Todaro M, Vurchio V, Yang SN, Moon J, Kwee I, Rinaldi A, Pan H, Crescenzo R, Cheng M, Cerchietti L, Elemento O, Riveiro ME, et al. Therapeutic efficacy of the bromodomain inhibitor OTX015/MK-8628 in ALK-positive anaplastic large cell lymphoma: an alternative modality to overcome resistant phenotypes. Oncotarget. 2016; 7: 79637-53. doi: 10.18632/oncotarget.12876.