
INTRODUCTION
Despite many novel therapeutic approaches, breast cancer remains one of the leading causes of cancer mortality among women. It has recently been proved that the onset of cancer depends on a very small pool of tumor cells with a phenotype similar to that of normal adult stem cells [1]. Cancer stem cells (CSCs) are rare, tumor-initiating cells that exhibit stem cell properties: capacity for self-renewal, pluripotency, high tumorigenic potential, and resistance to therapy [2]. CSCs have been isolated from most human solid tumor types, suggesting their central role in tumor development, progression, and recurrence [3]. The CSC pool is associated with aggressiveness and a negative prognosis in breast cancer patients and its presence has important implications in cancer treatment. Current anti-cancer therapy is effective for removing the tumor mass, but the effects are often transient, leading to relapses and metastatic disease. A possible explanation for the failure of anti-cancer therapies is that they fail to kill CSCs, and even therapies that cause complete tumor regression might spare enough CSCs to allow regrowth.
Moreover, CSCs have a great capacity to survive in hypoxic microenvironments. In a solid tumor, rapid growth and aberrant blood flow often generate areas of hypoxia [4]. Hypoxic tumors have a more aggressive phenotype, showing more propensity to metastasize, poorer prognosis, and increased resistance to radiotherapy and chemotherapy [5]. The main regulators of the hypoxia signaling pathway are the HIF proteins. The stabilization of HIF proteins in hypoxic cancer cells promotes tumor progression through the regulation of the expression of players such as VEGF, glycolytic enzymes, glucose transporters, and proteins regulating mobility and metastasis [6, 7]. Furthermore, HIFs regulate signaling pathways that control stem cell properties, metabolic reprogramming, epithelial mesenchymal transition (EMT), metastasis and resistance to therapy [8].
Recently, microRNAs (miRNAs) have emerged as critical players in the maintenance of pluripotency, control of self-renewal, and cell fate [9–12]. Embryonic stem cells express specific patterns of miRNAs [13, 14], and specific miRNAs regulate and are regulated by key stem cell genes [15, 16]. Most miRNAs important for embryonic stem cell biology participate also in oncogenesis, mainly by promoting EMT [17, 18], metastasis [19] and survival in hypoxia.
An abundant miRNA that is highly conserved in different species is miR-24. It is clustered with two other miRNAs on chromosome 9 (cluster-1: miR-23b, miR-27b, and miR-24-1) and on chromosome 19 (cluster-2: miR-23a, miR-27a, and miR-24-2). Recent evidence indicates that miR-24 is an oncomiR in several cancer formation processes, such as in breast carcinoma and glioma; however, it acts as a tumor suppressor in laryngeal carcinoma [20]. Similarly, the role of miR-24 in stemness is incongruous, since it can promoter either differentiation or stemness depending on the cell type [21–24]. Here, we show that miR-24 acts as a stress sensor in breast CSCs, interfering with chemotherapy-induced apoptosis and, more importantly, regulating CSC resistance to hypoxic conditions.
RESULTS
MiR-24 modulates the stemness properties of breast cancer cells
Microarray analysis has identified several miRNAs differently expressed in primary BCSCs compared to differentiated cells [25]. Among these, we found miR-24 to be up-regulated in mammospheres. Microarray data were confirmed by qRT-PCR (Figure 1A). Suspension cultures of T47D, MCF-7, MDA-MB-231, and BT-549 breast cancer cells were used to enrich for BCSCs. Some molecular characteristics, including expression of stem cell markers, were increased in suspension cultures compared to differentiated cells, as confirmed by Western blotting and qRT-PCR in T47D [25], MCF-7, MDA-MB-231 and BT-549 cells (Supplementary Figure 1A-1B). MiR-24 was upregulated in mammosphere populations originated from T47D, MCF-7, MDA-MB-231 and BT-549 cells, similarly to what was observed in primary BCSCs (Figure 1B).
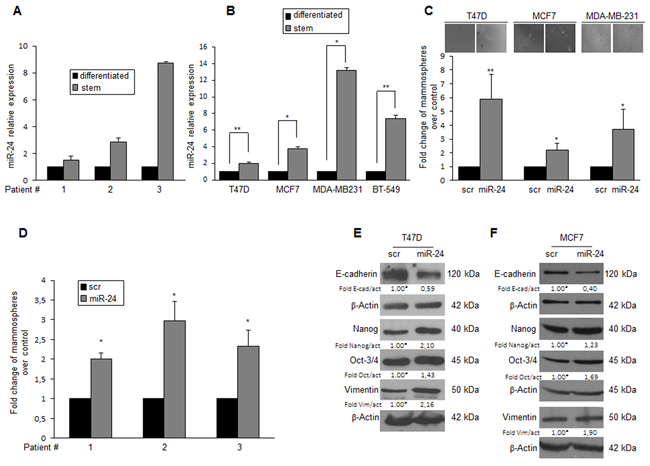
Figure 1: MiR-24 expression in BCSCs. A. miR-24 expression analyzed by qRT-PCR in primary breast cancer cells cultured in suspension (stem cell enriched) or adherent (differentiated) conditions. B. qRT-PCR levels of miR-24 in T47D, MCF-7, MDA-MB-231, and BT-549 breast cancer and in differentiated cells. C. T47D, MCF-7, and BT-549 cells were transfected with miR-24 or a scrambled oligonucleotide, grown as mammospheres, and counted after 6 days. D. Primary cells were transfected with miR-24 or scambled oligonucleotide, grown as mammospheres and counted after 6 days. E-F. Western blot showing that miR-24 overexpression in T47D and MCF-7 cells determines an upregulation of the stem markers Nanog, Oct-3/4, and Vimentin, and a downregulation of E-cadherin. In B, C, D, data are mean values ± SD of three independent experiments. Significance was calculated using Student’s t-test.*, p<0.05; **, p<0.01. Western blots are from representative experiments.
To investigate the biological implication of miR-24 upregulation in BCSCs, we modulated miR-24 levels in differentiated T47D, MCF-7 and MDA-MB-231 cells (Supplementary Figure 1C) and then assessed their capacity to form mammospheres. Overexpression of miR-24 boosted the formation of mammospheres in breast cancer cell lines (Figure 1C) and in cultures from primary breast tumor cells (Figure 1D). In addition, miR-24 overexpression led to the increase of several stemness genes, including Nanog, Oct-3/4, and Vimentin, and the reduction of E-cadherin, in T47D and MCF-7 cells (Figure 1E, 1F). Importantly, the expression of anti-miR-24 induced the opposite effect (Supplementary Figure 2A-2D). These findings suggest that miR-24 contributes to the regulation of key pathways implicated in the maintenance of the stem cell phenotype in breast cancer.
Since the stem cell pool is correlated with a more aggressive phenotype, we evaluated whether the up-regulation of miR-24 was associated with bad prognosis and survival. We used MIRUMIR [26] (http://www.bioprofiling.de/GEO/MIRUMIR/mirumir.html), an online tool that provides an analysis of miRNAs as potential biomarkers, to predict survival in cancer patients. We found that high expression of miR-24 was associated with poor outcome in breast cancer. Interestingly, statistical significance was also reached in another two publically available datasets (nasopharingeal carcinoma and osteosarcoma), supporting an oncogenic role for miR-24 (Supplementary Figure 3).
MiR-24 induces resistance to cisplatin
Chemoresistance plays a crucial role in breast cancer relapse and recurrence. This phenomenon is tightly linked to the characteristics of BCSCs that allow them further resistance to the treatment [27]. To confirm this observation, we compared apoptosis in T47D and MDA-MB-231 mammosphere and adherent cell cultures after 48h of cisplatin treatment. We found that BCSCs from T47D and MDA-MB-231 cells were more viable (data not shown) and exhibited a lower level of Caspase-3 activation after cisplatin treatment compared to differentiated cells (data not shown).
Cancer stem cells are more resistant than differentiated cells to cisplatin. In order to assess whether this property was in part mediated by the high levels of miR-24, we evaluated the effect of its overexpression on cells exposed to cisplatin. MiR-24 decreased the degree of apoptosis in breast cancer cells, as assessed by Caspase-3/7 activity (Figure 2A, 2B, 2C), PARP, and Caspase-3 cleavage (Figure 2D, 2E). To further confirm the effect of miR-24 as a chemotherapeutic protector, we transfected MDA-MB-231 stem cells with an anti-miR-24. We found that downregulation of miR-24 in cancer stem cells induced an increase of Caspase-3 activation and of PARP cleavage compared to anti-scrambled control (Figure 2F, 2G). Taken together, these results indicate that miR-24 induces enhanced resistance to apoptosis in BCSCs.
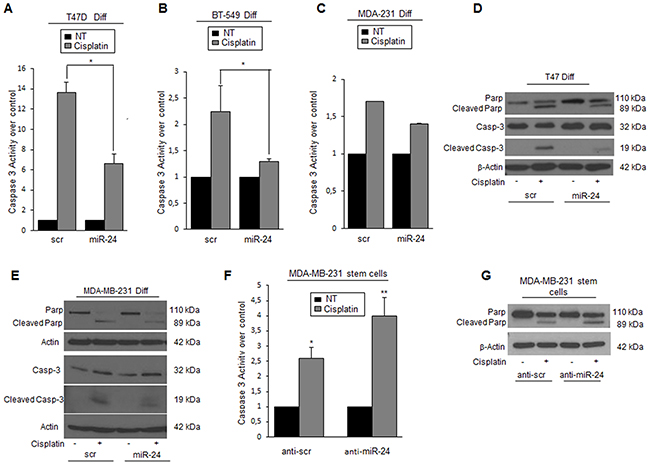
Figure 2: MiR-24 regulates the response to cisplatin in breast cancer cells. A, B, C. miR-24 transfection increased cisplatin resistance in T47D, BT-549, and MDA-MB-231 cells as assessed by Caspase-3/7 assay and D, E. by Western blot for cleaved PARP and Caspase-3. F, G. Anti-miR-24 transfection increased sensibility to cisplatin in MDA-MB-231 cancer stem cells. Western blots show representative experiments. Data are mean values ± SD of three independent experiments. In A, B, and F, significance was calculated using Student’s t-test.*, p<0.05; **, p<0.01.
Downregulation of BimL is involved in miR-24-mediated resistance to cisplatin
MiR-24 is known to bind to the 3’UTR of bimL, impairing translation of the mRNA [28]. BimL is an isoform generated by alternative splicing of bim, a member of the Bcl-2 family involved in the intrinsic apoptotic pathway. To confirm bimL as a target of miR-24 also in breast cancer cells, we transfected T47D cells with pre-miR-24 for 48 h and then analyzed BimL levels by qRT-PCR and Western blotting. Indeed, miR-24 downregulated bimL mRNA and protein levels (Figure 3A). Coherently, BimL protein expression was increased in T47D cells transfected with anti-miR-24 (Figure 3B). Furthermore, the expression of miR-24 was inversely correlated with BimL also in T47D mammosphere cultures (Figure 3C). To demonstrate the involvement of BimL in miR-24-induced cisplatin resistance, we transfected T47D cells with miR-24 or a scrambled control and then treated the cells with cisplatin for 48 h. As anticipated, increased expression of BimL was found in control cells but not in those overexpressing miR-24 (Figure 3D). To prove the direct links between miR-24, downregulation of BimL, and resistance to apoptosis, we carried out a rescue experiment by transfecting T47D cells with pre-miR-24 and a BimL cDNA lacking the 3’UTR. We found that overexpression of miR-24-resistant BimL had an effect on miR-24-mediated resistance to cisplatin (Figure 3E) and reverted miR-24’s effects also on mammosphere formation (Figure 3F). Similar results were obtained in the MDA-MB-231 cell line (Supplementary Figure 2E-2H).
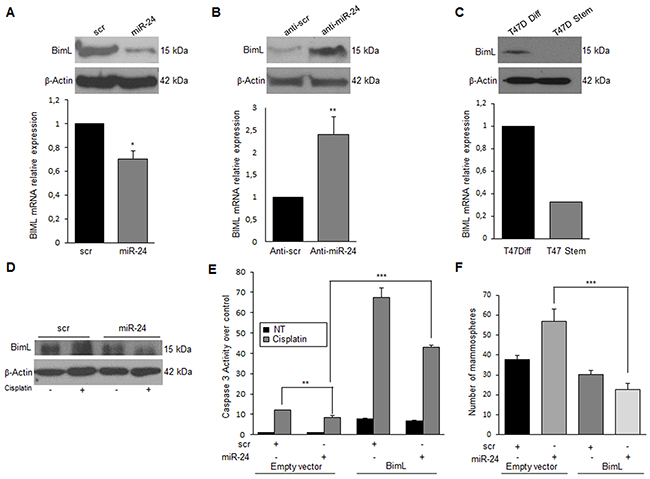
Figure 3: MiR-24 mediates cisplatin resistance by down-modulating BimL. MiR-24 downregulates BimL at mRNA and protein levels, as assessed by qRT-PCR and Western blotting in T47D cells A. On the contrary, anti-miR-24 increased in BimL expression B. Endogenous levels of BimL are higher in differentiated T47D cells compared to stem cells C. MiR-24 transfection reduced cisplatin’s effect on BimL levels D. Rescue experiment with the concomitant transfection of miR-24 and BimL cDNA lacking the 3’UTR, indicated that cisplatin resistance (assessed with a Caspase-3/7 assay) was in part reverted E. BimL also reverted the effect of miR-24 on mammosphere formation F. In A, B, E and F, data are mean values ± SD from three independent experiments. In A, B and E, significance was calculated using Student’s t-test.*, p<0.05; **, p<0.01; ***, p<0,001. In F, significance was calculated with ANOVA and Bonifaci correction.***, p<0,001. Western blotting analyses are from representative experiments.
Expression of miR-24 in hypoxic conditions
Stem cells reside in specialized microenvironments or niches that regulate their function. In vitro studies employing hypoxic culture conditions have revealed strong regulatory links between O2 availability and stem/precursor cell functions [29]. It has been reported that miR-24 has a HIF binding site on its promoter region [30]. Therefore, we assessed if miR-24 expression was induced under hypoxic conditions, thus contributing to stem cell survival. To this end, expression of miR-24 was analyzed in MCF-7, MDA-MB-231 and BT-549 cells cultured in an incubator with 1% O2 for 6h, and then miR-24 expression was analyzed by qRT-PCR. Indeed, miR-24 was induced by hypoxia in all breast cancer cells tested (Figure 4A). Moreover, MCF-7, MDA-MB-231, BT-549 and T47D cells transfected with miR-24 formed more mammospheres than control cells when cultured under hypoxic conditions (Figure 4B). Of note, we found that expression of Nanog and Oct-3/4 stemness genes was increased upon hypoxia, in particular in cells overexpressing miR-24 (Figure 4C, D). Interestingly, we also found that the level of BimL was decreased during hypoxia, and that this was more evident upon miR-24 transfection (Figure 4E).
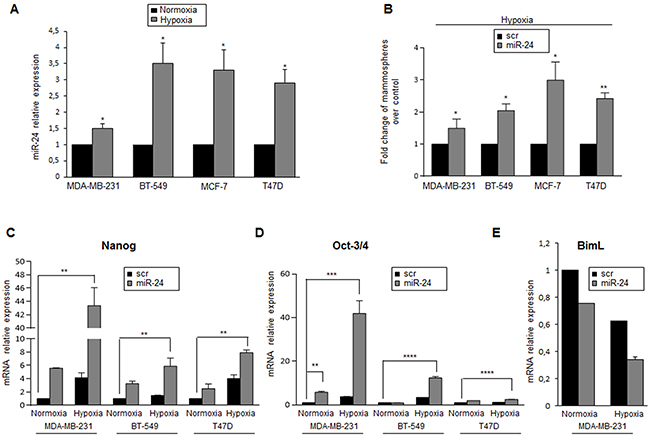
Figure 4: MiR-24 levels are regulated by hypoxia. A. MiR-24 levels, analyzed by qRT-PCR, were increased under hypoxic conditions in MDA-MB-231, BT-549, MCF-7 and T47D cells. B. MiR-24 overexpression upregulates mammosphere formation under hypoxia. C, D. MiR-24 up-regulates Nanog and Oct-3/4 expression in MDA-MB-231, BT-549 and T47D cells, as assessed by real time PCR. E. MiR-24 down-regulates BimL RNA in normoxic and hypoxic conditions. In A, B, C and D, data are mean values ± SD from two independent experiments. Significance was calculated using Student’s t-test.*, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0,001.
Analysis of genes involved in hypoxia and EMT pathways
Hypoxia regulates stem cell function through the direct activation of specific HIF target genes. HIF1α plays a key role in many crucial aspects of breast cancer biology, including stem cell maintenance, metabolic reprogramming, EMT, metastasis, and resistance to therapy. Therefore, we investigated miR-24’s effect on the expression of HIF1α. To this end, MCF-7, MDA-MB-231, BT-549 and T47D cells were transfected with either a scrambled oligonucleotide or a pre-miR-24 for 24h and then cultured under hypoxic conditions for 6h. We found that miR-24 upregulated HIF1α expression levels (Figure 5A). Furthermore, expression of two direct HIF1α targets, Snail and VEGFA, were increased (Figure 5B, 5C). These findings suggest that miR-24 induces an adaptive response to the toxic stimulus (i.e., low oxygen) by inducing expression of hypoxia inducible factors.
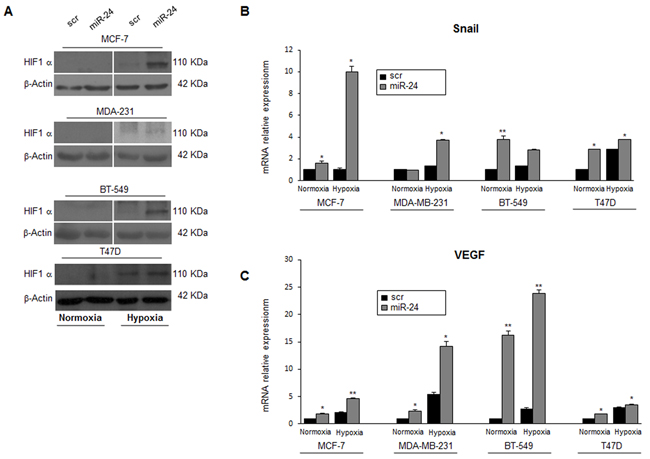
Figure 5: MiR-24 regulates HIF1α levels and hypoxia pathways. A. MiR-24 modulated HIF1α expression during hypoxia at the protein level in MCF-7, MDA-MB-231, BT549 and T47D cells. B, C. Real time PCR showing induction of Snail and VEGF mRNAs by miR-24 in hypoxic conditions. In B and C, data are mean values ± SD from two independent experiments. Significance was calculated using Student’s t-test.*, p<0.05;**, p<0.01; Western blots are representative experiments.
MiR-24 downregulates FIH1
We next investigated the mechanism of miR-24-mediated HIF1α protein stabilization. We found FIH1, an asparaginyl β-hydroxylase that promotes transcriptional repression of HIFs, among the potential miR-24 targets predicted by bioinformatics programs (Figure 6A). To verify whether miR-24 recognizes the 3’UTR of FIH1, this region was cloned downstream of a luciferase reporter gene. FIH1 3′UTR luciferase reporter activity was significantly repressed upon the addition of miR-24, while it was not affected by overexpression of miR-24 in the presence of a mutant construct in which the seed sequence was cloned back to front (Figure 6A). Real time-PCR and Western blotting of cells overexpressing miR-24 confirmed the decrease of FIH1 at mRNA (Figure 6B) and protein (Figure 6C, 6D) levels. Coherently, transfection of an anti-miR-24 oligonucleotide increased FIH1 expression (Figure 6C, 6D). Of note, hypoxia decreased FIH1 protein levels, an effect enhanced by overexpression of miR-24 (Figure 6E, 6F).
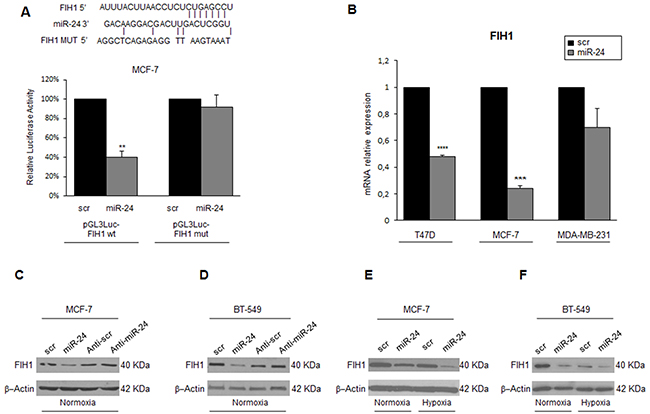
Figure 6: MiR-24 regulates hypoxia pathways by targeting FIH1. Predicted alignment between the miR-24 sequence and the 3′UTR of FIH1. Luciferase reporter assays show that reporter activity was inhibited in MCF-7 cells only in the presence of wild type and not mutated 3′UTR FIH1 A. miR-24 transfection downregulated FIH1 mRNA and protein levels, as assessed by qRT-PCR B. and Western blotting C, D. whereas transfection of anti-miR-24 upregulated FIH1 levels in MCF-7 and BT-549 cells C, D. Western blot showing that FIH1 is downregulated during hypoxia and upon miR-24 expression in MCF-7 and BT-549 cells E, F. Western blots are representative experiments. In A and B, data are mean values ± SD from two independent experiments. Significance was calculated using Student’s t-test. **, p<0.01; ***, p<0.001.
Overexpression of FIH1 reverts miR-24-mediated effects
To investigate whether miR-24-mediated downregulation of FIH1 was really responsible for the stemness features observed under hypoxia, we performed a rescue experiment. To this end, we transfected MDA-MB-231 and BT-549 breast cancer cells with pre-miR-24 and a FIH1 cDNA lacking the 3′UTR, and analyzed the effect on mammosphere formation, migration and HIF1α expression. Interestingly, miR-24’s effect on mammosphere formation was abolished by overexpression of FIH1 in both cancer cell lines (Figure 7A, 7B). In addition, under hypoxic conditions miR-24 significantly increased migration of MDA-MB-231 and BT-459 cells. In contrast, overexpression of FIH1 cDNA partially reverted cell motility (Figure 7C, 7D). Finally, we also verified that FIH1 overexpression was able to revert the effect of miR-24 on Nanog and Oct-3/4 expression in BT-549 (Figure 8A, 8B) and MDA-MB-231 (Supplementary Figure 4A, 4B) cells, and that it reverted the action of miR-24 on HIF1α upregulation as well as of its targets Snail and VEGF (Figure 8C, 8D, 8E, Supplementary Figure 4C, 4D, 4E).
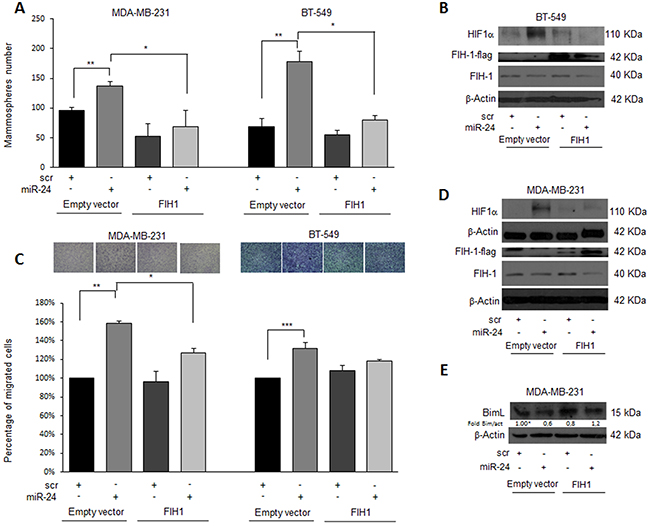
Figure 7: FIH1 rescues the effects of miR-24 on mammosphere formation and cell migration during hypoxia. FIH1 cDNA transfection in MDA-MB-231 and BT-459 cells reverted the effect of miR-24 on A. mammosphere formation and C. migration. B, D. Western blot analysis of HIF1α and FIH1 expression in the rescue experiment in MDA-MB-231 and BT-549 cells upon hypoxia. E. Western blot analysis of BimL expression in the rescue experiment in MDA-MB-231 cells upon hypoxia. In A and C, data are mean values ± SD from two independent experiments. Significance was calculated using Student’s t-test.*, p<0.05; **, p<0.01; ***, p<0.001.
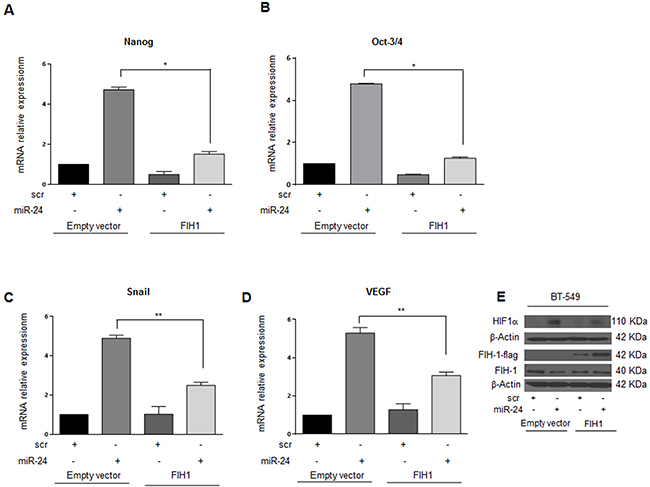
Figure 8: FIH1 reverts the effects of miR-24 on stem markers and on HIF1α in BT-549 cells during hypoxia. FIH1 cDNA transfection in BT-549 cells abolished the effect of miR-24 on Nanog and Oct-3/4 expression A, B. FIH1 also reverted the effect of the miR-24 on HIF1α E. and its targets Snail and VEGF C, D. In A, B, C, and D data are mean values ± SD from two independent experiments. Significance was calculated using Student’s t-test.*, p<0.05.
DISCUSSION
Many studies strongly support the concept that breast tumors originate from mammary stem cells that have become mutated during self-renewal or differentiation processes [31]. Poorly differentiated cancers display higher CSC content than well-differentiated tumors [32], in agreement with the concept that the proportion of CSCs reflects tumor malignancy [33]. CSCs may be responsible for poor prognosis, since they are resistant to current forms of therapy and, therefore, are responsible for the recurrence of the disease [34–36]. Therefore, further understanding of the molecular mechanisms underlying CSC maintenance is needed if we are to develop therapies that can eradicate breast cancer [37].
Recently, miRNAs have been found to be critical for various cellular processes, acting as major post-transcriptional regulators [38]. By their ability to target hundreds of mRNAs, they can induce rapid and simultaneous switches in cell fate and genome expression. miRNAs have been frequently found deregulated in different human cancers, acting either as oncogenes or tumor suppressors [39–41]. MiRNAs may regulate the stemness of embryonic stem cells, and at the same time may act as essential regulators of ESC differentiation [42, 43]. More interestingly, some miRNAs have been reported to regulate CSC phenotype, controlling self-renewal and differentiation [44].
The threat posed by CSCs derives from their unique properties: they have a slow rate of division compared to differentiated cells (i.e., they are quiescent); they overexpress drug transporters and DNA repair proteins that confer resistance to chemotherapeutics; and they have the ability to survive under adverse conditions, such as a hypoxic microenvironment. Areas of hypoxia are common in cancers in which the expanding tumor causes poor oxygen diffusion and chaotic vascularization. Moreover, hypoxia may contribute to CSC maintenance by promoting a pro-survival response and altering cellular behavior through multiple mechanisms affecting metabolism, angiogenesis, and invasion/migration [45]. Indeed, hypoxia increases self-renewal capacity as well as induction of essential stem cell factors, such as Oct-3/4, Nanog, and c-Myc [6]. Moreover, it promotes an immature phenotype in solid tumors such as human neuroblastoma, breast cancer [8, 46], and glioma [47].
The effects of hypoxia on CSCs seem to be primarily mediated by hypoxia inducible factors. HIF1, consisting of HIF1α and HIF1β subunits [48], is a key mediator of the cell response to hypoxia, and its high expression correlates with poor prognosis in various tumor types [4]. The transcriptional activity of HIF1 residing in the HIF1α subunit is partly controlled by asparaginyl hydroxylation by factor-inhibiting HIF hydroxylase 1(FIH1) [49]. FIH1 was originally found to be a negative regulator of HIF1 and was later shown to be an asparaginyl hydroxylase capable of hydroxylating N803 in the C-terminal activation domain of human HIF1 [50–52]. Recent studies have demonstrated that FIH1 may be regulated by microRNAs [53, 54].
In high-grade gliomas, a genetic alteration characterized by deletion of chromosome 10q23–q26 encompassing the FIH1 locus at 10q24 has been described [55]. FIH-1 loss of function may contribute to increased HIF1-mediated transactivation of downstream target genes, such as VEGF in gliomas and other human cancers [56], thus conferring a growth advantage to stem cells under hypoxic conditions.
In the present study, we identify miR-24 as an important player in the control of breast CSCs maintenance, at least in part through the regulation of FIH1 and Bim levels. We provide evidence that miR-24 is upregulated in the stem cell population of primary tumors and breast cancer cell lines compared to differentiated cells. Moreover, miR-24 was upregulated in hypoxic conditions, and overexpression of miR-24 increased mammospheres formation in normoxic, as well as in hypoxic conditions. MiR-24 increased HIF1α protein, in part by directly targeting FIH1. Moreover, miR-24 increased two common HIF target proteins, Snail and VEGF, which are involved in conferring a growth advantage to cancer cells under hypoxic conditions.
MiR-24 plays an important role in various types of cancer. Indeed, expression of the miR-23a~27a~24-2 cluster is upregulated in acute lymphoblastic leukemia [57], acute myeloid leukemia, chronic lymphocytic leukemia [58], breast cancer [59], gastric cancer [60], and hepatocellular carcinoma cells [61]. Moreover, antisense inhibition of miR-24 attenuated A549 cell growth [62]. Recently, overexpression of miR-24 was shown to promote cell proliferation and inhibit apoptosis in MDA-MB-435 and MDA-MB-468 mammary adenocarcinoma cell lines, with p27Kip1 identified as the direct miR-24-3p target mediating these effects [59]. Other reported targets of miR-24 include proapoptotic (FAF-1, Caspase-9, Bim, and Apaf-1) and cell cycle proteins [28, 63–65]; miR-24 was also shown to regulate XIAP, reducing the threshold for apoptosis in cancer cells [66].
Besides modulating survival when oxygen is scarce, miR-24 may affect stem cell behavior by hindering chemotherapy-induced apoptosis. Indeed, we demonstrate that overexpression of miR-24 increases the resistance to cisplatin, and that this effect is mediated by inhibition of the pro-apoptotic protein BimL, which has previously been described as a miR-24 target in cardiomyocytes [28]. We also show that differentiated cells overexpressing miR-24 have an enhanced capacity to form mammospheres, and that overexpression of BimL inhibits this effect, suggesting that the regulation of apoptosis is involved in the stemness phenotype. It is well known that stem cells are more resistant to anoikis, namely cell death induced by detachment from the extracellular matrix. This may suggest that miR-24 protects stem cells from this kind of death, favoring their survival. Furthermore, miR-24 has also been implicated in stem cell maintenance and resistance to therapy in other systems, for example it was found to be enriched in CD34+ HSPCs, where it played a role as a regulator of normal erythropoiesis via targeting of human activin receptor type 1, ALK4 [67]. Some reports have described that miR-24 inhibits erythroid differentiation of K562 cells, erythroid colony formation, and maturation of human CD34+ hematopoietic progenitor cells. Pan et al. have shown that miR-24-3p overexpression inhibited autophagy induction and reduced cell viability with VP16–DDP treatment, by targeting the autophagy-related gene ATG4A in small lung cancer cells [68].
In summary, we report here for the first time that miR-24 regulates stem cell maintenance in breast cancer, contributing to the preservation of the stem cell compartment from stressful conditions such as chemotherapy or hypoxia. MiR-24 regulates apoptosis by targeting BimL, contributing to resistance to cisplatin. Moreover, by targeting Fih1, miR-24 regulates HIF expression, conferring stem cells a growth advantage in hypoxic conditions (Figure 9). In conclusion, these findings suggest that inhibition of miR-24 might be a useful strategy for combating chemoresistance and survival of breast cancer cells in hypoxia.
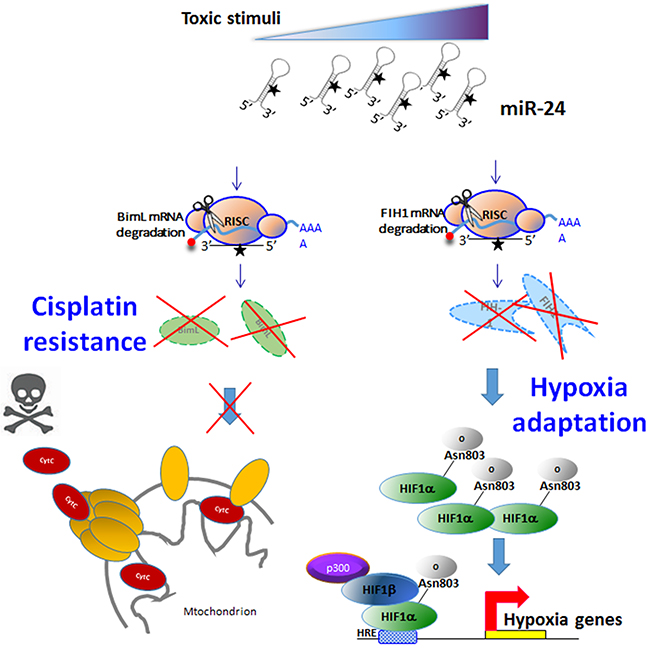
Figure 9: Mechanism of action of miR-24 in breast cancer stem cells. Toxic stimuli induce expression of miR-24. MiR-24 downregulates BimL (inducing resistance to chemotherapeutics) and FIH1 (inducing resistance under hypoxia).
MATERIALS AND METHODS
Cell and mammosphere cultures
Differentiated cells from breast tumors of three patients (#1, #2, #3) and breast cancer stem cells (BCSCs) were obtained and used for microRNA array as previously described [25, 69]. T47D, BT-549, and MDA-MB-231 cells were grown in RPMI 1640. MCF-7 cells were grown in DMEM. Media were supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine, and 100 U/ml penicillin/streptomycin. T47D, BT549, MDA-MB-231, and MCF-7 cells were from ATCC (LG Standards, Milan Italy). All media and supplements were from Sigma Aldrich (Milan, Italy) unless stated otherwise.
For mammosphere culture, T47D, BT-549, MDA-MB-231, and MCF-7cells were plated at a density of 1,000 cells/ml in non-adherent conditions. Cells were grown in serum-free DMEM-F12 supplemented with B27 (Life Technologies), 10 ng/ml EGF, 20 ng/ml β FGF (BD Biosciences, Milan, Italy), and 1X antibiotic-anti-mycotic (Life Technologies). After 5−7 days, mammospheres, which appeared as spheres of floating viable cells, were collected by gentle centrifugation (800 rpm) and dissociated with 0.25% trypsin for 5 min.
Transfection of cells and spheres
For transient transfection of miRNAs, cells at 50% confluence were transfected with either 100 nM pre-miR-24, scrambled pre-miR-24, or anti-miR-24 (Ambion, Life Technologies) in Oligofectamine (Thermo Fisher Scientific). To overexpress BimL, cells were transfected with 3 µg BimLcDNA, a kind gift of Dr. Douglas Green (St. Jude Children’s Research Hospital, Memphis, TN, USA), in Lipofectamine 2000 (Thermo Fisher Scientific). To overexpress FIH-1, cells were transfected with 3 μg FIH1 cDNA (OriGene Technologies, Rockville, USA) in Lipofectamine 2000.
Protein isolation and western blotting
Cells were washed twice in ice-cold PBS, and lysed in JS buffer (50 mM HEPES, pH 7.5, containing 150 mMNaCl, 1% glycerol, 1% Triton X100, 1.5mM MgCl2, 5mM EGTA, 1 mM Na3VO4, and 1X protease inhibitor cocktail). Protein concentration was determined by the Bradford assay (BioRad, Milan, Italy) using bovine serum albumin as the standard, and equal amounts of proteins were resolved by SDS-PAGE (12.5% acrylamide). Gels were electroblotted onto nitrocellulose membranes (G&E Healthcare, Milan, Italy), which were then blocked for 1h with 5% non-fat dry milk in Tris-buffered saline (TBS) containing 0.1% Tween-20, and incubated at 4°C overnight with a primary antibody. Detection was performed with peroxidase-conjugated secondary antibodies using the enhanced chemiluminescence system (Thermo, Euroclone, Milan, Italy). The primary antibodies used were: anti-Oct-3/4, anti-Nanog, anti-E-cadherin, anti-Vimentin (all from Santa Cruz Biotechnologies, MA, USA), anti-BimL, anti-PARP, anti-Caspase-3 (from Cell Signaling Technology, Milan, Italy), anti-human HIF1α (BD Biosciences, Milan, Italy), anti-FIH1(Abcam, MA, USA) and anti-βActin (Sigma Aldrich).
Mammosphere formation quantification
Mammospheres were resuspended in 0.5% agar (Bacto-Agar, Difco Laboratories, NSW, Australia) and layered onto 60 mm Petri dishes with a preformed 0.8% agar layer (BD Biosciences). Colonies were counted under an inverted microscope (Nikon, Milan, Italy) and then photographed.
RNA extraction and real-time PCR
Total RNAs (miRNA and mRNA) were extracted using Trizol (Thermo Fisher Scientific) according to the manufacturer’s protocol. Reverse transcription of total miRNA was performed using miScript reverse Transcription Kit (Qiagen, Milan, Italy), for mRNA was used SuperScript® III Reverse Transcriptase (Thermo Fisher Scientific). Quantitative analysis of Nanog, Oct-3/4, BimL, FIH1, VEGF, SNAIL, and Actin (as an internal reference), miR-24, and RNU6B (as an internal reference) was performed by RealTime PCR using specific primers (Qiagen), miScript SYBR Green PCR Kit (Qiagen), and iQTM SYBR Green Supermix (Bio-Rad), respectively.
Cell death and cell proliferation quantification
Cells were plated in 96-well plates in triplicate and incubated at 37°C in a 5% CO2 incubator. Cisplatin (Sigma Aldrich) was used for 48 h at 20 µg/ml. Apoptosis was analyzed with Caspase-Glo® 3/7 Assay Systems (Promega), according to the manufacturer’s protocol. Briefly, cells were incubated with medium supplemented with Caspase-3/7 reagent. Luminescence was measured following an incubation of 30 minutes at room temperature.
Rescue experiments
The effects of miR-24 were measured in the setting of overexpression of a BimL cDNA lacking the 3′UTR. Cells were transfected with miR-24 and with the BimL cDNA using Lipofectamine 2000. The effects of miR-24 were also measured under conditions of overexpression of a FIH1 cDNA lacking the 3′UTR.
Luciferase assay
The 3′UTR of the human FIH1 gene was PCR-amplified using Fw:5′GCTCTAGATGGGAAGGAGCATATGTCCT3′ and Rv:5′GCTCTAGATTTCAACATGCCTCCCTCCA3′ primers cloned downstream of the Renilla luciferase stop codon in a pGL3 control vector (Promega). An inverted sequence of the miRNA-binding sites was used as negative control. MCF-7 cells were co-transfected with 1.2 µg of plasmid and 400 µg of a Renilla luciferase expression construct, pRL-TK (Promega) with Lipofectamine 2000. Cells were harvested 24 h post-transfection and analyzed with the Dual Luciferase Assay (Promega), according to the manufacturer’s instructions. The experiments were performed in triplicate.
Hypoxic culture conditions
Hypoxic conditions were achieved using a Hypoxia Incubator Chamber (Stem Cell Technologies, Milan, Italy), which generates a 1% hypoxic environment for cell culture. MDA-MB-231, BT-549, MCF-7 and T47D cells were incubated in the chamber for 6h and then harvested for either RNA or protein extraction.
Migration assay
Transwells Permeable Support 8.0 μm polycarbonate membrane 6.5 mm Insert (Corning Incorporate, Corning, NY, USA) were used to carry out migration assay. MDA-MB-231 and BT-549 cells were grown as described above, then harvested by Trypsin-EDTA Solution (Sigma Aldrich, USA). 5x104 cells were washed with PBS, then resuspended in 1% fetal bovine serum containing RPMI 1640 medium and seeded in the upper chamber. The lower chamber of the transwell was filled with 600 μl of culture medium containing 10% fetal bovine serum. Cells were incubated at 37°C for 24h. The transwells were removed from the 24 wells plates and stained 0.1% Crystal Violet in 25% methanol. Non migrated cells were scraped off the top of the transwell with a cotton swab. Percentage of migrated cells was evaluated by eluting Crystal Violet with 1% SDS and reading the absorbance at λ 570nm.
ACKNOWLEDGMENTS
The authors thank Prof. Antonella Scorziello and Dr. Gaetano Iannello for technical advice during hypoxia experiments and Francesca De Micco for the patient samples recruitment.
CONFLICTS OF INTEREST
The authors disclose no potential conflicts of interest
GRANT SUPPORT
This work was partially supported by Associazione Italiana Ricerca sul Cancro, AIRC to G.C. IG 2016 Id. 18473; Fondazione Berlucchi to G.C.; the Plan Estatal of I+D+I 2013-2016 [PI14/01328] to M.V and co-financed by the ISCIII and FEDER Funds to M.V.
REFERENCES
1. Valent P, Bonnet D, De Maria R, Lapidot T, Copland M, Melo JV, Chomienne C, Ishikawa F, Schuringa JJ, Stassi G, Huntly B, Herrmann H, Soulier J, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nature reviews Cancer. 2012; 1211:767-775.
2. Czerwinska P and Kaminska B. Regulation of breast cancer stem cell features. Contemporary oncology. 2015; 19:A7-A15.
3. Brabletz T, Jung A, Spaderna S, Hlubek F and Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nature reviews Cancer. 2005; 59:744-749.
4. Li Z and Rich JN. Hypoxia and hypoxia inducible factors in cancer stem cell maintenance. Current topics in microbiology and immunology. 2010; 345:21-30.
5. Clarke L and van der Kooy D. Low oxygen enhances primitive and definitive neural stem cell colony formation by inhibiting distinct cell death pathways. Stem cells (Dayton, Ohio). 2009; 278:1879-1886.
6. Heddleston JM, Li Z, Lathia JD, Bao S, Hjelmeland AB and Rich JN. Hypoxia inducible factors in cancer stem cells. Br J Cancer. 2010; 1025:789-795.
7. Samanta D, Gilkes DM, Chaturvedi P, Xiang L and Semenza GL. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2014; 11150:E5429-5438.
8. Iriondo O, Rabano M, Domenici G, Carlevaris O, Lopez-Ruiz JA, Zabalza I, Berra E and Vivanco M. Distinct breast cancer stem/progenitor cell populations require either HIF1alpha or loss of PHD3 to expand under hypoxic conditions. Oncotarget. 2015; 631:31721-31739. doi: 10.18632/oncotarget.5564.
9. Liu C and Tang DG. MicroRNA regulation of cancer stem cells. Cancer research. 2003; 7118:5950-5954.
10. Garofalo M, Quintavalle C, Romano G, Croce CM and Condorelli G. miR221/222 in cancer: their role in tumor progression and response to therapy. Current molecular medicine. 2012; 121:27-33.
11. Quintavalle C, Garofalo M, Croce CM and Condorelli G. “ApoptomiRs” in vascular cells: their role in physiological and pathological angiogenesis. Vascular pharmacology. 2011; 554:87-91.
12. Garofalo M, Condorelli GL, Croce CM and Condorelli G. MicroRNAs as regulators of death receptors signaling. Cell death and differentiation. 2010; 172:200-208.
13. Houbaviy HB, Murray MF and Sharp PA. Embryonic stem cell-specific MicroRNAs. Developmental cell. 2003; 52:351-358.
14. Suh MR, Lee Y, Kim JY, Kim SK, Moon SH, Lee JY, Cha KY, Chung HM, Yoon HS, Moon SY, Kim VN and Kim KS. Human embryonic stem cells express a unique set of microRNAs. Developmental biology. 2004; 2702:488-498.
15. Tay Y, Zhang J, Thomson AM, Lim B and Rigoutsos I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature. 2008; 4557216:1124-1128.
16. Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, Guenther MG, Johnston WK, Wernig M, Newman J, Calabrese JM, Dennis LM, Volkert TL, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008; 1343:521-533.
17. Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y and Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nature cell biology. 2008; 105:593-601.
18. Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, Martin-Trevino R, Shang L, McDermott SP, Landis MD, Hong S, Adams A, D’Angelo R, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem cell reports. 2014; 21:78-91.
19. Huang HN, Chen SY, Hwang SM, Yu CC, Su MW, Mai W, Wang HW, Cheng WC, Schuyler SC, Ma N, Lu FL and Lu J. miR-200c and GATA binding protein 4 regulate human embryonic stem cell renewal and differentiation. Stem cell research. 2014; 122:338-353.
20. Chhabra R, Dubey R and Saini N. Cooperative and individualistic functions of the microRNAs in the miR-23a~27a~24-2 cluster and its implication in human diseases. Molecular cancer. 2010; 9:232.
21. Musto A, Navarra A, Vocca A, Gargiulo A, Minopoli G, Romano S, Romano MF, Russo T and Parisi S. miR-23a, miR-24 and miR-27a protect differentiating ESCs from BMP4-induced apoptosis. Cell death and differentiation. 2015; 226:1047-1057.
22. Roy L, Bikorimana E, Lapid D, Choi H, Nguyen T and Dahl R. MiR-24 is required for hematopoietic differentiation of mouse embryonic stem cells. PLoS genetics. 2015; 111:e1004959.
23. Amelio I, Lena AM, Viticchie G, Shalom-Feuerstein R, Terrinoni A, Dinsdale D, Russo G, Fortunato C, Bonanno E, Spagnoli LG, Aberdam D, Knight RA, Candi E, et al. miR-24 triggers epidermal differentiation by controlling actin adhesion and cell migration. J Cell Biol. 2012; 1992:347-363.
24. Amelio I, Lena AM, Bonanno E, Melino G and Candi E. miR-24 affects hair follicle morphogenesis targeting Tcf-3. Cell death & disease. 2013; 4:e922.
25. Roscigno G, Quintavalle C, Donnarumma E, Puoti I, Diaz-Lagares A, Iaboni M, Fiore D, Russo V, Todaro M, Romano G, Thomas R, Cortino G, Gaggianesi M, et al. MiR-221 promotes stemness of breast cancer cells by targeting DNMT3b. Oncotarget. 2016; 7:580-592. doi: 10.18632/oncotarget.5979.
26. Antonov AV, Knight RA, Melino G, Barlev NA and Tsvetkov PO. MIRUMIR: an online tool to test microRNAs as biomarkers to predict survival in cancer using multiple clinical data sets. Cell death and differentiation. 2013; 202:367.
27. Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J and Chang JC. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. Journal of the National Cancer Institute. 2008; 1009:672-679.
28. Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF and Srivastava D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. The Journal of experimental medicine. 2011; 2083:549-560.
29. Peng G and Liu Y. Hypoxia-inducible factors in cancer stem cells and inflammation. Trends in pharmacological sciences. 2015; 366:374-383.
30. Kulshreshtha R, Ferracin M, Wojcik SE, Garzon R, Alder H, Agosto-Perez FJ, Davuluri R, Liu CG, Croce CM, Negrini M, Calin GA and Ivan M. A microRNA signature of hypoxia. Molecular and cellular biology. 2007; 275:1859-1867.
31. Visvader JE and Stingl J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes & development. 2014; 2811:1143-1158.
32. Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, Bernard L, Viale G, Pelicci PG and Di Fiore PP. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010; 1401:62-73.
33. Vivanco M. Function follows form: defining mammary stem cells. Sci Transl Med. 2010; 231:31ps22.
34. Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, Fan C, Zhang X, He X, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proceedings of the National Academy of Sciences of the United States of America. 2009; 10633:13820-13825.
35. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006; 4447120:756-760.
36. Piva M, Domenici G, Iriondo O, Rabano M, Simoes BM, Comaills V, Barredo I, Lopez-Ruiz JA, Zabalza I, Kypta R and Vivanco M. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO molecular medicine. 2014; 61:66-79.
37. Simoes BM and Vivanco MD. Cancer stem cells in the human mammary gland and regulation of their differentiation by estrogen. Future Oncol. 2011; 78:995-1006.
38. He L and Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nature reviews Genetics. 2004; 57:522-531.
39. Garofalo M, Quintavalle C, Di Leva G, Zanca C, Romano G, Taccioli C, Liu CG, Croce CM and Condorelli G. MicroRNA signatures of TRAIL resistance in human non-small cell lung cancer. Oncogene. 2008; 2727:3845-3855.
40. Garofalo M, Leva GD and Croce CM. MicroRNAs as anti-cancer therapy. Current pharmaceutical design. 2014; 2033:5328-5335.
41. Calin GA and Croce CM. MicroRNA signatures in human cancers. Nature reviews Cancer. 2006; 611:857-866.
42. Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM and Rajewsky K. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes & development. 2005; 194:489-501.
43. Shcherbata HR, Hatfield S, Ward EJ, Reynolds S, Fischer KA and Ruohola-Baker H. The MicroRNA pathway plays a regulatory role in stem cell division. Cell cycle. 2006; 52:172-175.
44. Polytarchou C, Iliopoulos D and Struhl K. An integrated transcriptional regulatory circuit that reinforces the breast cancer stem cell state. Proceedings of the National Academy of Sciences of the United States of America. 2012; 10936:14470-14475.
45. Harris AL. Hypoxia--a key regulatory factor in tumour growth. Nature reviews Cancer. 2002; 21:38-47.
46. Axelson H, Fredlund E, Ovenberger M, Landberg G and Pahlman S. Hypoxia-induced dedifferentiation of tumor cells--a mechanism behind heterogeneity and aggressiveness of solid tumors. Seminars in cell & developmental biology. 2005; 164-5:554-563.
47. Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, Engh J, Iwama T, Kunisada T, Kassam AB, Pollack IF and Park DM. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene. 2009; 2845:3949-3959.
48. Keely S, Glover LE, Weissmueller T, MacManus CF, Fillon S, Fennimore B and Colgan SP. Hypoxia-inducible factor-dependent regulation of platelet-activating factor receptor as a route for gram-positive bacterial translocation across epithelia. Molecular biology of the cell. 2010; 214:538-546.
49. Coleman ML and Ratcliffe PJ. Signalling cross talk of the HIF system: involvement of the FIH protein. Current pharmaceutical design. 2009; 1533:3904-3907.
50. Mahon PC, Hirota K and Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes & development. 2001; 1520:2675-2686.
51. Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML and Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes & development. 2002; 1612:1466-1471.
52. Lando D, Peet DJ, Whelan DA, Gorman JJ and Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002; 2955556:858-861.
53. Umezu T, Tadokoro H, Azuma K, Yoshizawa S, Ohyashiki K and Ohyashiki JH. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood. 2014; 12425:3748-3757.
54. Peng H, Kaplan N, Hamanaka RB, Katsnelson J, Blatt H, Yang W, Hao L, Bryar PJ, Johnson RS, Getsios S, Chandel NS and Lavker RM. microRNA-31/factor-inhibiting hypoxia-inducible factor 1 nexus regulates keratinocyte differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2012; 10935:14030-14034.
55. Bigner SH and Vogelstein B. Cytogenetics and molecular genetics of malignant gliomas and medulloblastoma. Brain pathology. 1990; 11:12-18.
56. Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. Journal of applied physiology. 2000; 884:1474-1480.
57. Mi S, Lu J, Sun M, Li Z, Zhang H, Neilly MB, Wang Y, Qian Z, Jin J, Zhang Y, Bohlander SK, Le Beau MM, Larson RA, et al. MicroRNA expression signatures accurately discriminate acute lymphoblastic leukemia from acute myeloid leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2007; 10450:19971-19976.
58. Undi RB, Kandi R and Gutti RK. MicroRNAs as Haematopoiesis Regulators. Adv Hematol. 2013; 2013:695754.
59. Lu K, Wang J, Song Y, Zhao S, Liu H, Tang D, Pan B, Zhao H and Zhang Q. miRNA-24-3p promotes cell proliferation and inhibits apoptosis in human breast cancer by targeting p27Kip1. Oncology reports. 2015; 342:995-1002.
60. Lee EJ, Gusev Y, Jiang J, Nuovo GJ, Lerner MR, Frankel WL, Morgan DL, Postier RG, Brackett DJ and Schmittgen TD. Expression profiling identifies microRNA signature in pancreatic cancer. International journal of cancer. 2007; 1205:1046-1054.
61. Salvi A, Abeni E, Portolani N, Barlati S and De Petro G. Human hepatocellular carcinoma cell-specific miRNAs reveal the differential expression of miR-24 and miR-27a in cirrhotic/non-cirrhotic HCC. International journal of oncology. 2013; 422:391-402.
62. Cheng AM, Byrom MW, Shelton J and Ford LP. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic acids research. 2005; 334:1290-1297.
63. Chen Q, Xu J, Li L, Li H, Mao S, Zhang F, Zen K, Zhang CY and Zhang Q. MicroRNA-23a/b and microRNA-27a/b suppress Apaf-1 protein and alleviate hypoxia-induced neuronal apoptosis. Cell death & disease. 2014; 5:e1132.
64. Qin W, Shi Y, Zhao B, Yao C, Jin L, Ma J and Jin Y. miR-24 regulates apoptosis by targeting the open reading frame (ORF) region of FAF1 in cancer cells. PLoS One. 2010; 52:e9429.
65. Bueno MJ and Malumbres M. MicroRNAs and the cell cycle. Biochimica et biophysica acta. 2011; 18125:592-601.
66. Xie Y, Tobin LA, Camps J, Wangsa D, Yang J, Rao M, Witasp E, Awad KS, Yoo N, Ried T and Kwong KF. MicroRNA-24 regulates XIAP to reduce the apoptosis threshold in cancer cells. Oncogene. 2013; 3219:2442-2451.
67. Wang Q, Huang Z, Xue H, Jin C, Ju XL, Han JD and Chen YG. MicroRNA miR-24 inhibits erythropoiesis by targeting activin type I receptor ALK4. Blood. 2008; 1112:588-595.
68. Pan B, Chen Y, Song H, Xu Y, Wang R and Chen L. Mir-24-3p downregulation contributes to VP16-DDP resistance in small-cell lung cancer by targeting ATG4A. Oncotarget. 2015; 6:317-331. doi: 10.18632/oncotarget.2787.
69. Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA and Daidone MG. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer research. 2005; 6513:5506-5511.