
INTRODUCTION
Müllerian duct abnormality (MDA) is a complex and serious female reproductive tract malformation, which bring serious physical and psychological impact on patients [1]. Congenital absence of the uterus and vagina (CAUV), also known as Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome, is the most extreme MDA, characterized by the congenital aplasia of the uterus and the upper part of the vagina in women with normal ovaries, karyotypes and secondary sexual characteristics [2, 3]. It is the second most frequent cause of primary amenorrhea after Turner syndrome in females, and the estimated prevalence of the disease was about 1 in 4000 to 5000 female newborns [4]. Approximately 50% of women with CAUV have concomitant congenital cardiac, renal, skeletal and hearing abnormalities [3]. Notably, most of the reported patients are sporadic while limited familial cases suggested the disease was transmitted as an autosomal recessive inheritance with reduced penetrance and variable expressivity or a polygenic and multifactorial pathogenesis [3].
The underlying etiology of CAUV remains unclear to date. Though the genetic basis has been studied, limited informative CAUV-affected families prevented the identification of any loci through conventional genetic linkage analysis [5]. Previous genetic studies based on targeted mutagenesis in the mouse delineated the genes that involved in initial Müllerian duct formation [6]. Candidate genes can be classified into three categories according to their involvement in the initial formation, differentiation, or regression of Müllerian duct, respectively, which include members of the HOXA, WNT gene families, Emx2, Pax2, AMH (AMHR2) and LHX1 [7]. However, most mutations found in these candidate genes frequently showed contradicting evidence of replications. What’s more, hardly any candidate genes or mutations were verified by functional analysis to enhance the understanding of how and why they influenced the development of Müllerian ducts.
By performing whole exome sequencing (WES) on a cohort of Chinese Han CAUV patients, we identified a novel missense mutation in LHX1 as the prioritized causative gene of the malformation. Functional experiments confirmed our inference by showing the mutation could change the transcriptional activity of LHX1. This project might provide a new potential association between CAUV and LHX1.
RESULTS
Identification of a new LHX1 variant associated with CAUV
By performing whole-exome sequencing, quality control statistics, strict filtering criteria and bioinformatic analysis in a cohort of CAUV patients, we identified a novel heterozygous missense variant in the LHX1 gene (NM_005568: c.G1108A, p.A370T) in one patient. The patient was admitted to hospital due to primary amenorrhea at the age of 21. Clinical examinations using a transvaginal ultrasonography, hysteroscopy, laparoscopy showed the absence of the uterus and the upper part of vagina. She had normal kidney and 46XX karyotype. Candidate variants were prioritized based on their mutational effects, animal models and functions of affected genes. LHX1 is the only candidate gene that has been associated with CAUV in human with recurrent mutations.
The variant was absent from public databases (1000Genomes, ESP6500si, ExAC) and our internal database (98 MDA patients sequencing data). In silico analysis predicated that this variant would have a function in disease causing. The mutated amino acid at this site was highly conserved among different species, including human, chimpanzee, mouse, cattle and African clawed frog (Figure 1A), aligned by the CLC Free Workbench 4 software. The variant was confirmed by Sanger sequencing (Figure 1B).
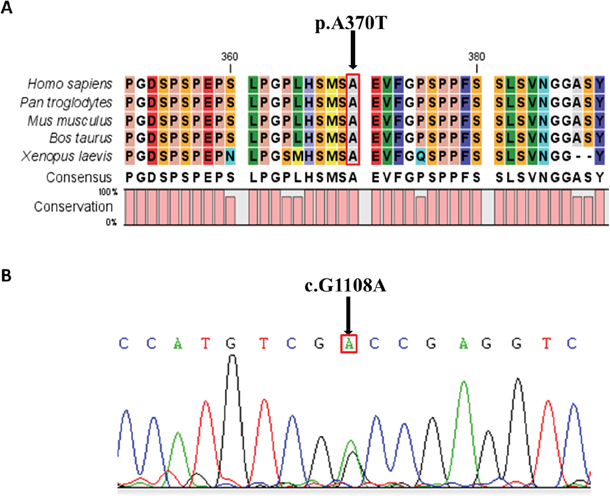
Figure 1: Conservative Prediction and Sequencing Validation. A. Alignments of the amino acid sequence of part of LHX1 protein among different species. The mutation indicated by the arrow is in the conserved region of all the species shown (human, chimpanzee, mouse, cattle, African clawed frog); B. Validation of p.Ala370Thr (c.G1108A) heterozygous variant. Sanger sequencing results are shown.
The variant decrease the transcriptional and biological activity of LHX1
Luciferase reporter assay was used to evaluate whether the mutation affect the transcriptional or biological activity of LHX1. As the Figure 2A showed, both wild-type and mutant pCMX-Gal4-DBD-CT266 plasmids led to a significant increase in luciferase activity compared with pCMX-Gal4 empty vector (T test, p< 0.01; p< 0.05). However, the mutant group showed weakened luciferase activity compared with wild-type (T test, p< 0.05). These findings indicated that the mutation may have an inhibitory effect on the transcriptional activity of LHX1.
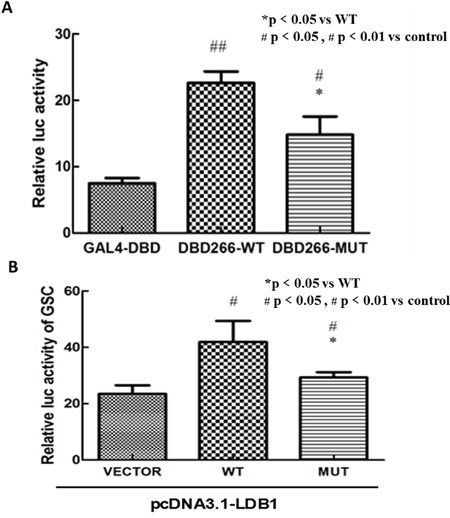
Figure 2: In vitro luciferase reporter assay. A. Both wild-type and mutant pCMX-Gal4-DBD-CT266 plasmids had a significant increase in luciferase activity compared with pCMX-Gal4-DBD vector (p value<0.01, p value<0.05). The vector carrying this mutation showed a weakened luciferase activity compared with wild-type (p value<0.05). B. The luciferase activity of GSC-promoter reporter plasmid was significantly decreased in mutant pcDNA3.1-LHX1 group compared with wild group. Note: The results represent the mean (±SD) of 3 determinations in three independent experiments.
Using similar methods, we detected whether the variant affect the gene’s ability of regulation of GSC transcription cooperate with LDB1. It turned out that the luciferase activity of GSC-promoter reporter plasmid was significantly decreased in mutant LHX1 group compared with wild group (Figure 2B). These findings also indicated that the mutation may have an inhibitory effect on the ability of regulation of GSC transcription.
DISCUSSION
LHX1, also known as LIM1, encodes a LIM-class homeodomain transcription factor that is essential for kidney and urogenital systems development [8]. Female LHX1-null mice present with normal ovaries but lack uterus, oviducts and the upper region of the vagina. Several human studies also confirmed its essential role in the development of the urogenital system [9, 10]. The recurrent 17q12 copy number losses was observed in 5~6% CAUV patients, which deleted one copy of LHX1 genes [11, 12]. Sandbacka et al. supported the relevance of LHX1 and copy number variations (CNVs) in the development of congenital malformation [13]. One missense single nucleotide variation (SNV) and a frameshift insertion of LHX1 were found in two CAUV patients [7, 11]. From all of these researches, there is strong evidence that the haplo-insufficiency of LHX1 is associated with CAUV.
The variant p.Ala370Thr we found in one CAUV patient was located in the transcriptional activation domain of carboxyl-terminal region of LHX1. In vitro luciferase reporter analysis indicated that the variant reduced the transcriptional activity of LHX1 compared with the wild type. It was consistent with the findings that the amino acids region 266-403 of LHX1, in addition to homeodomains (178-238), is important for its transcriptional activation and interactions with other proteins [14]. GSC is a direct target gene for protein LHX1 in the organizer and the prechordal plate of Xenopus [15]. Furthermore, it has been confirmed that microdeletion and homozygous predicted null mutations of GSC were involved in SAMS syndrome, which was characterized by unique rhizomelic skeletal anomalies, cryptorchidism (male), hypoplastic labia (female) and other urogenital anomalies [16]. So, we surmised that LHX1 might cooperate with GSC to affect the development of urogenital system. Our results were consistent with this prediction as the activity of GSC-promoter reporter was significantly decreased in mutant LHX1 group, cooperated with an important LIM-domain binding protein LDB1.
However, LHX1 was previously sequenced in 96 Chinese MDA patients reported by Xia et al., but they did not find any missense or nonsense mutations [17]. Similarly, LHX1 was also screened in 56 Caucasian MRKH syndrome patients and no mutation was identified [11]. Furthermore, in our project, rare missense SNV in LHX1 did not seem to be enriched in 98 typical MDA patients. These findings illustrated that variants in LHX1 may not be common genetic etiologic factors involved in female reproductive tract development malformations. More other pathogenic genes need to be found.
In summary, this is the first study to explore the pathogenic gene of CAUV by whole exome sequencing. In addition, we preliminary proposed the possible pathogenic mechanism of LHX1 in CAUV, which provided a new evidence for the follow-up research, prevention and treatment of the disease. However, we hold the opinion that more systematic and intensive experiments are needed to explore the precise mechanism of the influence of LHX1 on CAUV.
MATERIALS AND METHODS
Participants
This study included 10 Chinese Han unrelated CAUV patients that were recruited from Peking Union Medical College Hospital and the Fist Affiliated Hospital of Anhui Medical University. CAUV was diagnosed by experienced experts of gynecology and obstetrics according to medical history, results of karyotype analysis, androgen levels, and clinical examination using a transvaginal ultrasonography, hysteroscopy, laparoscopy and hysterosalpingogram.
This research was approved by the ethical committee of National Research Institute for Family Planning. The methods and experiments were carried out in accordance with approved guidelines. Informed written consents were obtained from all the participants.
Whole-exome sequencing and validation
We sequenced exomes of the ten unrelated CAUV patients. All exons were captured by NimbleGen Human All Exon Enrichment kit and then massively parallel sequenced on Illumina Hiseq 2500 platform. We used BWA v0.5.9 [18] to align reads to reference human genome (GRC build 37), Picardtools to mark and remove duplicates, Samtools to detect SNPs and indels, and ANNOVAR [19] to annotate all variants. We only focused on functional variants (missense, nonsense, splicing site and frame-shift) with a minor allele frequency (MAF) less than 1% in public and in House databases. Frequency-filtered variants were then predicted in silico to assess their impact on the function and structure of encoded proteins. The analysis process referred to the article we published before [20]. The candidate gene detected by whole-exome sequencing was validated by Sanger sequencing.
Site-directed mutagenesis and plasmids construction
The full CDS sequence of human LHX1 (1221bp, 406 amino acids, NM_005568) was amplified by polymerase chain reaction (PCR) from cDNA. The variant p.A370T was constructed by site-directed mutagenesis using the Quick Change II Site-Directed Mutagenesis kit (Agilent Technologies, USA).
The wild and mutant truncated open reading frame (ORF) of human LHX1, CT266, were also amplified by PCR and inserted respectively into the pCMX-Gal4 vector [14]. The pCMX-Gal4 vector containing GAL4-DBD were provided by Dr. Ronald M. Evans (Salk Institute for Biological Studies, USA).
The wild and mutant LHX1 were inserted respectively into the pcDNA3.1 vector (Invitrogen, Carlsbad, USA) to create the expression plasmid pcDNA3.1-LHX1. LDB1 (NM_003893.4) was subcloned into the pcDNA3.1 vector as well. The promoter sequence of goosecoid homeobox (GSC) was amplified based on (http://switchdb.switchgeargenomics.com/productinfo/id_704725/), and then inserted into the luciferase reporter PLG3-basic vector.
Transcriptional activity analysis and in vitro luciferase reporter assay
HEK 293 cells were seeded in 24-well tissue culture plates 18h prior to transfection (Lipofectamine 2000) at approximately 80% confluency. Cells were co-transfected with wild-type or mutant pCMX-Gal4-DBD-CT266 plasmids containing LHX1 truncated ORF (300 ng/well) and the TK-promoter reporter plasmids (500 ng/well). The pCMX-Gal4-DBD vector was used as negative control. pREP7-RLu plasmid containing the Renilla luciferase gene cotransfection medium (10 ng/well) was used as an internal control. Thirty hours after transfection, cells were lysed with Passive Lysis Buffer (Promega, USA) and the Dual-luciferase Reporter Assay System (Promega) was used to assay the luciferase activity. Luminescence in relative light units (RLU) was measured for 10 s by luminescence detector (Promega). Firefly luciferase activity was first normalized to the level of Renilla luciferase activity. Results were then calculated as fold induction relative to pCMX-Gal4-DBD-CT266 wild-type vector.
HEK 293 cells were seeded in 24-well tissue culture plates as well. Luciferase assays plasmids including pcDNA3.1-LDB1 (200 ng/well), pGL3-GSC promoter reporter vector (300 ng/well), and pcDNA3.1-LHX1 (wild-type or mutant) or pcDNA3.1 empty vector (200 ng/well) were co-transfected into cells. Thirty hours after transfection, cells were treated the same way as above. Results were eventually calculated as fold induction relative to pcDNA3.1-LHX1 wild-type vector as well.
Three independent experiments were performed in triplicate. The independent samples T test was adopted to determine statistical significance of unpaired samples. All data were analyzed by Excel (Microsoft 2010) and GraphPad Prism 5.
Abbreviations
CAUV, Congenital absence of the uterus and vagina; WES, whole-exome sequencing; MDA, Müllerian duct abnormality; SNV, single nucleotide variation; MAF, minor allele frequency; PCR, polymerase chain reaction; ORF, open reading frame.
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (81370691), the National Basic Research Program of China (2012CB944704) and Natural science research project of Anhui Province (KJ2013Z140).
CONFLICTS OF INTEREST
The authors have no conflicts of interest relevant to this article to disclose.
REFERENCES
1. Rackow BW, Arici A. Reproductive performance of women with mullerian anomalies. Curr Opin Obstet Gynecol. 2007; 19:229-237.
2. Lalwani S, Wu HH, Reindollar RH, Gray MR. HOXA10 mutations in congenital absence of uterus and vagina. Fertility and sterility. 2008; 89:325-330.
3. Klipstein S, Bhagavath B, Topipat C, Sasur L, Reindollar RH, Gray MR. The N314D polymorphism of the GALT gene is not associated with congenital absence of the uterus and vagina. Molecular human reproduction. 2003; 9:171-174.
4. Londra L, Chuong FS, Kolp L. Mayer-Rokitansky-Kuster-Hauser syndrome: a review. Int J Womens Health. 2015; 7:865-870.
5. Resendes BL, Sohn SH, Stelling JR, Tineo R, Davis AJ, Gray MR, Reindollar RH. Role for anti-Mullerian hormone in congenital absence of the uterus and vagina. Am J Med Genet. 2001; 98:129-136.
6. Guerrier D, Mouchel T, Pasquier L, Pellerin I. The Mayer-Rokitansky-Kuster-Hauser syndrome (congenital absence of uterus and vagina)--phenotypic manifestations and genetic approaches. J Negat Results Biomed. 2006; 5:1.
7. Ledig S, Brucker S, Barresi G, Schomburg J, Rall K, Wieacker P. Frame shift mutation of LHX1 is associated with Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome. Hum Reprod. 2012; 27:2872-2875.
8. Kobayashi A, Shawlot W, Kania A, Behringer RR. Requirement of Lim1 for female reproductive tract development. Development. 2004; 131:539-549.
9. Cheroki C, Krepischi-Santos AC, Szuhai K, Brenner V, Kim CA, Otto PA, Rosenberg C. Genomic imbalances associated with mullerian aplasia. J Med Genet. 2008; 45:228-232.
10. Bernardini L, Gimelli S, Gervasini C, Carella M, Baban A, Frontino G, Barbano G, Divizia MT, Fedele L, Novelli A, Bena F, Lalatta F, Miozzo M, Dallapiccola B. Recurrent microdeletion at 17q12 as a cause of Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: two case reports. Orphanet J Rare Dis. 2009; 4:25.
11. Ledig S, Schippert C, Strick R, Beckmann MW, Oppelt PG, Wieacker P. Recurrent aberrations identified by array-CGH in patients with Mayer-Rokitansky-Kuster-Hauser syndrome. Fertil Steril. 2011; 95:1589-1594.
12. Nik-Zainal S, Strick R, Storer M, Huang N, Rad R, Willatt L, Fitzgerald T, Martin V, Sandford R, Carter NP, Janecke AR, Renner SP, Oppelt PG, Oppelt P, Schulze C, Brucker S, et al. High incidence of recurrent copy number variants in patients with isolated and syndromic Mullerian aplasia. J Med Genet. 2011; 48:197-204.
13. Sandbacka M, Laivuori H, Freitas E, Halttunen M, Jokimaa V, Morin-Papunen L, Rosenberg C, Aittomaki K. TBX6, LHX1 and copy number variations in the complex genetics of Mullerian aplasia. Orphanet J Rare Dis. 2013; 8:125.
14. Breen JJ, Agulnick AD, Westphal H, Dawid IB. Interactions between LIM domains and the LIM domain-binding protein Ldb1. J Biol Chem. 1998; 273:4712-4717.
15. Mochizuki T, Karavanov AA, Curtiss PE, Ault KT, Sugimoto N, Watabe T, Shiokawa K, Jamrich M, Cho KW, Dawid IB, Taira M. Xlim-1 and LIM domain binding protein 1 cooperate with various transcription factors in the regulation of the goosecoid promoter. Dev Biol. 2000; 224:470-485.
16. Parry DA, Logan CV, Stegmann AP, Abdelhamed ZA, Calder A, Khan S, Bonthron DT, Clowes V, Sheridan E, Ghali N, Chudley AE, Dobbie A, Stumpel CT, Johnson CA. SAMS, a syndrome of short stature, auditory-canal atresia, mandibular hypoplasia, and skeletal abnormalities is a unique neurocristopathy caused by mutations in Goosecoid. Am J Hum Genet. 2013; 93:1135-1142.
17. Xia M, Zhao H, Qin Y, Mu Y, Wang J, Bian Y, Ma J, Chen ZJ. LHX1 mutation screening in 96 patients with mullerian duct abnormalities. Fertil Steril. 2012; 97:682-685.
18. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754-1760.
19. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010; 38:e164.
20. Chen H, Zhou X, Wang J, Wang X, Liu L, Wu S, Li T, Chen S, Yang J, Sham PC, Zhu G, Zhang X, Wang B. Exome Sequencing and Gene Prioritization Correct Misdiagnosis in a Chinese Kindred with Familial Amyloid Polyneuropathy. Sci Rep. 2016; 6:26362.