
INTRODUCTION
Osteoblasts are important for the bone formation and remodeling [1, 2]. Yet, these mesenchymal progenitor cells-derived cells are also the main target cells of oxidative stress [1, 2, 5]. Increased reactive oxygen species (ROS) production will lead to oxidative stress, causing osteoblast cell damage and apoptosis [6, 7]. Hydrogen peroxide (H2O2) is often added to cultured osteoblasts to establish a cellular model of osteonecrosis [8–11]. For many years, our group [12–15] has been focusing on indentifying novel molecular targets to promote osteoblast cell survival.
AMP-activated protein kinase (AMPK) is a master regulator of cellular metabolism and energy [16]. It plays a pivotal function in maintaining cell energy balance [16]. Existing studies have suggested that AMPK activation could also promote cell survival [17]. Recent literatures investigated the potential functions of AMPK in osteoblasts, and demonstrated that activating AMPK, either genetically or pharmacologically, could protect osteoblasts from oxidative stress and dexamethasone [12, 18–20]. Therefore, AMPK is a valuable pro-survival target at least in osteoblasts [12, 18–20].
Multiple AMPK activators of different mechanisms of actions have been developed thus far, many of them activate AMPK though increasing the AMP:ATP ratio, such as AICAR [21, 22]. Others, however, provoke AMPK activation by directly inducing AMPKα1 phosphorylation at Thr-172, i.e. Compound 13 [21–24]. Recent studies have developed GSK621 as a novel AMPK activator [25]. Its potential activity in osteoblasts has not been tested thus far. In this study, we show that GSK621 activates AMPK signaling and potentially inhibits H2O2-induced oxidative damages in cultured osteoblasts.
RESULTS
GSK621 protects osteoblasts from H2O2
The current study aims to understand the potential effect of GSK621 on oxidative-stressed osteoblasts. CCK-8 viability results in Figure 1A demonstrated that H2O2 (250 μM, 24 hours) treatment in MC3T3-E1 osteoblastic cells [15] induced over 50% cell viability reduction. Significantly, co-treatment with GSK621 at 2.5-25 μM dramatically attenuated H2O2-induced MC3T3-E1 cell viability reduction (Figure 1A). LDH release results in Figure 1B confirmed H2O2 (250 μM)-induced MC3T3-E1 cell death, which was again largely attenuated with co-treatment of GSK621 (2.5-25 μM). Meanwhile, H2O2 (250 μM)-induced MC3T3-E1 cell apoptosis, tested by Histone DNA ELISA assay [12, 13], was also significantly alleviated by GSK621 co-treatment (Figure 1C). The anti-H2O2 activity of GSK621 in MC3T3-E1 cells was dose-dependent (Figure 1A-1C). At a low concentration (1 μM), GSK621 was invalid to inhibit H2O2 damages (Figure 1A-1C). Notably, treatment with GSK621 alone at tested concentrations failed to induce survival change (Figure 1D) and apoptosis (Data not shown) in MC3T3-E1 cells.
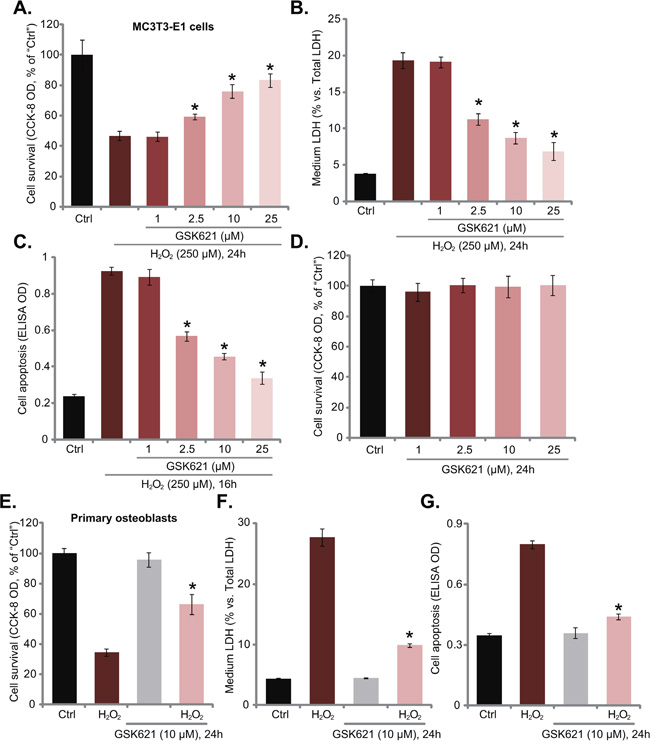
Figure 1: GSK621 protects osteoblasts from H2O2. MC3T3-E1 osteoblastic cells A-D. or the primary murine osteoblasts E-G. were treated with hydrogen peroxide (“H2O2”, 250 μM) with/out GSK621 at applied concentration, cells were then cultured for additional 16/24 hours; Cell survival (CCK-8 assay, A, D and E), cell death (LDH release assay, B and F) and apoptosis (Histone DNA ELISA assay, C and G) were tested. Data are shown as mean (n=5) ± standard deviation (SD). “Ctrl” stands for medium treatment control (Same for all figures). Experiments in this figure were repeated for three times, and similar results were obtained. *p<0.05 vs. H2O2 only group.
Using the methods described [12–15], we also established primary murine osteoblasts. H2O2 (250 μM) treatment in these primary cells also induced viability reduction (Figure 1E), cell death (Figure 1F) and apoptosis (Figure 1G). Remarkably, GSK621 (10 μM) co-administration significantly alleviated H2O2-induced damages of the primary osteoblasts (Figure 1E-1G). GSK621 (10 μM) alone again didn’t affect survival and apoptosis of the primary cells (Figure 1E-1G). These results show that GSK621 indeed protects osteoblasts from H2O2.
GSK621-mediated osteoblast cytoprotection requires AMPK activation
GSK621 is a newly-developed AMPK activator [25–27], we therefore tested AMPK signaling in GSK621-treated cells. As shown in Figure 3A, treatment with GSK621 (at 2.5-25 μM, 2 hours) in MC3T3-E1 cells induced significant AMPK activation, which was tested by phosphorylation (“p”) of AMPKα1 (Thr-172) and its major downstream target protein ACC (acetyl-CoA carboxylase, Ser-79) [12]. Expression of total AMPKα1 and ACC was not changed following the GSK621 treatment (Figure 3A). To study the link between GSK621-induced AMPK activation and osteoblast cytoprotection, shRNA strategy [12] was applied to silence AMPK signaling. In the study, a total of three different lentiviral AMPKα1 shRNAs (“Seq-1/-2/-3”) were designed (See Methods), and each of them potently downregulated AMPKα1 in MC3T3-E1 cells (Figure 2B). Consequently, GSK621-induced AMPK activation, or p-AMPKα1/p-ACC, was almost abolished in AMPKα1-silenced cells (Figure 2B). Importantly, although the AMPKα1 shRNAs alone didn’t affect MC3T3-E1 cell survival (Figure 2C), they almost abolished GSK621-mediated osteoblast cytoprotection against H2O2 (250 μM) (Figure 2D and 2E). In another words, GSK621 was pretty much invalid against H2O2 when AMPKα1 was silenced (Figure 2D and 2E).
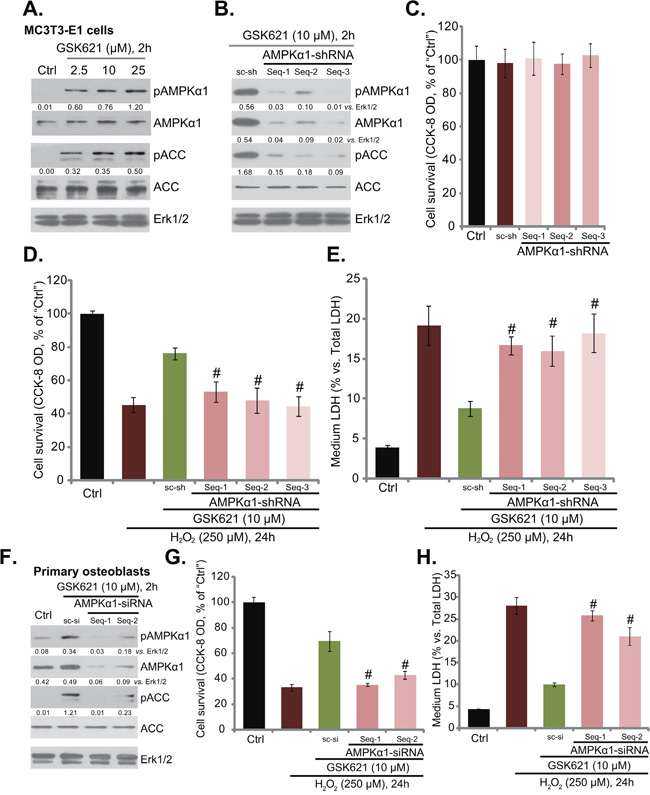
Figure 2: GSK621-mediated osteoblast cytoprotection requires AMPK activation. MC3T3-E1 cells were treated with GSK621 at applied concentrations for 2 hours, expression of listed proteins was shown A. MC3T3-E1 cells, expressing listed lentiviral AMPKα1 shRNA (“Seq-1/-2/3”) or scramble control shRNA (“sc-sh”), were subjected to Western blotting assay of listed proteins B.; These cells were also treated with/out H2O2 (250 μM) or plus GSK621 (10 μM) for 24 hours; Cell viability (CCK-8 assay, C. and D.) and cell death (LDH release assay, E. were tested. Primary murine osteoblasts, transfected with indicated AMPKα1 siRNA (“Seq-1/-2”) or scramble control siRNA (“sc-si”), were treated with GSK621 (10 μM) for 2 hours, expression of listed proteins was tested F. Cells were also stimulated with H2O2 (250 μM) for 24 hours, and cell survival G. and cell death H. were tested. AMPKα1 (“p-” and total) or p-ACC were quantified. Data are shown as mean (n=6) ± SD. Experiments in this figure were repeated for three times, and similar results were obtained. #p<0.05 vs. “sc-sh”/“sc-si” group.
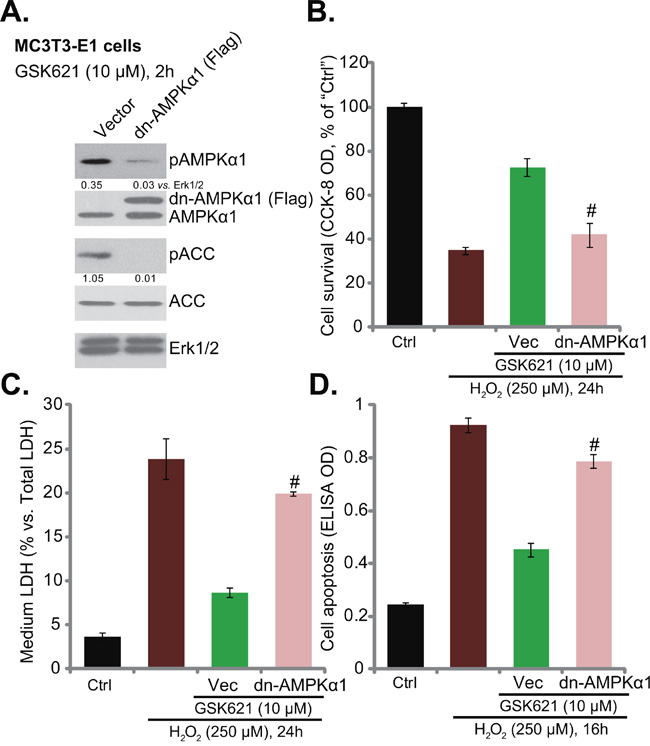
Figure 3: AMPKα1 dominant negative mutation abolishes GSK621-induced osteoblast cytoprotection. Neomycin-selected stable MC3T3-E1 cells expressing dominant negative AMPKα1 (T172A) construct (“dn-AMPKα1”, Flag-tagged) or the empty vector (pSuper-Flag, “Vec”) were treated with GSK621 (10 μM) for 2 hours, expression of listed proteins was tested A. Cells were also subjected to H2O2 (250 μM) stimulation; Cell survival (CCK-8 assay, B. Cell death (LDH release assay, C. and apoptosis (Histone-DNA ELISA assay, D. were tested. Data are shown as mean (n=4) ± SD. p-AMPKα1 and p-ACC were quantified. Experiments in this figure were repeated for three times, and similar results were obtained. #p<0.05 vs. “Vec” group.
In the primary murine osteoblasts, two non-overlapping AMPKα1 siRNAs (“Seq-1/-2”) were utilized to knockdown AMPKα1. As demonstrated, the two applied siRNAs knocked down AMPKα1 in the primary cells (Figure 2F). GSK621 (10 μM, 2 hours)-induced AMPK activation, or p-AMPKα1/p-ACC, was also dramatically inhibited with AMPKα1 siRNA knockdown (Figure 2F). Similarly, GSK621 was also largely ineffective when AMPKα1 was silenced (Figure 2G and 2H). Seq-1 AMPKα1 siRNA was slightly more efficient than Seq-2 siRNA in downregulating AMPKα1 (Figure 2F). It was also more efficient in shutting down GSK621-mediated cytoprotection (Figure 2G and 2H). Together, these results indicate that AMPK activation is required for GSK621-induced osteoblast cytoprotection against H2O2.
AMPKα1 dominant negative mutation abolishes GSK621-induced osteoblast cytoprotection
To further confirm the requirement of AMPK activation in GSK621-induced actions in osteoblasts, a dominant negative AMPKα1 (T172A) construct (“dn-AMPKα1”, Flag-tagged) [12] was utilized. The mutant AMPKα1 was introduced to MC3T3-E1 cells. Via neomycin selection, stable MC3T3-E1 cells with the mutant AMPKα1 were established. Western blotting assay results in Figure 3A confirmed dn-AMPKα1 expression in the stable cells. Further, GSK621 (10 μM, 2 hours)-induced AMPK activation (p-AMPKα1/p-ACC) was almost abolished in dn-AMPKα1-expressing cells (Figure 3A). More importantly, GSK621-induced osteoblast protection against H2O2 was also largely inhibited with AMPKα1 mutation (Figure 3B-3D). GSK621-induced pro-survival (Figure 3B), anti-death (Figure 3C) and anti-apoptosis (Figure 3D) actions in H2O2-treated cells were significantly attenuated. These results again confirm that AMPK activation is required for GSK621-mediated osteoblast cytoprotection against H2O2.
Forced-activation of AMPK protects osteoblasts from H2O2, taking over GSK621’s actions
Next, a constitutively-active AMPKα1 (T172D) construct (“ca-AMPKα1”, Flag-tagged) [12] was introduced to MC3T3-E1 cells, and stable cell line with the ca-AMPKα1 was selected by puromycin. Western blotting results in Figure 4A confirmed expression of ca-AMPKα1 in the stable cells. As expected, AMPK was constitutively active in these cells, and p-AMPKα1 and p-ACC level was high (Figure 4A). Compared to the vector control MC3T3-E1 cells, the ca-AMPKα1-expressing cells were protected from H2O2, presenting with significantly less viability reduction (Figure 4B) and cell death (Figure 4C) after H2O2 treatment. Significantly, in ca-AMPKα1-expressing MC3T3-E1 cells, GSK621 (10 μM) could not further protect cells from H2O2 (Figure 4B and 4C). Thus, ca-AMPKα1 expression took over GSK621’s actions and inhibited H2O2 damages in MC3T3-E1 cells. These results against indicate that activation of AMPK is required for GSK621-mediated osteoblast cytoprotection.
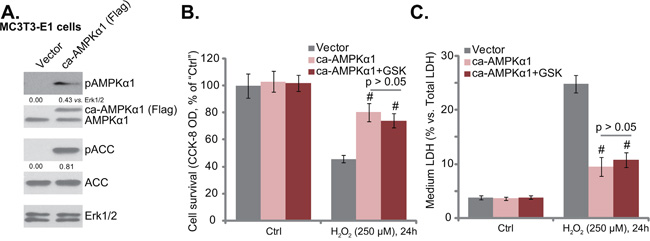
Figure 4: Forced-activation of AMPK protects osteoblasts from H2O2, taking over GSK621’s actions. Puromycin-selected stable MC3T3-E1 cells, expressing the constitutively-active AMPKα1 (T172D) construct (“ca-AMPKα1”, Flag-tagged) or the empty vector (pSuper-Flag, “Vector”), were subjected to Western blotting assay of listed proteins A. Above cells were also treated with H2O2 (250 μM) or plus GSK621 (10 μM) for 24 hours; Cell survival (CCK-8 assay, B. and cell death (LDH release assay, C. were tested. Data are shown as mean (n=4) ± SD. p-AMPKα1 and p-ACC were quantified. Experiments in this figure were repeated for four times, and similar results were obtained. #p<0.05 vs. “Vector” group.
GSK621 increases NAPDH content and inhibits H2O2-induced oxidative stress
Recent studies have proposed an anti-oxidant function of AMPK under many stress conditions. Activated AMPK may increase nicotinamide adenine dinucleotide phosphate (NADPH) content to inhibit ROS production and accumulation [19, 20, 28, 29]. Here, GSK621 treatment also increased NADPH level in MC3T3-E1 cells (Figure 5A), which was blocked by AMPKα1 shRNA knockdown or dominant negative mutation (Figure 5A). Intriguingly, H2O2-induced ROS production was also largely inhibited with co-treatment of GSK621 (Figure 5B). More importantly, the anti-oxidant activity by GSK621 was nullified with AMPKα1 shRNA or dominant negative mutation (Figure 5B). Therefore, GSK621 inhibits H2O2-induced ROS production in an AMPK-dependent manner. In the primary murine osteoblasts, GSK621 (10 μM) similarly increased NAPDH content (Figure 5C) and inhibited H2O2-induced ROS production (Figure 5C). These results show that GSK621 increases NAPDH content and inhibits H2O2-induced oxidative stress in osteoblasts.
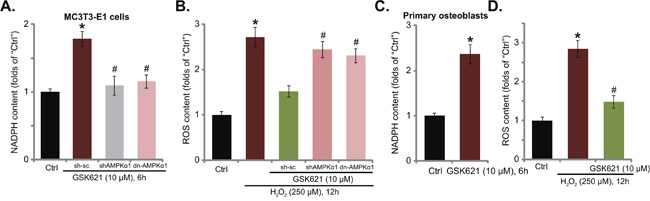
Figure 5: GSK621 increases NAPDH content and inhibits H2O2-induced oxidative stress. MC3T3-E1 cells, expressing scramble control shRNA (“sc-sh”), AMPKα1 shRNA (Seq-1, “sh AMPKα1”) or dominant negative AMPKα1 (T172A) (“dn-AMPKα1”), were treated with GSK621 (10 μM) or plus H2O2 (250 μM) for applied time; Relative NAPDH level A. and ROS content B. were tested by the listed assays. The primary murine osteoblasts were treated with GSK621 (10 μM) or plus H2O2 (250 μM) for applied time, relative NAPDH level C. and ROS content D. were tested. Data are shown as mean (n=3) ± SD. Experiments in this figure were repeated for four times, and similar results were obtained. *p<0.05 vs. “Ctrl” group. #p<0.05 vs. “H2O2” only group.
GSK621 activates cytoprotective autophagy in osteoblasts
AMPK could also provoke autophagy to promote cell survival [19, 30, 31]. AMPK is shown to directly phosphorylate and activate its key downstream Ulk1 to initiate autophagy, which is cytoprotective [19, 30, 31]. Here, in both MC3T3-E1 cells (Figure 6A) and primary murine osteoblasts (Figure 6B), GSK621 (10 μM, 2 hours) induced significant Ulk1 phosphorylation at Ser-317 (Results were quantified), the site that can only be activated by AMPK [30]. Subsequently, expression of autophagy-associated proteins, including Beclin-1, autophagy-related homologue 5 (ATG-5) and light chain 3B-II (LC3B-II), was significantly increased, while p62 was degradated (See quantified results in Figure 6C-6D). These results suggested significant autophagy activation in GSK621-treated cells [32–34]. Remarkably, as shown in Figure 6E and 6F, GSK621 (10 μM)-induced osteoblast cytoprotection against H2O2 was compromised in the presence of autophagy inhibitor 3-methyaldenine (3-MA) and chloroquine (Cq) [32, 35]. Therefore, GSK621’s actions in MC3T3-E1 cells was weakened when autophagy was pharmacologically inhibited (Figure 6E and 6F). These results indicate that GSK621 activates cytoprotective autophagy in osteoblasts.
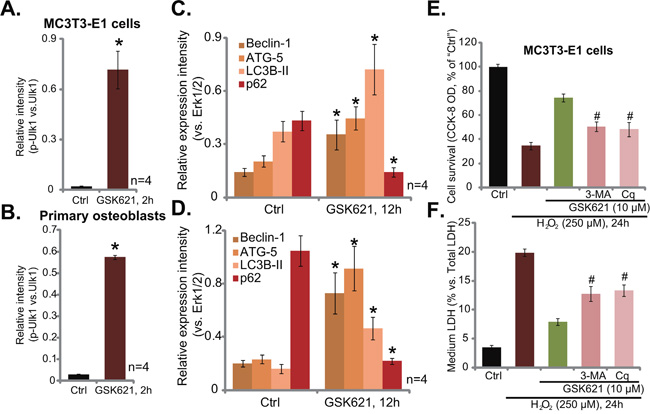
Figure 6: GSK621 activates cytoprotective autophagy in osteoblasts. MC3T3-E1 cells (A and C) or the primary murine osteoblasts (B. and D.) were treated with GSK621 (10 μM) for designated time; Expression of listed proteins was tested by Western blotting assay, results were quantified (A-D). MC3T3-E1 cells, pre-treated for 1 hour with 3-methyladenine (3-MA, 0.5 mM) or chloroquine (Cq, 10 μM), were then treated with H2O2 (250 μM) or plus GSK621 (10 μM) for 24 hours; Cell viability (CCK-8 assay, E) and cell death (LDH release assay, F) were tested. Data are shown as mean (n=4) ± SD. Experiments in this figure were repeated for four times, and similar results were obtained. #p<0.05 vs. “GSK621” only group.
DISCUSSION
The results of the current study suggest that AMPK activation is required for GSK621-induced osteoblast cytoprotection against H2O2. shRNA stable knockdown of AMPKα1 almost completely blocked GSK621-induced AMPK activation and osteoblast cytoprotection. Similarly, AMPK in-activation via intruding the dominant negative mutant AMPKα1 (T172A) also nullified GSK621-mediated anti-H2O2 actions in osteoblasts. Reversely, forced-activation of AMPK via introducing the ca-AMPKα1 mimicked GSK621’s actions and alleviated H2O2-induced osteoblast cell death. Remarkably, GSK621 was unable to further protect osteoblasts with ca-AMPKα1 expression. All these results imply that activation of AMPK is indispensable for GSK621-induced cytoprotection in osteoblasts.
Several mechanisms are responsible for AMPK-mediated cytoprotection. For example, AMPK could activate cytoprotective autophagy, which recycles cellular components to provide nutrition for cell survive [36, 37]. AMPK is shown to directly phosphorylate Ulk1 to initiate cell autophagy [36, 37]. Further, AMPK could also function as an anti-oxidant protein, via increasing NADPH content and limiting ATP consumption [28]. In addition, under starvation condition, AMPK activation could also protect cells through in-activating mTOR complex 1 (mTORC1) [38].
Here, we found that GSK621 induced NADPH production to inhibit H2O2-induced ROS production in osteoblasts. Such effects by GSK621 were dependent on AMPK activation. As AMPKα1 dominant negative mutation (T172A) or shRNA stable knockdown almost abolished GSK621-induced NADPH production and ROS scavenging activity. Our results here are consistent with recent findings showing ROS clearance by several different AMPK activators in osteoblasts [12, 18, 19]. Meanwhile, we showed that GSK621 treatment in the osteoblasts also activated cytoprotective autophagy, which was evidenced by Ulk1 phosphorylation at Ser-317, upregulation of Beclin-1, ATG-5 and LC3B-II, as well as degradation of p62. Importantly, autophagy inhibitors alleviated GSK621-mediated osteoblast cytoprotection. Together, ROS clearance and autophagy activation could be two major downstream targets of AMPK in mediating GSK621’s cytoprotection in osteoblasts. Although, the detailed underlying mechanisms may warrant further investigations.
MATERIALS AND METHODS
Chemicals, regents and antibodies
GSK621 was purchased from Gao-Chem (Shanghai, China). H2O2, puromycin, 3-methyaldenine (3-MA) and chloroquine (Cq) were purchased from Sigma Chemicals (St. Louis, MO). Anti-AMPKα1, acetyl-CoA carboxylase (ACC) and Beclin-1, autophagy-related homologue 5 (ATG-5), light chain 3B-II (LC3B-II), p62 and Erk1/2 antibodies were purchased from Santa Cruz Biotech (Santa Cruz, CA). Antibodies against phospho(p)-AMPKα1 (Thr 172), p-ACC (Ser 79), p-S6K1 (Thr-389), p-S6 (Ser-235/236) and total S6K1 were obtained from Cell Signaling Tech (Denver MA).
Culture of MC3T3-E1 osteoblastic cells
The murine calvaria-derived osteoblastic-like MC3T3-E1 cells were cultured and differentiated as described in our previous studies [13, 14].
Isolation and primary culture of murine osteoblasts
The isolation and primary culture of murine osteoblasts were described previously [13, 14]. The animal protocols were approved by Institutional Animal Care and Use Committee (IACUC) and Institutional Ethics Committee.
Cell survival assay
Cell Counting Kit-8 (CCK-8, Dojindo Laboratories, Kumamoto, Japan) was applied to test survival of osteoblasts with applied treatment/s. The detailed protocol was described in our previous studies [13, 14].
Cell apoptosis assay
To test cell apoptosis, the histone-DNA ELISA plus kit (Roche, Palo Alto, CA) [13, 14] was applied. ELISA OD at 450 nm was recorded as the indicator of cell apoptosis [13, 14].
Cell death assay
Analyzing cell death by measuring lactate dehydrogenase (LDH) content in the conditional medium was through a two-step enzymatic reaction LDH assay kit (Takara, Tokyo, Japan), as described [13, 14].
Western blotting assay
As described previously [13, 14], 30 μg protein lysates per sample were separated by SDS-PAGE gel, and were transferred to the PVDF membranes. These blots were then blocked, and were subsequently incubated with primary and specific secondary antibodies. Protein bands were visualized via ECL reagents (Pierce, Shanghai, China). Band intensity of total gray was quantified via the ImageJ software [13, 14].
AMPKα1 stable knockdown
A set of three different murine AMPKα1 shRNAs with non-overlapping sequences (“Seq-1/-2/-3”, Genepharm, Shanghai, China) were constructed into the GV428 lentiviral vector (Genepharm), containing a puromycin-resistance gene and the Flag-tag. MC3T3-E1 cells were cultured with 50% confluence. The lentiviral shRNA (5 μL/mL) was added directly to the cultured cells for 24 hours. Cells were then subjected to puromycin (0.5 μg/ml) selection for the other 24 hours. Afterwards, AMPKα1 expression was detected by Western blotting assay. Control cells were infected with scramble control shRNA (Genepharm).
AMPKα1 mutation
As described previously [12], the dominant negative AMPK-α1 (dn-AMPK-α1, T172A) construct or the constitutively-active mutant AMPKα1 (T172D, ca-AMPKα1) was transfected to the MC3T3-E1 cells (0.20 μg/mL \ each) via the Lipofectamine 2000 reagent [39]. Stable cells were selected via neomycin (1 μg/mL, for dn-AMPK-α1) or puromycin (0.5 μg/mL, for ca-AMPKα1) for a total of two weeks. AMPKα1 expression and phosphorylation in the stable cells were detected by Western blotting assay.
AMPKα1 siRNA knockdown
The two non-overlapping murine AMPKα1 siRNAs (“Seq-1/Seq-2”) and the scramble control siRNA were synthesized by Genepharm. Transient transfection (100 nM, 24 hours) was performed by the Lipofectamine 2000 reagents according to the manufacturer’s instructions. Transfection efficiency was determined by Western blotting assay.
Assay of intracellular ROS level
The cellular reactive oxygen species (ROS) content was detected via the 2′, 7′-dichlorofluorescein diacetate (H2-DCFDA; Abcam, Shanghai, China) FACS method as described [18]. ROS level in treatment group was normalized to that of control group.
NADPH content assay
The intracellular NADPH content was tested via the previously described enzymatic cycling method [18, 19, 28]. Briefly, after treatment, twp million cells per sample were lysed, and the supernatant was incubated at 60 °C for 30 min before NADP-cycling buffer plus glucose-6-phosphate dehydrogenase (G6PD, Sigma) [19] were added. Afterwards, glucose 6-phosphate (G6P, Sigma) was added to the mixture, and the change in absorbance at 570 nm was measured at 30 °C. NADPH level in treatment group was normalized to that of control group.
Statistical analysis
Comparisons between groups were performed via one-way ANOVA and the Newman-Keuls test (SPSS 18.0). p values < 0.05 were considered statistically significant.
CONCLUSIONS
Together, we conclude that targeted activation of AMPK by GSK621 significantly alleviates H2O2-induced damages in osteoblasts. GSK621 and other AMPK activators might have translational value for treatment oxidative stress-associated osteonecrosis.
ACKNOWLEDGMENTS
This work is supported by the National Natural Science Foundation.
CONFLICTS OF INTEREST
The listed authors have no conflicts of interests.
Author contributions
All authors carried out the experiments, participated in the design of the study and performed the statistical analysis, participated in its design and coordination and helped to draft the manuscript.
REFERENCES
1. Souttou B, Raulais D, Vigny M. Pleiotrophin induces angiogenesis: involvement of the phosphoinositide-3 kinase but not the nitric oxide synthase pathways. J Cell Physiol. 2001; 187:59-64.
2. Himburg HA, Muramoto GG, Daher P, Meadows SK, Russell JL, Doan P, Chi JT, Salter AB, Lento WE, Reya T, Chao NJ, Chute JP. Pleiotrophin regulates the expansion and regeneration of hematopoietic stem cells. Nat Med. 2010; 16:475-482.
3. Baek KH, Oh KW, Lee WY, Lee SS, Kim MK, Kwon HS, Rhee EJ, Han JH, Song KH, Cha BY, Lee KW, Kang MI. Association of oxidative stress with postmenopausal osteoporosis and the effects of hydrogen peroxide on osteoclast formation in human bone marrow cell cultures. Calcif Tissue Int. 2010; 87:226-235.
4. Tare RS, Oreffo RO, Sato K, Rauvala H, Clarke NM, Roach HI. Effects of targeted overexpression of pleiotrophin on postnatal bone development. Biochem Biophys Res Commun. 2002; 298:324-332.
5. Fan JB, Liu W, Zhu XH, Yuan K, Xu DW, Chen JJ, Cui ZM. EGFR-AKT-mTOR activation mediates epiregulin-induced pleiotropic functions in cultured osteoblasts. Mol Cell Biochem. 2015; 398:105-113.
6. Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004; 59:21-26.
7. Talasila KM, Soentgerath A, Euskirchen P, Rosland GV, Wang J, Huszthy PC, Prestegarden L, Skaftnesmo KO, Sakariassen PO, Eskilsson E, Stieber D, Keunen O, Brekka N, Moen I, Nigro JM, Vintermyr OK, et al. EGFR wild-type amplification and activation promote invasion and development of glioblastoma independent of angiogenesis. Acta Neuropathologica. 2013; 125:683-698.
8. Liang D, Xiang L, Yang M, Zhang X, Guo B, Chen Y, Yang L, Cao J. ZnT7 can protect MC3T3-E1 cells from oxidative stress-induced apoptosis via PI3K/Akt and MAPK/ERK signaling pathways. Cell Signal. 2013; 25:1126-1135.
9. Rigel DS, Friedman RJ, Kopf AW. The incidence of malignant melanoma in the United States: issues as we approach the 21st century. J Am Acad Dermatol. 1996; 34:839-847.
10. Salopek TG, Marghoob AA, Slade JM, Rao B, Rigel DS, Kopf AW, Bart RS. An estimate of the incidence of malignant melanoma in the United States. Based on a survey of members of the American Academy of Dermatology. Dermatol Surg. 1995; 21:301-305.
11. Koh HK. Cutaneous melanoma. N Engl J Med. 1991; 325:171-182.
12. Guo S, Mao L, Ji F, Wang S, Xie Y, Fei H, Wang XD. Activating AMP-activated protein kinase by an alpha1 selective activator compound 13 attenuates dexamethasone-induced osteoblast cell death. Biochem Biophys Res Commun. 2016; 471:545-552.
13. Guo S, Xie Y, Fan JB, Ji F, Wang S, Fei H. alpha-Melanocyte stimulating hormone attenuates dexamethasone-induced osteoblast damages through activating melanocortin receptor 4-SphK1 signaling. Biochem Biophys Res Commun. 2016; 469:281-287.
14. Ji F, Mao L, Liu Y, Cao X, Xie Y, Wang S, Fei H. K6PC-5, a novel sphingosine kinase 1 (SphK1) activator, alleviates dexamethasone-induced damages to osteoblasts through activating SphK1-Akt signaling. Biochem Biophys Res Commun. 2015; 458:568-575.
15. Zhao S, Chen C, Wang S, Ji F, Xie Y. MHY1485 activates mTOR and protects osteoblasts from dexamethasone. Biochem Biophys Res Commun. 2016; 481:212-218.
16. Carling D, Thornton C, Woods A, Sanders MJ. AMP-activated protein kinase: new regulation, new roles? Biochem J. 2012; 445:11-27.
17. Wang S, Song P, Zou MH. AMP-activated protein kinase, stress responses and cardiovascular diseases. Clin Sci (Lond). 2012; 122:555-573.
18. Zhu Y, Zhou J, Ao R, Yu B. A-769662 protects osteoblasts from hydrogen dioxide-induced apoptosis through activating of AMP-activated protein kinase (AMPK). Int J Mol Sci. 2014; 15:11190-11203.
19. She C, Zhu LQ, Zhen YF, Wang XD, Dong QR. Activation of AMPK protects against hydrogen peroxide-induced osteoblast apoptosis through autophagy induction and NADPH maintenance: New implications for osteonecrosis treatment? Cell Signal. 2014; 26:1-8.
20. Fan JB, Ruan JW, Liu W, Zhu LQ, Zhu XH, Yi H, Cui SY, Zhao JN, Cui ZM. miR-135b expression downregulates Ppm1e to activate AMPK signaling and protect osteoblastic cells from dexamethasone. Oncotarget. 2016; 7:70613-70622. doi: 10.18632/oncotarget.12138.
21. Faubert B, Vincent EE, Poffenberger MC, Jones RG. The AMP-activated protein kinase (AMPK) and cancer: many faces of a metabolic regulator. Cancer Lett. 2015; 356:165-170.
22. Hardie DG, Ross FA, Hawley SA. AMP-Activated Protein Kinase: A Target for Drugs both Ancient and Modern. Chem Biol. 2012; 19:1222-1236.
23. Zhao H, Zhu H, Lin Z, Lin G, Lv G. Compound 13, an alpha1-selective small molecule activator of AMPK, inhibits Helicobacter pylori-induced oxidative stresses and gastric epithelial cell apoptosis. Biochem Biophys Res Commun. 2015; 463:510-517.
24. Hu X, Jiang F, Bao Q, Qian H, Fang Q, Shao Z. Compound 13, an alpha1-selective small molecule activator of AMPK, potently inhibits melanoma cell proliferation. Tumour Biol. 2016; 37:1071-1078.
25. Sujobert P, Poulain L, Paubelle E, Zylbersztejn F, Grenier A, Lambert M, Townsend EC, Brusq JM, Nicodeme E, Decrooqc J, Nepstad I, Green AS, Mondesir J, Hospital MA, Jacque N, Christodoulou A, et al. Co-activation of AMPK and mTORC1 Induces Cytotoxicity in Acute Myeloid Leukemia. Cell Rep. 2015; 11:1446-1457.
26. Jiang H, Liu W, Zhan SK, Pan YX, Bian LG, Sun B, Sun QF, Pan SJ. GSK621 Targets Glioma Cells via Activating AMP-Activated Protein Kinase Signalings. PLoS One. 2016; 11:e0161017.
27. Chen L, Chen Q, Deng G, Kuang S, Lian J, Wang M, Zhu H. AMPK activation by GSK621 inhibits human melanoma cells in vitro and in vivo. Biochem Biophys Res Commun. 2016; 480:515-521.
28. Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012; 485:661-665.
29. Balteau M, Van Steenbergen A, Timmermans AD, Dessy C, Behets-Wydemans G, Tajeddine N, Castanares-Zapatero D, Gilon P, Vanoverschelde JL, Horman S, Hue L, Bertrand L, Beauloye C. AMPK activation by glucagon-like peptide-1 prevents NADPH oxidase activation induced by hyperglycemia in adult cardiomyocytes. Am J Physiol Heart Circ Physiol. 2014.
30. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011; 13:132-141.
31. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011; 331:456-461.
32. Zheng B, Zhu H, Gu D, Pan X, Qian L, Xue B, Yang D, Zhou J, Shan Y. MiRNA-30a-mediated autophagy inhibition sensitizes renal cell carcinoma cells to sorafenib. Biochem Biophys Res Commun. 2015.
33. Zheng B, Mao JH, Qian L, Zhu H, Gu DH, Pan XD, Yi F, Ji DM. Pre-clinical evaluation of AZD-2014, a novel mTORC1/2 dual inhibitor, against renal cell carcinoma. Cancer Lett. 2015; 357:468-475.
34. Zhang Q, Yang M, Qu Z, Zhou J, Jiang Q. Autophagy prevention sensitizes AKTi-1/2-induced anti-hepatocellular carcinoma cell activity in vitro and in vivo. Biochem Biophys Res Commun. 2016.
35. Huo HZ, Zhou ZY, Wang B, Qin J, Liu WY, Gu Y. Dramatic suppression of colorectal cancer cell growth by the dual mTORC1 and mTORC2 inhibitor AZD-2014. Biochem Biophys Res Commun. 2014; 443:406-412.
36. Kim I, He YY. Targeting the AMP-Activated Protein Kinase for Cancer Prevention and Therapy. Front Oncol. 2013; 3:175.
37. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011; 13:1016-1023.
38. Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, You M, Williams BO, Guan KL. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006; 126:955-968.
39. Wang B, Wang XB, Chen LY, Huang L, Dong RZ. Belinostat-induced apoptosis and growth inhibition in pancreatic cancer cells involve activation of TAK1-AMPK signaling axis. Biochem Biophys Res Commun. 2013; 437:1-6.