
Introduction
The personalized therapeutic strategy targeting specific molecular alterations involved into tumorigenesis is the new frontier of cancer treatment. Dysfunction of DNA damage repair from cancer, impairing the integrity of the genome, could be a valuable target for anticancer therapy.
BRCA germline mutations represent the molecular basis for the majority of hereditary breast and ovarian cancer (HBOC) syndrome. BRCA1 and BRCA2 are tumor suppressor genes encoding proteins essential for DNA repair, involved in key processes for the maintenance of genomic stability [1].
Specifically BRCA1/2 are essential for the accurate repair of DNA double-strand breaks (DSBs) through homologous recombination (HR) [2]. This mechanism of DNA repair is conservative and it allows to restore the original sequence of the DNA [3].
Mutations in BRCA1 or BRCA2 result in protein isoforms unable to repair DSBs by HR. The HR-deficient cells use alternative DNA-damage response (DDR) pathway. This compensatory repair mechanism is non-conservative, causes genomic instability and make the tumor cells subordinated to their proper functioning to survival [2, 3].
Germline mutations in tumor suppressors BRCA1/2 result in an increased risk of developing breast, ovarian and others, such as pancreas, stomach and prostate cancers [4, 5]: the neoplastic process will rise if the wild-type gene is lost or inactivated.
Other genes and low penetrance alleles can be involved in HBOC susceptibility including some genes encoding for proteins involved in HR DNA repair pathway, such as RAD50, RAD51C, RAD51D, PALB2, CHEK2, MRE11A, BARD1, BRIP 1, NBS1 and ATM [3]. Furthermore alterations in such genes have been identified also in sporadic breast tumors with early age of onset, hormone receptors (ER/PgR) negativity and a high tumor grade, but in absence of germline BRCA1/2 mutations, which define a BRCA-like phenotype defined as “BRCAness” [5].
All these findings suggest that DNA repair process and specially the HR are associated with the onset of many tumors and have today important therapeutic implications for patients: the loss of a functional copy of BRCA1/2 is a workable target for using a synthetic lethal approach to treat BRCA-deficient cancers [6].
The concept of synthetic lethality between two genes imply that the concomitant damage of both produces the cell death. Conversely, the loss of either gene allows cell survival. BRCA1/2 and Poly(ADPribose) polymerase 1 (PARP1) genes are a model of synthetic lethality [7]. PARP1 encodes an enzyme with an important role in DNA single-strand break repair (SSBs) [8].
PARP inhibitors (PARPi), an emerging class of drugs, are synthetically lethal against BRCA1/2-deficient cancer cells. In these cells the PARP inhibition produces accumulation of genetic aberrations that cannot be restored through BRCA1/2-homologous recombination [9]. The PARPi target is, therefore, the dysregulation of DNA repair pathway and the subsequent addiction of the BRCA-mutated tumors by the function of compensatory repair mechanism.
PARPi have been developed and tested in clinical trials to treat patients carrying BRCA1/2 mutations. These genetic alterations have been detected in about 17% of patients with ovarian cancer, mainly high grade serous adenocarcinoma. These findings prompted the approval of the first PARPi olaparib in Dicember 2014 as monotherapy for the treatment of patients with advanced ovarian cancer and BRCA1/2 germline mutations who have received three or more prior lines of chemotherapy [10-12].
Currently, several clinical trials are investigating the effectiveness of new PARPi, alone or in combination with chemotherapy or targeted therapy [13]. Moreover, the discovery of somatic BRCA mutations also in other cancer provides new and promising applications for PARPi.
Most data from clinical trials suggests that some patients initially treated with PARPi eventually develop resistance [14]. The mechanisms of resistance are multiple and they typically restore the function of DDR pathway [15, 16].
Biology of PARP inhibition
Several genotoxic agents, including reactive oxygen species, ultraviolet light, ionizing radiation, and endogenous and synthetic compounds may lead to single strand break (SSB) or double strands breaks (DSB) at the DNA helix, which are physiologically repaired by different systems that provide the restoration and maintenance of DNA integrity [17].
During replication (cell cycle S phase) polymerases usually replace the incorrect single nucleotides insertions by proofreading and exonuclease activity, whereas if the new synthesized strand presents deletions, insertions or wrong bases incorporations, mismatch repair mechanisms (MMR) will be activated.
Both polymerases and MMR are able to repair only “simple” replication errors consisting of single nucleotides alterations. If more “complex alterations” occur, such as chromosomal rearrangements and transposons, the final effect will be the DNA damage.
In this case the cells put in place two main strategies aiming to correct these negative events: first stopping the cell cycle, and subsequently activating the repair cellular pathways. If the damage cannot be corrected cells will die by apoptosis. After the detection of DNA lesions several check-point proteins are recruited, including DNA-dependent protein kinase (DNA-PK), ataxia-telangiectasia mutated (ATM) and and Rad3-related kinase (ATR). All these are serine threonine-kinase enzymes responsible for the activation of specific damage-related repair systems during the check-point response. The activation of repair mechanisms is correlated to the specific DNA damage causes and extent.
The two main cellular systems responsible for the correction of the SSB are called: Base Excision Repair (BER) and Nucleotide Excision Repair (NER).
BER occurs after X rays, oxygen radicals, alkylating agents or spontaneous reactions which alter the nucleotide bases during S and G2 phases; while NER complex is activated by UV light, which arises pyridine dimers or large DNA adducts, and has an important role during G1 phase [18, 19].
BER system activity is initiated by a DNA glycosylase which recognizes the damaged site and removes the nucleotide base; after that, X-ray repair cross-complementing gene 1 (XRCC1) protein, DNA polymerase B (polβ), exo and endo-nucleases enzymes are recruited and provide to the repair, synthesis and ligation of DNA [20, 21]. Another enzyme involved in the BER pathway, is the poly ADP-ribose polymerases (PARP). PARP is a post-translation protein that synthesizes ADP-ribose polymers (PAR) on target proteins by the nicotinamide adenine dinucleotide NAD+ substrate, facilitating the annealing of system repairs. These enzymes have a pleiotropic function in response to genotoxic stressors aiming to maintain the genomic stability. They take part to different processes: cellular metabolism and death, chromatin modification, insulator function, mitotic apparatus response, and transcriptional regulation. PARP1 was the first member of the members of PARP’s family proteins identified. PARP 1 is a multi-domain protein: the amino-terminal DNA-binding domain (DBD) contains zinc finger domains and a bipartite nuclear localization signal (NLS); the auto-modification domain (AD), and the carboxyl-terminal PARP homology domain, which includes the catalytic domain (CAT) responsible for PAR formation. The damage in DNA is recognized by PARP1 through zinc fingers FI and FII domains. PARP1 early recognizes nicked DNA and organizes the recruitment of the different repair systems, accumulating PAR protein targets. PARP1 is itself the major acceptor of PAR, used to attract and assist the assembling multi-protein complexes in chromatin remodeling, DNA repair and damage checkpoint signaling [22, 23]. DNA repair scaffolds as XRCC1 may be directly recruited by auto-modified PARP1. PARP1 also recruits the DNA check-point protein ATM activating the signaling cascade for DNA damage and cell cycle arrest.
NER system acts by several enzymes, including XPA, RPA, XPG, TFIIH. Among them there are endonucleases, DNA helicases, DNA synthesis and ligation enzymes. NER system can be divided into global repair and transcription-coupled repair (TCR). The last one specifically aims to repair lesions of transcribed genes.
Similarly there are two main pathways regulating DSB repair: homologous recombination repair (HRR), and non-homologous end-joining (NHEJ). NHEJ may be activated during G1-phase acting without an homologous template to guide repair process. Therefore it’s defined as an “error-prone” system which often causes the occurrence of further mutations.
NHEJ includes a multistep reaction starting with both Ku 70 and Ku 80 binding proteins which rapidly constitutes a complex with DNA-dependent kinase and ligase proteins, in order to repair the breaks at DNA ends.
Recent studies have shown a determinant role of NHJE systems in the repair to DNA damage caused by radiomimetic drugs, ionizing radiation (IR), or ultraviolet (UV) light, suggesting that the recruitment of an “error-prone” system, like NHJE, may increase the cells’ genetic instability [24].
HRR is the most accurate mechanism among the DSBs during S - G2 phases because the source of information is represented by the sister chromatids which are used like a template to repair the damaged DNA. HRR is introduced mainly by the so-called MNR complex, including MRE11, RAD50, and NBS1 proteins, which regulates the generation of single strand ends of DNA (ssDNA), at DSB site, thus mediating the ATR recruitment. Furthermore MNR-complex interacts with ATM, playing a crucial role for the beginning of HRR cascade, by the activation of CHEK1, CHEK2, P53, NSB1, and BRCA proteins and downstream signaling pathways [25]. The HRR mechanism begins with 5′ to 3′ end-processing at broken ends of the DNA fragments which need to be repaired. The 3’ end of ssDNA, arised by exonuclease, are stabilized, through the intervention of RAD (RAD 51, RAD52) and RAP proteins. BRCA2 then mediates the substitution of RAP1 with RAD51, and leads the pre-synaptic filament towards the homologous DNA template. Following the complementary strands pairing the DNA polymerase can extend the DNA strand, finally leading to the repaired heteroduplexed DNA which is characterized by the formation of the “Holliday junctions”.
HRR defective seems to be a major contributor to tumorigenesis in individuals carrying ATM, CHEK2, RAD51, MNR, and germline and somatic mutations of BRCA, defining a tumor cells phenotype known as homologous recombination deficiency (HRD).
The crucial role of the breast cancer susceptibility proteins, BRCA1 and BRCA2 in the DNA repair has been known since 1995 [26]. These proteins are considered the product of two distinct tumor suppressor genes which play a crucial role in the response to cellular stressors and in the activation of DNA repair processes. BRCA1 and BRCA2 germline mutations predispose to develop breast and ovarian cancer and also increases the risk to develop other cancer types including pancreatic cancer, prostate cancer, and melanoma [27-29].
In details, BRCA1 protein provides to: G1-S and G2-M transition checkpoints, transcriptional regulation and chromatin remodeling. BRCA1 is rapidly phosphorylated after DNA damage in dividing cells by different kinases. The different roles of BRCA1 is guaranteed by its several domains that allow to regulate various processes by specific interactions with other cellular factors. Among them are included: nuclear localization signals NLS, binding sites for proteins p53, cMyc, RB and a transcriptional repressor ZBRK1. These proteins are known as “guardian of genome” for their crucial role in cancer development prevention and cell cycle progression restoration. Another site is the binding domain to recruit the chromatin remodeling SW1-SNF system, which is able to create a free access to other factors involved in the repair mechanisms. BRCA1 fulfills its transcriptional regulation role interacting by BRCA C - Terminal binding site (BRCT), with RNA polymerase II, the complex p300-CBP transcriptional co-activating proteins, transcription regulator protein BACH1, the histone complexes - deacetylase HDAC1 and HDAC2. These activated complexes are needed to maintain chromatins relaxed and to promote gene expression through the recruitment of the basal transcriptional machinery. BRCT also favors the binding between BRCA and CtIP, an enzyme which together with MRN complex allows the beginning of HRR pathway. Recent studies suggested that a large group of cellular proteins interact with BRCA1 favoring the activity of HRR as the predominant mechanism to ensure the genomic stability. In contrast, HRR is not efficient in BRCA1 deficient cells, and the chromatin breaks are often rejoined by components of NHEJ in aberrant fusion between heterologous chromosomes [30].
As regards BRCA2 protein it seems to act directly in HRR, by interacting with RAD 51 protein into the nucleus. The interactions between BRCA2 and RAD 51 seem to be negatively regulated by cycline A-CDK2, and a recent study has shown that cycline D1-CDK4 destablished the cycline A-CDK2 complex, promoting BRCA2-RAD51 activity, and ultimately improving the HHR.
BRCA2 seems to interact also with the Fanconi’s anemia protein ubiquitylation pathway, contributing to regulate protein-protein interactions and/or accessibility of repair factors to the damage sites. When FA genes are deficient the BRCA-mediated HHR doesn’t work correctly and NHEJ will be recruited determining an increase of the genomic instability [31, 32].
A deeper understanding of the molecular pathways underlying the DNA repair mechanisms have led to the advent of a new family of targeted drugs able to inhibit PARPs proteins. The clinical development of these drugs was based on several evidences provided by preclinical studies, which attested the possibility to induce cancer cells die by “synthetic lethality”. This model consists of determining the simultaneous block of two main DNA repair pathways in the same cancer cell, ultimately inducing cell death by apoptosis. The first study in xenograft models has shown that once inactivated by inhibitors, PARP1 lost the ability to interact with the single strand repair system preventing DNA repair via the BER pathway. The accumulation of SSB leads to the collaps of the replication forks translating into DSB, which if not immediately repaired by HRR system, results into the cell death. This is precisely what happened in cancer cells defective for HRR pathways, as a result of BRCA1/2 mutations [33-35]. Indeed, in absence of a functional HRR system, these lesions are usually repaired by alternative error-prone pathways, such as NHEJ and single strand annealing (SSA) resulting in gross genomic instability and ultimately leading to the cell death.
However all the potential interactions between PARP1 and HRR pathway, have not been well-elucidated [36]. Experimental data suggest that PARP inhibition increases spontaneous HRR , but it had no effects on DSB-induced HRR [37]. Other studies have shown a possible relationship between PARP enzymes and BRCA proteins, likely involving BRCA2. Indeed BRCA2 contains three tandem oligonucleotide oligosaccharide binding folds (OB-folds ) involved in DNA binding during DNA DBS repair. This domain recognizes PAR and mediates the fast recruitment of BRCA2 to the DNA lesion needed for the first steps of HRR [38].
Subsequently several both pre-clinical and clinical studies confirmed that the sensitivity to PARPi is not limited to cells harboring BRCA 1/2 mutations, but occurs also in cells carrying mutated genes encoding other proteins involved in HRR system. The hypermethylation of the BRCA1 promoter or the loss of function of other genes as ATM, CHEK2, RAD51, BRIP1, and PALB2 belonging at DNA repair machinery, define a specific phenotype with features and behavior similar to BRCA-related cancers, defined as “BRCAness”. These patients might benefit from platinum-based therapies and/or PARP inhibition like BRCA mutated [39-42].
Clinical development of PARP inhibitors
To date there are about ten molecules, including olaparib, veliparib, rucaparib, niraparib, among those in more advanced stages of experimental development. The majority of such studies have been focused on solid tumors harboring germ-line BRCA1-2 mutations, mainly ovarian and breast, but also prostate or pancreatic cancers. However the use of PARP inhibitors as single agent was extended also to “BRCA-like” sporadic tumors with suspected homologous recombination (HR) genes defects, including high grade serous ovarian cancer (HGSOC) and triple negative breast cancer (TNBC). Furthermore the peculiar mechanisms of action of such compounds has led to evaluate potential combinations with both DNA damaging cytotoxic agents, including chemotherapy and radiotherapy, and targeted agents able to induce HR dysfunctions, such as CDK1, PI3K, PTEN and HSP90 inhibitors or anti-angiogenic drugs.
Ovarian cancer
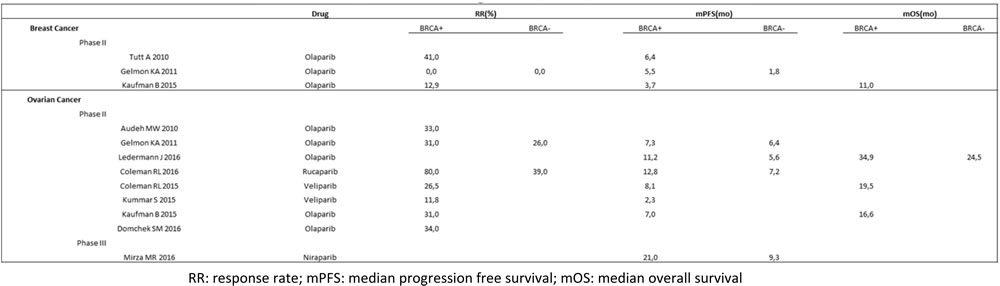
Fong et al have has first demonstrated the activity of single agent PARP inhibitor olaparib in BRCA-deficient advanced ovarian, breast and prostate cancers [43], with grater activity observed in platinum sensitive ovarian cancer patients [44]. Response rates of 30% have been reported in a subsequent phase II studies conducted in refractory advanced BRCA-mutant ovarian cancer [45], suggesting BRCA1-2 mutations as potential predictive biomarkers for clinical use. However another phase II study including both BRCA mutant and wild type patients with breast cancer and HGSOC showed encouraging activity also in the cohort of wild type, platinum-sensitive HGSOC patients [46], likely due to the acquired defects of HR genes responsible for “BRCA-like phenotype”, which conferred the same sensitivity to both platinum-chemotherapy and PARP inhibition. All such findings have led to the design of a randomized phase III, placebo controlled trial, of olaparib as single agent maintenance therapy in patients with recurrent HGSOC who responded to prior platinum-based chemotherapy, showing a significant improvement of 3.6 months progression free survival (PFS) in the overall population (HR: 0.35, p < 0·001), with the greatest increment of 6.9 months PFS (HR 0.18, p < 0.0001) occurring in the subgroup of patients with BRCA mutations [47]. The updated analysis of the study 19 has recently shown a significant overall survival benefit limited to the BRCA mutant patients (34.9 vs 30.2 months; HR: 0.62, p = 0.025), which was not extended to the wild type population (HR 0.83, p = 0.37). As expected adverse events like fatigue, anemia, nausea and vomiting were significantly higher with olaparib than placebo [48]. On the basis of such positive results olaparib was the first PARP-inhibitor receiving the approval by the European Medical Agency (EMA) at doses of 400 mg twice daily as maintenance therapy for platinum-sensitive patients with advanced HGSOC, fallopian tube, or primary peritoneal cancer, harboring BRCA-mutations. A companion diagnostic test has been also approved by FDA to identify mutations in BRCA1/2 genes using DNA obtained from a blood sample. Along with olaparib, several other PARP inhibitors, including veliparib, rucaparib and niraparib have shown encouraging activity and acceptable safety profile in early phase I-II studies. In particular the phase II randomized ARIAL 2 study of rucaparib has shown an objective response rate (ORR) of 80% and median PFS of 12.8 months in BRCA-mutant platinum sensitive patients with recurrent ovarian cancer and ORR 39% with and median PFS of 7.2 months in BRCA wild type patients with a BRCA-like signature, compared to ORR of 13% and median PFS of 5 months in biomarker negative patients. Rucaparib was associated with a manageable safety profile, including nausea, asthenia/fatigue and ALT/AST elevations among the most common treatment-related AEs [49, 50]. These impressive results led to the recent Breakthrough Therapy designation status of rucaparib by the FDA for the treatment of ovarian cancer, while the ARIEL3 randomized study is currently recruiting patients. Veliparib has recently shown a significant activity and tolerable safety profile as single agent in a phase II single arm trial including ovarian cancer patients carrying a germline BRCA1-2 mutation who progressed to prior chemotherapy regimens, reporting ORR of 35% and 20% in platinum-sensitive and platinum-resistant patients, respectively [51]. A phase 3 trial is currently ongoing in order to further elucidate the potential of this drug in such setting. Niraparib 300 mg/day has shown a good safety profile and a promising activity with ORR 40% in pre-treated ovarian cancer patients with BRCA 1-2 mutations [52]. The phase III randomized ENGOT-OV16/NOVA trial investigated the PARP inhibitor niraparib as single agent maintenance therapy in patients with recurrent, platinum sensitive HGSOC, stratified by BRCA-mutation status. The study has met its primary end-point showing a significant PFS improvement both in BRCA-mutant (HR: 0.27; p < 0.001) and in BRCA-wild type (HR: 0.45; p < 0.0001) populations. A further analysis of BRCA-wild type patients identified the subgroup of HRD-positive patients who receive more benefit (HR: 0.38; p < 0.001) compared to HRD-negative patients (HR: 0.58; p < 0.0226) [53]. The large benefit observed in the overall population included in the NOVA study could led to a fast approval of niraparib in the treatment of platinum sensitive recurrent ovarian cancer, regardless of BRCA-status. An alternative approach has been investigated by another phase II randomized trial comparing olaparib plus chemotherapy (paclitaxel and carboplatin), followed by olaparib single agent maintenance versus chemotherapy alone in platinum-sensitive recurrent HGSOC patients. The results of such trial showed that combining olaparib 200 mg twice daily for 10 days of each cycle with lower dose of carboplatin (AUC 4) plus paclitaxel, followed by olaparib 400 mg twice daily as maintenance treatment is an effective and tolerable option leading to a significant 2.6-month PFS advantage (HR 0.51, p = 0.0012), with the greatest clinical benefit in BRCA-mutated patients (HR: 0.21, p = 0.0015) [54]. The addition of veliparib to cyclophosphamide didn’t improve RR and PFS in patients with recurrent HGSOC [55], while another phase II randomized study comparing veliparib plus temozolomide vs PLD in the same setting of patients has just completed recruitment (NCT01113957, https://clinicaltrials.gov/ ). A very promising strategy emerged from the phase II randomized trial by Liu et al. which showed a further PFS benefit from adding the anti-angiogenic agent cediranib to olaparib for platinum-sensitive, recurrent, HGSOC, fallopian tube, or primary peritoneal cancer (median PFS 17.7 vs 9 months; (HR: 0.42; p = 0·005) [56], warranting investigation in a phase III ongoing trial. Similarly a phase I-II study is currently comparing tolerability and efficacy of niraparib alone versus niraparib-bevacizumab combination versus sequential bevacizumab and niraparib in platinum sensitive relapsed ovarian cancer [57], while the PAOLA1 trial is currently investigating first-line chemotherapy with bevacizumab plus olaparib or placebo as maintenance treatment. Finally the combination of olaparib and PD-L1 checkpoint inhibitor Durvalumab has shown promising durable long-term responses and a tolerable safety profile in pre-treated ovarian cancer and triple negative breast cancer (TNBC) patients [58].
Breast cancer
As for ovarian cancer, first proof-of-concept trials have shown a significant activity of PARP inhibitors in women with advanced breast cancer and BRCA1-2 mutations [43], reporting ORR to olaparib nearly to 40% and median PFS 5.7 months in the overall population and ORR of 54% in TNBC patients [59]. Simultaneously another phase II study including both BRCA mutant and wild type patients with breast cancer and HGSOC showed no confirmed clinical responses in breast cancer patients according to RECIST criteria but only a partial reduction in tumor size for 5 (50%) of patients with BRCA mutations [46]. Recently the results of a multicenter phase II study confirmed that encouraging responses to olaparib were observed across different tumor types associated with germline BRCA1/2 mutations including breast, ovarian, prostate, and pancreatic cancer who received prior chemotherapies, and AEs most frequently reported were fatigue, nausea, and vomiting [60]. All these evidences have led to the design of phase III Olympia trials which are currently investigating the activity of olaparib as single agent both in metastatic and adjuvant treatment of breast cancer patients with germ-line BRCA1-2 mutations. Rucaparib has shown preliminary activity in a phase II study including patients with BRCA1-2-mutated metastatic breast cancer [61], and it will be early under evaluation in advanced breast cancer patients with a BRCA-like profile, including a specific genomic signature or somatic BRCA mutations (NCT02505048, https://clinicaltrials.gov/). However the addition of rucaparib to cisplatin failed to improve the DFS in patients with TNBC who had residual disease after preoperative chemotherapy [61]. Recently early phase I-II studies investigated both tolerability and activity of veliparib in combination with different chemotherapy regimens, including temozolomide, carboplatin, irinotecan, cisplatin, vinorelbine and cyclophosphamide in pre-treated advanced breast cancer patients with or without BRCA mutations, showing RR between 20% and 40%. Veliparib is currently under investigation in two phase III randomized trials comparing veliparib plus carboplatin-based regimens both in advanced and neoadjuvant setting, including BRCA-mutated and TNBC, respectively (NCT02163694; NCT02032277, https://clinicaltrials.gov/). Niraparib 300 mg/day has shown a good safety profile and a promising activity with ORR of 50% in pre-treated breast cancer patients with BRCA1-2 mutations [52], and is currently under investigation as single agent in the phase III randomized BRAVO trial in this setting of patients. Finally talazoparib is a PARP inhibitor currently under investigation as single agent in BRCA mutated breast cancer both in a phase III study in metastatic disease and in a pilot phase II study as neoadjuvant treatment (NCT02282345, https://clinicaltrials.gov/).
Other malignancies
According to the first evidences reported by Fong et al, the results of a multicenter phase II study by Kaufman et al, have recently showed encouraging responses to olaparib across different tumor types associated with germline BRCA1/2 mutations including breast, ovarian, prostate, and pancreatic cancer who received prior chemotherapy regimens [60], confirming that BRCA1/2 germline status defines a target population responsive to PARP inhibition, regardless of tumor anatomic origin and histotype. Currently phase II-III studies are investigating the potential activity of PARP inhibitors in genetically defined advanced solid tumors harboring BRCA1/2 mutations. Particularly the phase III randomized POLO study is evaluating single agent olaparib as ‘switch maintenance’ therapy in patients with BRCA1/2 mutated advanced pancreatic cancer who have not progressed on first-line platinum chemotherapy (NCT02184195, https://clinicaltrials.gov/). However, except for ovarian and breast cancers, BRCA1/2 germline mutations are less frequently observed in other malignancies, including gastric and pancreatic cancers (5%-10%), melanoma (5%) [62], prostate and NSCLC (1%-2%) [63, 64]. Beyond BRCA1/2, anti-tumor activity of PARP inhibitors was also reported in sporadic (BRCA1/2 wild type) cancers with BRCA-like phenotype. Recently a phase II study by Mateo et al. has shown promising antitumor activity of olaparib in patients with sporadic advanced CRPC who progressed to prior anticancer treatments and who had DNA-repair genes defects identified by NGS, accounting for about 25-30% of all sporadic CRPC patients [65]. Another phase II study is currently evaluating olaparib plus abiraterone in advanced sporadic CRPC including the assessment of activity in ETS fusion-positive tumors, which represent about 50% of all CRPC patients, in order to demonstrate its predictive role for clinical setting (NCT01972217, https://clinicaltrials.gov/). Similarly clinical studies are investigating the activity of PARP inhibitors in Ewing’s sarcoma with either EWS-FLI1 or EWS-ERG genomic fusions. Since pre-clinical studies demonstrated the potential role of PARP inhibitors as chemo/radio-sensitizers several conducted/ongoing studies investigated the addition of such agents to chemotherapy regimens in different sporadic tumor types. Interesting activity has been recently observed with veliparib plus chemotherapy combinations both in melanoma patients with unknown BRCA/HR status [66, 67], and in untreated NSCLC patients with squamous histology, reaching an hazard ratio of 0.50 for PFS, and 0.71 for OS [68], and leading to a randomized phase III ongoing study in this subgroup of patients (NCT02264990, https://clinicaltrials.gov/), while the addition of iniparib to first-line platinum-gemcitabine did not improve ORR in advanced NSCLC patients [69]. The addition of olaparib to paclitaxel as second-line therapy in patients with recurrent/metastatic gastric cancer was well tolerated and showed a statistically significant improvement in OS in the overall included population, with a larger benefit in the subgroup of patients with low ATM expression [70], and is currently under investigation in a phase III randomized study (NCT01924533, https://clinicaltrials.gov/). Randomized phase II and III studies are ongoing to evaluate the efficacy of veliparib plus chemotherapy regimens as first-line treatment in advanced colorectal cancer (NCT02305758, https://clinicaltrials.gov/) and glioblastoma (NCT02106546, https://clinicaltrials.gov/), respectively, while a phase II randomized study is comparing olaparib plus gefitinib vs gefitinib alone in advanced NSCLC patients with EGFR activating mutations. Finally several studies are also exploring the role of PARP inhibitors in combination with radiotherapy in different tumor types, including NSCLC, head and neck cancer, esophageal cancer, and pancreatic cancer. Recently a meta-analysis including randomized controlled trials (RCTs) of PARP inhibitors in cancer has shown that such agents significantly improve the PFS of overall included population, and that such benefit was even greater in the subgroup of patients harboring BRCA 1/2 mutations, but failed to improve OS of cancer patients [12], and this need to be taken into account to optimize the design of further clinical studies.
Known mechanisms of primary resistance
It has been already demonstrated that the activity of PARP inhibitors depends from the different levels of HR deficiency [71]. Studies in vitro and in vivo allowed to identify different genes involved in the HR pathway, whose alterations are responsible for different levels of HR deficiency finally influencing the sensitivity to PARP inhibition [72-75].
Certainly a well known mechanism of primary resistance to PARP inhibitors is represented by the mutation status of BRCA-1 and BRCA-2 genes. Indeed it has been shown that breast cancer cells with BRCA1/2 dysfunctions have a greater sensitivity to the PARP inhibitors because of the simultaneous ineffectiveness of both BER and HR mechanisms, respectively linked to the single (PARP inhibitors) and double (loss of function of BRCA1 and/or BRCA2) strand DNA damage, finally producing a chromosomal instability, cell cycle arrest and apoptosis [35, 71].
Another important gene involved in the HR pathway is PTEN. PTEN encodes a phosphatase protein regulating also the PI3K signaling pathway. Mutations and loss of PTEN function have been widely correlated with human carcinogenesis [76]. In glioblastoma cells, the deficiency of PTEN is of crucial importance since it contributes to an increased sensitivity to a temozolamide derivate (N-methylN’-nitro-N-nitrosoguanidine - MNNG). Indeed double strand DNA damages can’t be repaired because of the deficiency PTEN function, therefore resulting in cells death by apoptosis [77, 78]. The same mechanism is responsible for an increased activity of PARP inhibitors also in endometrial or prostate cancer cells with loss of PTEN function compared to wild-type cells, suggesting a potential role of PTEN as a marker of primary resistance to PARP inhibitors [79, 80].
PI3K is another important protein involved in the resistance mechanisms, also because of its close relationship with the above mentioned PTEN signaling pathway. PI3K is a member of a family of lipid kinases, and its activation is required for the functionality of HR pathway [81]. Recent studies have shown that an increased BRCA gene down-regulation, caused by PI3K protein target inhibition, makes triple-negative breast cancer cells more sensitive to PARP inhibitors, suggesting as PI3K functioning could represent a possible mechanism involved in the occurrence of primary resistance [82, 83]. The discovery of the correlation between DNA damage and PI3K activity has also led to the investigation of different PARP inhibitors in solid tumors characterized by PI3K gene alterations, such as NSCLC and colorectal, whereas its prognostic and/or predictive role is currently under investigation [84, 85].
The ATM gene also seems to have a role in the HR mechanisms, therefore on the sensitivity/resistance to PARP inhibitors. The protein kinase ATM shares the chemical group -COOH with PI3K protein and it is particularly involved in the repair of double strand DNA damage caused by ionizing radiation. The dysfunction of such protein leads to an accumulation of DNA damage by oxidative stress. Previous studies have shown that the functionality of ATM and PARP are absolutely necessary for embryonic development of mice, while a functional deficiency of both proteins led to their death within the first few days of life[86]. Subsequently, other studies clarified that PARP1 is more active in cancer cells that, due to the lack of function of ATM, accumulated DNA damage (most represented and studied in prostate cancer, breast cancer, mantle cell lymphoma) [65, 87, 88]. Being PARP-1 and ATM both involved in DNA repair mechanisms (BER and HR respectively), the synergic action between these two proteins could explain the PARP inhibitors resistance of cancer cells leading wild type ATM [86, 89].
As we know NHEJ is defective in about 40% of ovarian cancers, and a recent preclinical study has shown that it is associated with resistance to PARP inhibitors in ex vivo primary cultures independently from HR function [90] (Figure 1).
Although the validation of the above mentioned primary resistance processes is still under investigation, because of the recent advent of PARP inhibitors, it is clear how an important goal in the near future should be represented by the identification of predictive markers of response and /or resistance to PARP inhibitors to better select ideal patients to treat with these drugs. In this sense, a potential role could be played by miRNAs, fractions of non-coding RNA that regulate the post-trascriptional gene expression [91].
Indeed there are several data that suggest that miRNAs take part in modulating the cancer cells’ response to PARP inhibitors. Among the most common miRNAs identified in both ovarian cancer and triple-negative breast cancer, mir-9, mir-506, mir-96, mir-182 and mir-206 are worthy of mention. Liquid biopsy could represent an ideal tool to study this interesting but at the same time greatly complex and confounding scenario [92-96].
New findings on acquired resistance
Since the advent of PARP inhibitors for cancer treatment, there are growing evidences showing that not all patients with BRCAness genes alterations report the same treatment responses. Commonly to other targeted treatments the majority of tumors will develop acquired resistance within 1 year of therapy, leading to the disease progression and the subsequent discontinuation of cancer treatment [73]. It’s likely that the different emerging mechanisms of resistance may depend from the original BRCAness gene alterated, ultimately leading to different patterns of treatment response observed in clinical setting [97].
Considering the recent introduction of PARP inhibitors for clinical use, there is currently very limited understanding about the molecular mechanisms underlying the occurrence of acquired resistance to this family of drugs. The majority of available data have emerged from pre-clinical studies, showing several alterations responsible for the partial or complete restoration of the HR repair function, including: secondary mutations in BRCA1/2, Pgp overexpression, and the loss of 53BP1 protein function. Several studies have shown the occurrence of secondary mutations in the alterated BRCA1-2 genes during PARP inhibitors therapy, leading to a partial restoration of both BRCA protein and the related HR repair function, with the subsequent induction of resistance [97-99]. Mutations of C-terminal domain of BRCA1 (BRCT) may promote PARP inhibitor resistance preventing the recruitment of protein complexes which are useful in the site of the damaged DNA [100]. The same mechanism has been shown in human tumor cell lines harboring BRCA2 6174delT frameshift mutation. The result of such alteration is a truncated, non functional protein, because of the loss of C-terminal portion, BRC repeats, DNA-binding/DSS1 interaction domain, finally resulting in both platinum and PARP-inhibitors resistant phenotype [97].The over-expression of Glicoprotein-P (Pgp) as mechanism of resistance has been first observed in ovarian cell-lines receiving combination chemotherapy treatment with Paclitaxel plus Doxorubicin. Recently a significant up-regulation of Pgp associated mdr1 a/b genes has been identified in ovarian cell lines undergone prolonged treatment with olaparib. In-vivo studies in mouse models have clearly shown as such drug may be immediately ejected by tumor cells by overexpression of Pgp on the cell surface, confirming a potential role of such protein in the development of acquired resistance to Olaparib [101]. However such kind of resistance could be overcome by the addition of Pgp inhibitors to olaparib, as well as using other members of PARP inhibitors family which are less sensitive to Pgp mechanism of action, such as veliparib.
In 2010, in vitro and in vivo-studies by Bouwman and Bounting, respectively, have first demonstrated that loss of P53-binding protein 1 (53BP1) function was associated with the partial restoration of the HR repair function in BRCA1 mutated models [102, 103]. Indeed 53BP1 protein is involved in the control of DNA clivage at DSBs mediating BRCA-mutated cells death during PARP-inhibitors treatment. Recent evidences have shown also decreased levels of 53BP1 in patients with ovarian cancer who developed acquired resistance to both platinum-based chemotherapy and PARP-inhibitors, confirming a potential role of such protein in the occurrence of resistance. Other data emerging from in vivo-studies have shown the simultaneous occurrence of different alterations responsible for the development of resistance to PARP inhibition in mouse models, including BRCA1 secondary mutations, Pgp over-expression and loss of 53BP1 protein function [102].
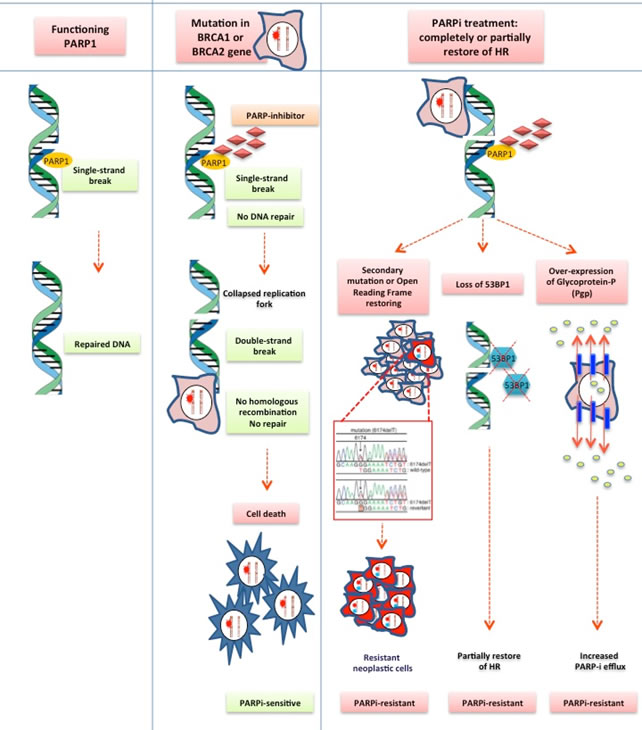
Figure 1: PARP inhibitor acquired resistance mechanism. Based from pre-clinical studies, several alteration are responsible of partial or complete restoration of the HR repair function: secondary mutations in BRCA1-2; loss of 53BP1 protein function; Pgp over-expression and PARPi efflux.
Table 1: Phase II and III clinical trials investigated PARP inhibitors in breast and ovarian cancer.
Conclusions
BRCA mutations increased tumor-cells sensitivity to PARP inhibitors by synthetic lethality and several compounds targeting the PARP enzymes system have been evaluated in clinical trials. In this review we have summarized the most relevant clinical trials carried out in patients affected by advanced breast or ovarian cancer or other malignancies and treated with PARP inhibitors alone or in combination with chemotherapy. The positive results of such studies have recently led to the approval of the first PARPi olaparib as maintenance therapy for platinum-sensitive patients with advanced, recurrent HGSOC, fallopian tube, or primary peritoneal cancer, harboring BRCA-mutations. Therefore offering to test for germline BRCA1/2 mutations should now be considered a routine part of clinical practice, in order to provide the best treatment strategy to each patient. Even if BRCA1/2 mutations have shown to be highly predictive of PARPi activity in the majority of studies, PARPi have shown also activity in patients without BRCA-mutations, especially in HRD-positive population. Testing for somatic BRCA mutations and HRD-signature is currently a developing area with interesting implications for clinical practice. There are some mechanisms that have been reported as responsible of primary resistance to these molecules. BRCA wild type status, PTEN, PI3K and ATM have been evaluated as markers of primary resistance. Many studies aim to identify positive predictive markers to select patients who may benefit most from PARP inhibition. In particular it has been hypothesized that the different action of PARP inhibitors may depend on the expression of some miRNAs. All patients initially responding to PARP inhibition at some time later develop acquired resistance. The suggested mechanisms of acquired resistance include secondary mutations in BRCA1-2, Pgp over-expression and loss of 53BP1 protein function. Further pre-clinical and clinical studies are required to better define the role of these innovative targeted agents in cancer treatment. It seems reasonable that for the peculiar mechanisms of action, PARP inhibitors could be usefully combined with both DNA damaging cytotoxic agents, radiotherapy, and with other targeted therapies. Initial results of combining PARPi with anti-angiogenic drugs are promising, leading to several randomized studies in different lines of treatment. Finally PARPi and PD1/PDL1 checkpoint inhibitors combinations have recently shown durable responses, emerging as another promising strategy to expand the treatment arsenal against cancer.
Conflicts of Interest
The authors declared no potential financial conflicts of interest.
GRANT SUPPORT
This work was supported by the Consorzio Interuniversitario Nazionale per la Bio-Oncologia (CINBO).
References
1. Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene 2006; 25: 5864-5874
2. O’Donovan PJ, Livingston DM. BRCA1 and BRCA2: breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis 2010; 31: 961-967.
3. Walsh CS. Two decades beyond BRCA1/2: Homologous recombination, hereditary cancer risk and a target for ovarian cancer therapy. Gynecol Oncol 2015; 137: 343-350.
4. Cavanagh H, Rogers KM. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered Cancer Clin Pract 2015; 13: 16.
5. Kobayashi H, Ohno S, Sasaki Y, Matsuura M. Hereditary breast and ovarian cancer susceptibility genes (review). Oncol Rep 2013; 30: 1019-1029.
6. Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol 2011; 5: 387-393.
7. Yap TA, Sandhu SK, Carden CP, de Bono JS. Poly(ADP-ribose) polymerase (PARP) inhibitors: Exploiting a synthetic lethal strategy in the clinic. CA Cancer J Clin 2011; 61: 31-49.
8. Beck C, Robert I, Reina-San-Martin B, Shreiber V, Dantzer F. Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp Cell Res 2014; 329: 18-25.
9. Lord CJ, Ashworth A. Targeted therapy for cancer using PARP inhibitors. Curr Opin Pharmacol 2008; 8: 363-369.
10. Ricks TK, Chiu HJ, Ison G, Kim G, McKee AE, Kluetz P, Padzur R. Successes and Challenges of PARP Inhibitors in Cancer Therapy. Front Oncol 2015; 5: 222.
11. Tangutoori S, Baldwin P, Sridhar S. PARP inhibitors: A new era of targeted therapy. Maturitas 2015; 81: 5-9.
12. Bao Z, Cao C, Geng X, Tian B, Wu Y, Zhang C, Chen Z, Li W, Shen H and Ying S. Effectiveness and safety of poly (ADP-ribose) polymerase inhibitors in cancer therapy: A systematic review and meta-analysis. Oncotarget. 2016; 7:7629-7639. doi: 10.18632/oncotarget.5367.
13. Livraghi L, Garber JE. PARP inhibitors in the management of breast cancer: current data and future prospects. BMC Med 2015; 13: 188.
14. Lupo B, Trusolino L. Inhibition of poly(ADP-ribosyl)ation in cancer: old and new paradigms revisited. Biochim Biophys Acta 2014; 1846: 201-215.
15. Bouwman P, Jonkers J. Molecular pathways: how can BRCA-mutated tumors become resistant to PARP inhibitors? Clin Cancer Res 2014; 20: 540-547.
16. Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, Fenwick K, Assiotis I, Rodrigues DN, Reis Filho JS, Moreno V, Mateo J, Molife LR, De Bono J, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol 2013; 229: 422-429.
17. Jackson SP. Detecting, signalling and repairing DNA double-strand breaks. Biochem Soc Trans. 2001; 29:655-661.
18. Wilson SH. Mammalian base excision repair and DNA polymerase beta. Mutat Res. 1998; 407:203-215.
19. Lindahl T. DNA repair enzymes. Annu Rev Biochem. 1982; 51:61-87.
20. Seeberg E, Eide L and Bjørås M. The base excision repair pathway. Trends Biochem Sci. 1995; 20:391-397.
21. Krokan HE and Bjørås M. Base excision repair. Cold Spring Harb Perspect Biol. 2013; 5:a012583.
22. Ogata N, Ueda K, Kawaichi M and Hayaishi O. Poly(ADP-ribose) synthetase, a main acceptor of poly(ADP-ribose) in isolated nuclei. J Biol Chem. 1981; 256:4135-4137.
23. Krishnakumar R and Kraus WL. PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol Cell. 2010; 39:736-749.
24. Robu M, Shah RG, Petitclerc N, Brind’Amour J, Kandan-Kulangara F and Shah GM. Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc Natl Acad Sci U S A. 2013; 110:1658-1663.
25. Smith J, Tho LM, Xu N and Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010; 108:73-112.
26. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM and Ding W. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994; 266:66-71.
27. King MC, Marks JH, Mandell JB and Group NYBCS. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003; 302(5645):643-646.
28. Zweemer RP, van Diest PJ, Verheijen RH, Ryan A, Gille JJ, Sijmons RH, Jacobs IJ, Menko FH and Kenemans P. Molecular evidence linking primary cancer of the fallopian tube to BRCA1 germline mutations. Gynecol Oncol. 2000; 76:45-50.
29. Mersch J, Jackson MA, Park M, Nebgen D, Peterson SK, Singletary C, Arun BK and Litton JK. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015; 121:269-275.
30. Hoa NN, Kobayashi J, Omura M, Hirakawa M, Yang SH, Komatsu K, Paull TT, Takeda S and Sasanuma H. BRCA1 and CtIP Are Both Required to Recruit Dna2 at Double-Strand Breaks in Homologous Recombination. PLoS One. 2015; 10:e0124495.
31. Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, Martinez-Perez E, Boulton SJ and La Volpe A. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell. 2010; 39:25-35.
32. Burdak-Rothkamm S, Rothkamm K, McClelland K, Al Rashid ST and Prise KM. BRCA1, FANCD2 and Chk1 are potential molecular targets for the modulation of a radiation-induced DNA damage response in bystander cells. Cancer Lett. 2015; 356:454-461.
33. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ and Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434:913-917.
34. Curtin NJ and Szabo C. Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol Aspects Med. 2013; 34:1217-1256.
35. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434:917-921.
36. Schultz N, Lopez E, Saleh-Gohari N and Helleday T. Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res. 2003; 31:4959-4964.
37. Morgan WF and Cleaver JE. 3-Aminobenzamide synergistically increases sister-chromatid exchanges in cells exposed to methyl methanesulfonate but not to ultraviolet light. Mutat Res. 1982; 104:361-366.
38. Metzger MJ, Stoddard BL and Monnat RJ. PARP-mediated repair, homologous recombination, and back-up non-homologous end joining-like repair of single-strand nicks. DNA Repair (Amst). 2013; 12:529-534.
39. Sandhu SK, Omlin A, Hylands L, Miranda S, Barber LJ, Riisnaes R, Reid AH, Attard G, Chen L, Kozarewa I, Gevensleben H, Campbell J, Fenwick K, et al. Poly (ADP-ribose) polymerase (PARP) inhibitors for the treatment of advanced germline BRCA2 mutant prostate cancer. Ann Oncol. 2013; 24:1416-1418.
40. Paul I, Savage KI, Blayney JK, Lamers E, Gately K, Kerr K, Sheaff M, Arthur K, Richard DJ, Hamilton PW, James JA, O’Byrne KJ, Harkin DP, et al. PARP inhibition induces BAX/BAK-independent synthetic lethality of BRCA1-deficient non-small cell lung cancer. J Pathol. 2011; 224:564-574.
41. O’Sullivan CC, Moon DH, Kohn EC and Lee JM. Beyond Breast and Ovarian Cancers: PARP Inhibitors for BRCA Mutation-Associated and BRCA-Like Solid Tumors. Front Oncol. 2014; 4:42.
42. Ang JE, Gourley C, Powell CB, High H, Shapira-Frommer R, Castonguay V, De Greve J, Atkinson T, Yap TA, Sandhu S, Banerjee S, Chen LM, Friedlander ML, et al. Efficacy of chemotherapy in BRCA1/2 mutation carrier ovarian cancer in the setting of PARP inhibitor resistance: a multi-institutional study. Clin Cancer Res. 2013; 19:5485-5493.
43. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009; 361:123-134.
44. Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, De Greve J, Lubinski J, Shanley S, Messiou C, A’Hern R, Tutt A, Ashworth A, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010; 28:2512-2519.
45. Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, Lu K, Schmutzler RK, Matulonis U, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010; 376:245-251.
46. Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, Yerushalmi R, Macpherson E, Carmichael J et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011; 12:852-861.
47. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira-Frommer R, Safra T, Matei D, Fielding A, Spencer S, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014; 15:852-861.
48. Ledermann JA, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira-Frommer R, Safra T, Matei D, Fielding A, Spencer S, et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol. 2016; 17:1579-1589.
49. McNeish IA, Oza AM, Coleman RL, Scott CL, Konecny GE, Tinker A, O’Malley DM, Brenton J, Kristeleit RS, Bell-McGuinn K, Oaknin A, Leary A, Lin K, et al. Results of ARIEL2: A Phase 2 trial to prospectively identify ovarian cancer patients likely to respond to rucaparib using tumor genetic analysis. J Clin Oncol. 2015; 33, suppl, abstr 5008.
50. Coleman RL, Swisher EM, Oza AM, Scott CL, i Giordano H,. Lin KK,. Konecny GE, Tinker A, O’Malley DM, Kristeleit RS, Ma L, Bell-McGuinn KM, Brenton JD, et al. Refinement of prespecified cutoff for genomic loss of heterozygosity (LOH) in ARIEL2 part 1: A phase II study of rucaparib in patients (pts) with high grade ovarian carcinoma (HGOC). J Clin Oncol. 2016; 34, suppl, abstr 5540.
51. Coleman RL, Sill MW, Bell-McGuinn K, Aghajanian C, Gray HJ, Tewari KS, Rubin SC, Rutherford TJ, Chan JK, Chen A and Swisher EM. A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation - An NRG Oncology/Gynecologic Oncology Group study. Gynecol Oncol. 2015; 137:386-391.
52. Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, Hylands L, Riisnaes R, Forster M, Omlin A, Kreischer N, Thway K, Gevensleben H, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013; 14:882-892.
53. Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I, Ben-Baruch NE, Marth C, Mądry R, et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med. 2016; 375:2154-2164.
54. Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RH, Sonke GS, Colombo N, Špaček J, Vuylsteke P, Hirte H, Mahner S, Plante M, Schmalfeldt B, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol. 2015; 16:87-97.
55. Kummar S, Oza AM, Fleming GF, Sullivan DM, Gandara DR, Naughton MJ, Villalona-Calero MA, Morgan RJ, Szabo PM, Youn A, Chen AP, Ji J, Allen DE, et al. Randomized Trial of Oral Cyclophosphamide and Veliparib in High-Grade Serous Ovarian, Primary Peritoneal, or Fallopian Tube Cancers, or BRCA-Mutant Ovarian Cancer. Clin Cancer Res. 2015; 21:1574-1582.
56. Liu JF, Barry WT, Birrer M, Lee JM, Buckanovich RJ, Fleming GF, Rimel B, Buss MK, Nattam S, Hurteau J, Luo W, Quy P, Whalen C, et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol. 2014; 15:1207-1214.
57. Mirza MR, Mortensen CE, Avall-Lundqvist E, Bjorge L, Berek JS, Herrstedt J, Holm AJ, Kirkegaard T. and Maenpaa J. ENGOT-OV24-NSGO/AVANOVA: Niraparib versus bevacizumab-niraparib combination versus bevacizumab and niraparib as sequential therapy in women with platinum-sensitive epithelial ovarian, fallopian tube, or peritoneal cancer. J Clin Oncol . 2015; 33: (suppl; abstr TPS5607).
58. Lee J-M, Zimmer ADS, Lipkowitz S, Annunziata CM, Ho TW, Chiou VL, Minasian LM, Houston ND, Ekwede I, Kohn EC. Phase I study of the PD-L1 inhibitor, durvalumab (MEDI4736; D) in combination with a PARP inhibitor, olaparib (O) or a VEGFR inhibitor, cediranib (C) in women’s cancers (NCT02484404). J Clin Oncol. 2016; 34: (suppl; abstr 3015).
59. Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010; 376:235-244.
60. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, Mitchell G, Fried G, Stemmer SM, Hubert A, Rosengarten O, Steiner M, Loman N, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015; 33:244-250.
61. Dwadasi S, Tong Y, Walsh T, Danso MA, Ma CX, Silverman P, King M-C,. Perkins SM, Badve SS, Miller K. Cisplatin with or without rucaparib after preoperative chemotherapy in patients with triple-negative breast cancer (TNBC): Hoosier Oncology Group BRE09-146. J Clin Oncol. 2014; 32:5s, (suppl; abstr 1019^)
62. Lucas AL, Shakya R, Lipsyc MD, Mitchel EB, Kumar S, Hwang C, Deng L, Devoe C, Chabot JA, Szabolcs M, Ludwig T, Chung WK and Frucht H. High prevalence of BRCA1 and BRCA2 germline mutations with loss of heterozygosity in a series of resected pancreatic adenocarcinoma and other neoplastic lesions. Clin Cancer Res. 2013; 19:3396-3403.
63. Leongamornlert D, Mahmud N, Tymrakiewicz M, Saunders E, Dadaev T, Castro E, Goh C, Govindasami K, Guy M, O’Brien L, Sawyer E, Hall A, Wilkinson R, et al. Germline BRCA1 mutations increase prostate cancer risk. Br J Cancer. 2012; 106:1697-1701.
64. Marks JL, Golas B, Kirchoff T, Miller VA, Riely GJ, Offit K and Pao W. EGFR mutant lung adenocarcinomas in patients with germline BRCA mutations. J Thorac Oncol. 2008; 3:805.
65. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, Boysen G, Porta N, Flohr P, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015; 373:1697-1708.
66. Middleton MR, Friedlander P, Hamid O, Daud A, Plummer R, Falotico N, Chyla B, Jiang F, McKeegan E, Mostafa NM, Zhu M, Qian J, McKee M, et al. Randomized phase II study evaluating veliparib (ABT-888) with temozolomide in patients with metastatic melanoma. Ann Oncol. 2015; 26:2173-2179.
67. Plummer R, Lorigan P, Steven N, Scott L, Middleton MR, Wilson RH, Mulligan E, Curtin N, Wang D, Dewji R, Abbattista A, Gallo J and Calvert H. A phase II study of the potent PARP inhibitor, Rucaparib (PF-01367338, AG014699), with temozolomide in patients with metastatic melanoma demonstrating evidence of chemopotentiation. Cancer Chemother Pharmacol. 2013; 71:1191-1199.
68. Ramalingam S, Blais N, Mazieres J, Reck M, Jones CM, Juhasz E, Urban L, Orlov S, Barlesi F, Kio E, Keilholz U, Qin Q, Qian J, et al. A randomized, double-blind, phase 2 trial of veliparib (ABT-88) with carboplatin and paclitaxel in previously untreated metastatic or advanced non-small cell lung cancer. (Chicago Multidisciplinary Symposium in Thoracic Oncology. Abstract 8. Presented October 30, 2014.
69. Novello S, Besse B, Felip E, Barlesi F, Mazieres J, Zalcman G, von Pawel J, Reck M, Cappuzzo F, Ferry D, Carcereny E, Santoro A, Garcia-Ribas I, et al. A phase II randomized study evaluating the addition of iniparib to gemcitabine plus cisplatin as first-line therapy for metastatic non-small-cell lung cancer. Ann Oncol. 2014; 25:2156-2162.
70. Bang YJ, Im SA, Lee KW, Cho JY, Song EK, Lee KH, Kim YH, Park JO, Chun HG, Zang DY, Fielding A, Rowbottom J, Hodgson D, et al. Randomized, Double-Blind Phase II Trial With Prospective Classification by ATM Protein Level to Evaluate the Efficacy and Tolerability of Olaparib Plus Paclitaxel in Patients With Recurrent or Metastatic Gastric Cancer. J Clin Oncol. 2015; 33:3858-3865.
71. Davar D, Beumer JH, Hamieh L and Tawbi H. Role of PARP inhibitors in cancer biology and therapy. Curr Med Chem. 2012; 19:3907-3921.
72. Bouwman P and Jonkers J. Molecular pathways: how can BRCA-mutated tumors become resistant to PARP inhibitors? Clin Cancer Res. 2014; 20:540-547.
73. McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, Smith GC and Ashworth A. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006; 66:8109-8115.
74. Martin SA, Lord CJ and Ashworth A. DNA repair deficiency as a therapeutic target in cancer. Curr Opin Genet Dev. 2008; 18:80-86.
75. Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008; 26:3785-3790.
76. Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, Waldman T, Lord CJ and Ashworth A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009; 1:315-322.
77. McEllin B, Camacho CV, Mukherjee B, Hahm B, Tomimatsu N, Bachoo RM and Burma S. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res. 2010; 70:5457-5464.
78. Majuelos-Melguizo J, Rodríguez MI, López-Jiménez L, Rodríguez-Vargas JM, Martí Martín-Consuegra JM, Serrano-Sáenz S, Gavard J, de Almodóvar JM and Oliver FJ. PARP targeting counteracts gliomagenesis through induction of mitotic catastrophe and aggravation of deficiency in homologous recombination in PTEN-mutant glioma. Oncotarget. 2015; 6:4790-4803. doi: 10.18632/oncotarget.2993.
79. Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, Vatcheva R, Savage K, Mackay A, Lord CJ, Ashworth A and Reis-Filho JS. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2010; 2:53ra75.
80. Zhang J. Poly (ADP-ribose) polymerase inhibitor: an evolving paradigm in the treatment of prostate cancer. Asian J Androl. 2014; 16:401-406.
81. Kumar A, Fernandez-Capetillo O, Fernadez-Capetillo O and Carrera AC. Nuclear phosphoinositide 3-kinase beta controls double-strand break DNA repair. Proc Natl Acad Sci U S A. 2010; 107:7491-7496.
82. Ibrahim YH, García-García C, Serra V, He L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzmán M, Grueso J, Rodríguez O, Calvo MT, Aura C, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012; 2:1036-1047.
83. Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmañà J, Rajendran A, Papa A, Spencer K, Lyssiotis CA, Nardella C, Pandolfi PP, Baselga J, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012; 2:1048-1063.
84. Cardnell RJ, Feng Y, Diao L, Fan YH, Masrorpour F, Wang J, Shen Y, Mills GB, Minna JD, Heymach JV and Byers LA. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin Cancer Res. 2013; 19:6322-6328.
85. Soncini D, Caffa I, Patrone F, Ballestrero A and Nencioni A. Synthetic lethality-based therapeutics: perspectives for applications in colorectal cancer. Curr Cancer Drug Targets. 2012; 12:329-338.
86. Aguilar-Quesada R, Muñoz-Gámez JA, Martín-Oliva D, Peralta A, Valenzuela MT, Matínez-Romero R, Quiles-Pérez R, Menissier-de Murcia J, de Murcia G, Ruiz de Almodóvar M and Oliver FJ. Interaction between ATM and PARP-1 in response to DNA damage and sensitization of ATM deficient cells through PARP inhibition. BMC Mol Biol. 2007; 8:29.
87. Williamson CT, Muzik H, Turhan AG, Zamò A, O’Connor MJ, Bebb DG and Lees-Miller SP. ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Mol Cancer Ther. 2010; 9:347-357.
88. Gilardini Montani MS, Prodosmo A, Stagni V, Merli D, Monteonofrio L, Gatti V, Gentileschi MP, Barilà D and Soddu S. ATM-depletion in breast cancer cells confers sensitivity to PARP inhibition. J Exp Clin Cancer Res. 2013; 32:95.
89. Liang Y, Lin SY, Brunicardi FC, Goss J and Li K. DNA damage response pathways in tumor suppression and cancer treatment. World J Surg. 2009; 33:661-666.
90. McCormick A, Donoghue P, Dixon M, O’Sullivan R, O’Donnell RL, Murray J, Kaufmann A, Curtin NJ and Edmondson RJ. Ovarian Cancers Harbour Defects in Non-Homologous End Joining Resulting in Resistance to Rucaparib. Clin Cancer Res. 2016.
91. Neijenhuis S, Bajrami I, Miller R, Lord CJ and Ashworth A. Identification of miRNA modulators to PARP inhibitor response. DNA Repair (Amst). 2013; 12:394-402.
92. Sun C, Li N, Yang Z, Zhou B, He Y, Weng D, Fang Y, Wu P, Chen P, Yang X, Ma D, Zhou J and Chen G. miR-9 regulation of BRCA1 and ovarian cancer sensitivity to cisplatin and PARP inhibition. J Natl Cancer Inst. 2013; 105:1750-1758.
93. Liu G, Yang D, Rupaimoole R, Pecot CV, Sun Y, Mangala LS, Li X, Ji P, Cogdell D, Hu L, Wang Y, Rodriguez-Aguayo C, Lopez-Berestein G, et al. Augmentation of response to chemotherapy by microRNA-506 through regulation of RAD51 in serous ovarian cancers. J Natl Cancer Inst. 2015; 107.
94. Wang Y, Huang JW, Calses P, Kemp CJ and Taniguchi T. MiR-96 downregulates REV1 and RAD51 to promote cellular sensitivity to cisplatin and PARP inhibition. Cancer Res. 2012; 72:4037-4046.
95. Moskwa P, Buffa FM, Pan Y, Panchakshari R, Gottipati P, Muschel RJ, Beech J, Kulshrestha R, Abdelmohsen K, Weinstock DM, Gorospe M, Harris AL, Helleday et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol Cell. 2011; 41:210-220.
96. Sasaki A, Tsunoda Y, Tsuji M, Udaka Y, Oyamada H, Tsuchiya H and Oguchi K. Decreased miR-206 expression in BRCA1 wild-type triple-negative breast cancer cells after concomitant treatment with gemcitabine and a Poly(ADP-ribose) polymerase-1 inhibitor. Anticancer Res. 2014; 34:4893-4897.
97. Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS and Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008; 451:1111-1115.
98. Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP, Wurz K, Higgins J, Villegas E and Taniguchi T. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009; 69:6381-6386.
99. Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, Fenwick K, Assiotis I, Rodrigues DN, Reis Filho JS, Moreno V, Mateo J, Molife LR, De Bono J, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013; 229:422-429.
100. Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, Wang Y, Capelletti M, Sarosiek KA, Moreau LA, Chowdhury D, Wickramanayake A, Harrell MI, et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci U S A. 2013; 110:17041-17046.
101. Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A, Doroshow JH, Cranston A, Martin NM, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013; 3:68-81.
102. Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, Haffty BG, Tommiska J, Blomqvist C, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010; 17:688-695.
103. Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, Xu X, Deng CX, Finkel T, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010; 141:243-254.