
INTRODUCTION
Renal cell carcinoma (RCC) is one of the most common cancer types worldwide with clear cell RCC (ccRCC) being the most frequent and aggressive RCC subtype [1, 2]. In ccRCC the von Hippel-Lindau tumor suppressor gene (VHL) is frequently altered by deletion of one allele (90%) and promoter methylation (up to 20%) or mutations (70-80%) of the other. VHL inactivation is considered as a critical part of tumor initiation [3-5]. In addition to its well-known function as E3 ubiquitin ligase for ubiquitination and proteasomal degradation of hypoxia-inducible factor subunits (HIF1α and HIF2α) [6-8], the VHL protein (pVHL) has been recently identified as a multiadaptor protein involved in a variety of cellular processes such as microtubule stability, activation of p53, neuronal apoptosis, cellular senescence and aneuploidy, ubiquitination of RNA polymerase II and regulation of NFkB activity [1].
Given different types of VHL mutations, a deeper insight in the biological effects of VHL mutations may allow a better prediction of ccRCC prognosis. In particular, VHL loss-of-function mutations (LOF) (frameshift, nonsense and splice site mutations) highly likely abrogate pVHL function, whereas the consequences of missense mutations on pVHL stability and target binding ability are rather unclear. Missense mutations may provoke diverse effects on pVHL interactions with binding partners, thus exerting different impact on pathways normally regulated by pVHL. This was shown for HIF1α and HIF2α degradation [9] as well as for other pVHL binding partners, including Jade1, RPB1, VDU1, EEF1A1 and CCT-ζ-2, for which loss of binding capability upon missense mutations was demonstrated [10-15].
p53 is a well-known tumor suppressor gene, whose activation by hypoxia or DNA damage leads to cell cycle arrest, DNA repair and apoptosis. Under cellular stress, p53 level is increased by inhibition of its interaction with MDM2 and activated by post-translational modifications through different regulators which lead to transactivation of its downstream target genes p21 (alias CDKN1A, growth arrest), Bax and Noxa (apoptosis) [16]. The role of p53 in ccRCC and its relation to pVHL is yet unclear. Two previous studies showed that pVHL can stabilize p53 and enhance its transcriptional activity [17, 18] whereas another study found that p53 expression is not pVHL-dependent [19]. In addition, pVHL inactivation in RCC cells lead to decreased apoptosis [20], which may be explained by the lack of phosphorylation of pVHL by checkpoint-kinase 2, impairing the recruitment of p53 coactivators (such as p300 and Tip60) [21].
Tumors with p53 mutations are known to be associated with chemoresistance [22]. p53 is one of the most frequently mutated genes in several cancers [23], but p53 mutations are rare in ccRCC [24-26]. Interestingly, ccRCC is resistant to chemotherapy and Gurova et al. suggested that p53 signaling is repressed by mechanisms independent of p53 mutations [27]. ccRCC is currently treated with anti-angiogenic drugs, such as the Tyrosine-Kinase-Inhibitors (TKI) Sorafenib and Sunitinib, to counter the effects of the HIF1/2α accumulation occurring upon pVHL inactivation. The efficiency of this therapeutic strategy is still suboptimal [28]. As shown for colorectal cancer where p53 negative cells were less responsive to anti-angiogenic treatment than wild-type p53 cells [29], alteration of p53 signaling may also be an explanation for the low response rate in ccRCC.
We hypothesized that VHL missense mutations occurring in the p53 binding domain of pVHL lead to deficient p53 transactivation and/or promote HIF1α and HIF2α accumulation, thus impacting tumor behavior and response to treatment. In this study, we investigated four different missense mutations located in the p53 binding site (codons 154-163), which is overlapping with the ElonginC binding domain (codons 157-171). Due to this overlap, the missense mutations investigated could have an impact on p53 signaling and/or on HIF1/2α degradation through an altered binding to ElonginC. Our goal was to evaluate the selected missense mutations effectiveness in HIF1/2α degradation, p53 transactivation, and their response to chemotherapy and TKI.
RESULTS
Expression of p53 in ccRCC
Pathological and molecular characteristics of renal cell cancers on the tissue microarray (TMA) were previously described [9, 30]. In brief, the TMA consisted of 262 clear cell, 48 papillary (24 type I, 24 type II), 15 chromophobe RCC, eight non specified RCC, 22 oncocytoma and 28 normal tissue cores. 181 of 262 ccRCC were VHL mutated (69%).
TMA analysis revealed absent or only low p53 expression in most ccRCC (76%), and chromophobe RCC (80%), whereas nuclear p53 positivity was high in about 60% of papillary RCC (Figure 1A). By separating the 262 ccRCC in VHL wild-type and VHL mutated tumors, we observed that p53 expression was less frequent in tumors with VHL alterations (p=0.0212) (Figure 1B). Notably, the p53 expression frequency decreased with the predicted impact of mutations on the function of pVHL (Figure 1C).

Figure 1: A. p53 protein expression in RCC tissue; B. p53 protein expression in ccRCC and VHL mutation status; and C. p53 protein expression in ccRCC and mutation impact on pVHL stability. Wt: VHL wild type; Mild impact: Destabilizing or neutral or stabilizing; High impact: highly stabilizing or highly destabilizing; LOF (Loss of Function): Frameshift and nonsense mutations. p-value: *<0.05, **<0.01, ***<0.001
Selection of mutations in the p53/EloC binding domains of pVHL
The binding domains of p53 and EloC are located between codons 154 to 163 and 157 to 171, respectively [34]. Among 254 VHL mutations found in 360 ccRCC tissue specimen (70.6%), 25 (9.8%) resided in these binding domains. Eleven (4.3%) were missense mutations causing an amino acid exchange. Eight missense mutations were predicted to destabilize pVHL and three missense mutations had no or little impact on pVHL stability.
For this study, we selected four out of 11 missense mutations occurring in the p53/EloC binding domain for further analysis. Three were predicted to have mild impact on pVHL (Leu158Val, Arg161Gln, and Cys162Arg) and one was predicted to highly destabilize pVHL (Arg161Pro). Two other missense mutations occurring in the HIF1/2α binding domain (Tyr98His, Tyr98Asn), as well as one nonsense mutation located in the p53/EloC binding domain (Arg161X) were predicted to differently impair HIF1/2α degradation pathway and were used as controls for the cell line experiments (summarized in Table 1).
Table 1: Cell lines with VHL-expressing vector constructs, predicted effects on pVHL stability and affected binding domains of pVHL
RCC4 cell lines stably expressing: |
Predicted effect |
Binding domain affected |
|---|---|---|
Babe (Ser65Trp) |
destabilizing and cause protein malfunction |
others |
VHL30 |
none |
none |
Leu158Val |
slightly destabilizing |
p53/EloC |
Arg161Gln |
neutral |
p53/EloC |
Cys162Arg |
neutral |
p53/EloC |
Arg161Pro |
destabilizing and cause protein malfunction |
p53/EloC |
Tyr98His |
neutral |
HIF1/2α |
Tyr98Asn |
destabilizing |
HIF1/2α |
Arg161X |
Loss of function |
HIF1/2α/p53/EloC |
Effects of selected VHL mutations on HIF1/2α and p53
The established stable cell lines were investigated by Western Blot for the effects of the VHL missense mutations on HIF1/2α degradation and the two HIF1/2α targets CAIX and Glut1 (Figure 2). As expected, pVHL was undetectable in RCC4 Babe and RCC4 Arg161X. HIF1α and 2α were stabilized in RCC4 Babe, Arg161Pro, Arg161X, and also in RCC4 Cys162Arg. RCC4 Arg161Gln, Tyr98His and Tyr98Asn showed partial HIF1α and 2α degradation. HIF1/2α strong degradation was seen in Leu158Val which was similar to RCC4 VHL30 (wild-type). CAIX and Glut1 expression correlated with HIF1/2α. No clear difference in p53 expression was seen between VHL wild-type and mutated cell lines.
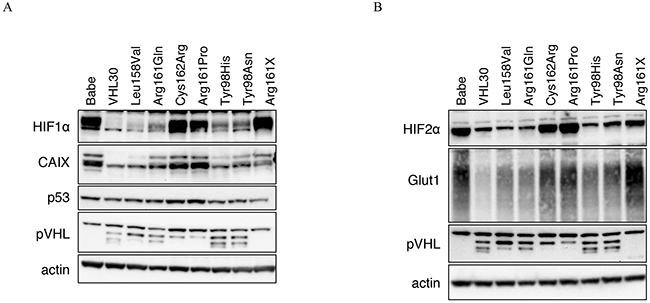
Figure 2: A. and B. Western blots showing pVHL, p53, HIF1/2α and HIF1/2α targets expression (CAIX and Glut1) in the established stable cell lines.
Impact of VHL mutations on p53 downstream targets in cell lines
Previous studies showed that pVHL enhances p53 transcriptional activity which was reduced by Ser111Arg or Ser111Cys missense mutations [19, 23]. Here we studied the effects of our selected VHL mutations on the RNA level of TP53 and its effectors p21, Bax and Noxa. The transcription levels of TP53, p21 and Noxa in cells expressing the different mutant forms were generally lower compared to the wild-type form of pVHL (Figure 3). Transcription levels of p21 and Noxa were significantly reduced in most VHL mutants compared to RCC4 expressing VHL wild-type. Bax RNA levels were lower only in RCC4 Leu158Val and Arg161Gln. Among the missense mutations in the p53 binding domain, no significant difference in RNA levels was seen between the three non-destabilizing/slightly destabilizing mutants and the one with destabilizing effects on pVHL.
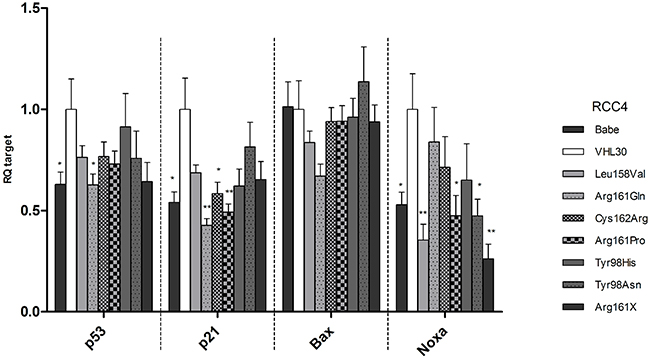
Figure 3: mRNA levels of TP53, p21, Bax, and Noxa in the established RCC4 stable cell lines. For each gene the mRNA level of the VHL30 sample was used as reference and compared to the mRNA levels in the other cell lines. p-value: *<0.05, **<0.01 (data are presented as mean +SEM).
Impact of VHL mutations on p53 downstream targets in tissues
Nine ccRCC tissues (one VHL wild-type, two with mutation Leu158Val (#1, #2), one with Arg161Gln, one with Cys162Arg, one with Arg161Pro, one with Tyr98Asn and two with Arg161X (#1, #2)) and three normal kidney tissues were investigated for RNA expression of VHL, TP53 and its downstream targets. TP53, p21, Bax and Noxa RNA levels relative to VHL were compared in three normal tissues and in one VHL wild-type ccRCC. TP53, p21, Bax and Noxa expression levels in tumor tissue were similar to the ones seen in normal tissues (Figure 4). The RNA levels of TP53 and its downstream targets relative to VHL transcription levels were assessed in ccRCC samples expressing the mutant forms of VHL (Figure 5). Similarly to the established cell lines, wild-type VHL tumor showed generally higher RNA levels of TP53 and its downstream targets than VHL mutated tumors. All eight mutant samples showed a decrease of p21, Bax and Noxa RNA levels compared to VHL wild-type except for Noxa in the Cys162Arg mutated tumor. VHL missense mutations located in the p53 binding domain and VHL mutations in the HIF1/2α binding domain were similarly affected in p53 signaling. Notably, no significant differences in the RNA levels were observed between the mutation Arg161Pro which highly destabilize pVHL and the other mutations which are predicted to have a lower impact on pVHL stability.
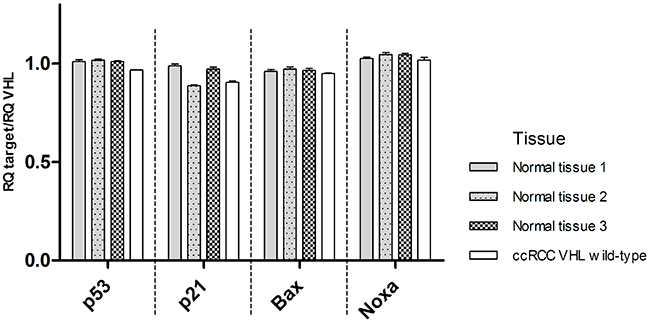
Figure 4: mRNA levels of TP53, p21, Bax, Noxa relative to VHL transcription levels in three normal kidney tissues and one VHL wild-type ccRCC (data are presented as mean +SEM).
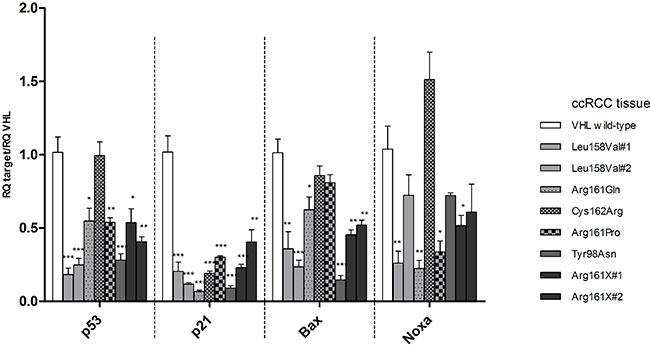
Figure 5: mRNA levels of TP53, p21, Bax, Noxa relative to VHL wild type transcription levels in ccRCC tissue samples carrying the VHL mutations selected for cell line experiments. No tissue with VHL mutation Ser65Trp (endogenous mutation of RCC4) and Tyr98His was available in our cohort. For each target the mRNA level of the VHL wild-type tumor is used as reference and compared to the mRNA levels in the other tumors. p-value: *<0.05, **<0.01, ***<0.001 (data are presented as mean +SEM)
Influence of HIF1α on p53 signaling
Since it is known that HIF1α can negatively regulate p53 activity [31, 32], we next asked whether impaired p53 signaling was directly linked to VHL mutation status or was a consequence of the deregulated pVHL/HIF1α axis. For this purpose, RCC4 cells with re-expressing VHL wild-type and with deficient VHL were transfected with a small hairpin RNA for HIF1α knockdown (Figure 6). HIF1α knockdown led to upregulation of TP53, p21, Bax and Noxa transcription in VHL wild-type and VHL deficient cell lines. This effect was significant for all targets in the absence of functional pVHL and no or little difference was seen in VHL expressing cell lines. Cell lines expressing functional pVHL showed higher RNA levels for all targets compared to pVHL deficient cell lines. These results suggest that the functional integrity of pVHL has a dominating impact on p53 signaling.
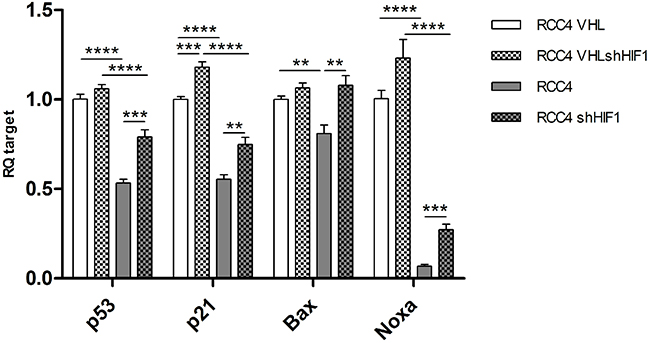
Figure 6: mRNA levels of TP53, p21, Bax and Noxa in RCC4 expressing VHL (white), VHL deficient (grey), and corresponding HIF1α knockdown (patterned). p-value: *<0.05, **<0.01, ***<0.001, ****<0.0001 (data are presented as mean +SEM).
p21 expression in ccRCC tumor tissues
The RCC TMA which was analyzed for p53 expression was also immunostained against p21 to correlate p21 expression with the VHL mutation status and p53 expression. ccRCC with LOF mutations expressed less p21 than VHL wild-type tumors (not significant) and missense mutated tumors (p<0.05) (Figure 7A). About 10% of the p53 negative and 52% of the p53 positive cores showed a high content of p21 (p<0.0001). The expression pattern of p53 and p21 is significantly different in VHL LOF ccRCC compared to missense (p=0.0288) and wild-type cases (p<0.0001) (Figure 7B). The tumors positive for both p53 and p21 were less frequent in VHL LOF ccRCC than in wild-type cases (p=0.0327).
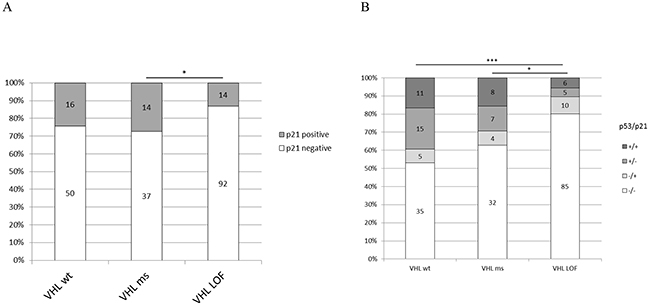
Figure 7: A. p21 protein expression in ccRCC tissue and VHL mutation type; B. p53/p21 combined expression and VHL mutation type. Wt: wild-type, ms: missense, LOF: Loss of function. p-value: *<0.05, **<0.01, ***<0.001
Effects of p53 and HIF1/2α binding site-specific VHL mutations on apoptosis and cell proliferation
The apoptotic behavior of cells expressing different VHL missense mutations was evaluated by Caspase 3/7 assay. All pVHL mutants were deficient in apoptosis compared to VHL wild- type (Figure 8A). Cells with p53 binding site-specific VHL mutations showed significantly lower apoptotic activity than those with mutations that affected the HIF1/2α binding domain (p=0.0088) (Figure 8B). In contrast, cell proliferation was not influenced by the different binding site-specific VHL mutants (Figure 8C-8D).
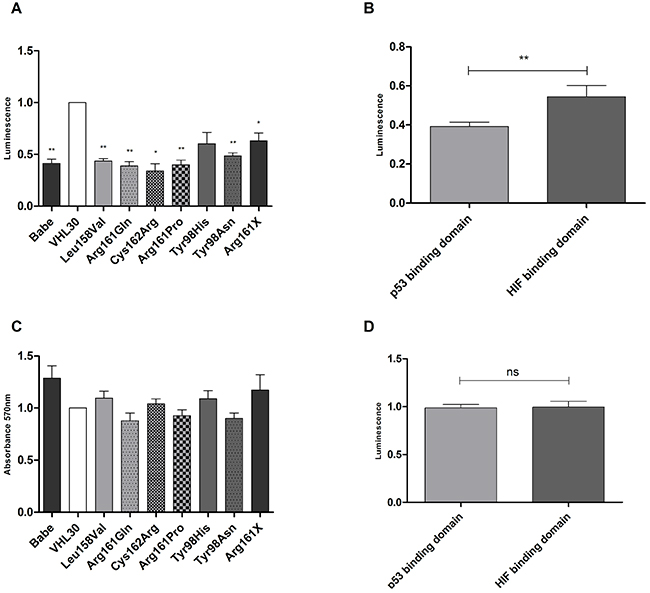
Figure 8: Effects of VHL mutations on apoptosis and cell proliferation. A. Apoptotic behavior of VHL wild type and mutated RCC4 cells; B. apoptotic behavior of RCC4 with missense mutations in the p53 and HIF1/2α binding domains; C. Proliferative behavior of VHL wild type and mutated RCC4 cells; D: Proliferative behavior of RCC4 with missense mutations in the p53 and HIF1/2α binding domains. VHL30 expression was used as reference and compared to the other cell lines. p-value: *<0.05, **<0.01 (data are presented as mean +SEM).
Apoptotic and proliferative behavior of cells upon treatment with Camptothecin and/or Sunitinib
Camptothecin, which stabilizes and activates p53, was applied to the stable cell lines alone or in combination with Sunitinib. Whereas Sunitinib affects the proliferation pathway, Camptothecin is known to affect both apoptosis and proliferation [33-35]. As expected, treatment with Camptothecin alone or in combination with Sunitinib highly increased apoptosis in all cell lines, whereas Sunitinib alone had no effect on apoptosis. Cells with Arg161Gln, Arg161Pro and Tyr98Asn showed the highest response to Camptothecin alone. Apoptosis was even increased with combined treatment in Arg161Gln and Tyr98Asn cells (Figure 9A).
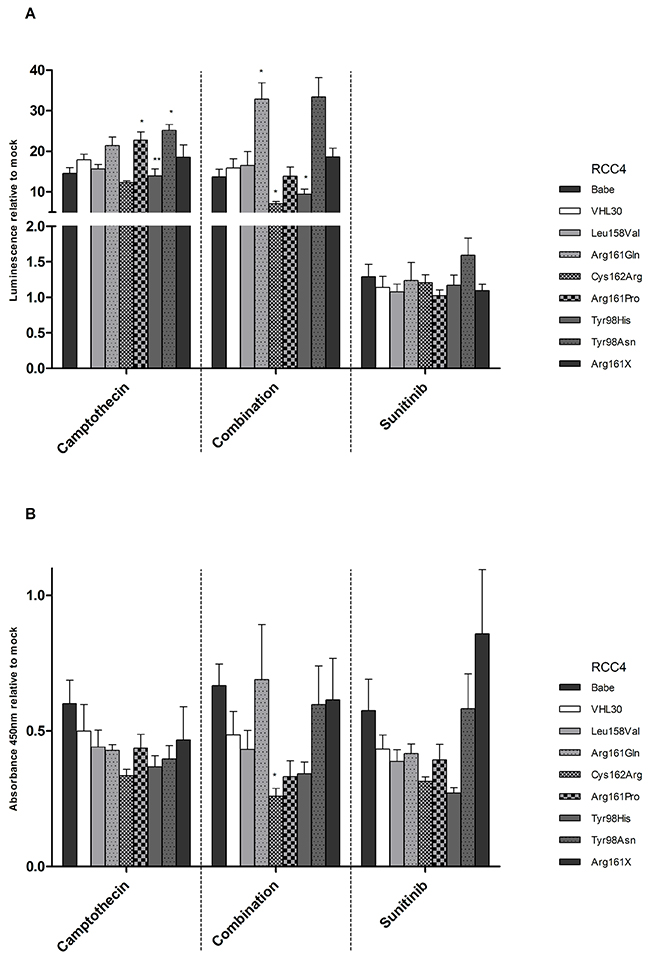
Figure 9: A. Apoptotic and B. proliferative behavior of VHL wt and mutated RCC4 cells after treatment with Camptothecin, Camptothecin and Sunitinib (Combination), and Sunitinib. Signals after treatment of each cell line were normalized with corresponding mock signals (without treatment). VHL30 was used as reference and compared to the other cell lines. p-value: *<0.05, **<0.01 (data are presented as mean +SEM).
An effect on cell proliferation was observed when the cells were treated with all three treatment strategies. With the exception of Arg161X, cell proliferation was decreased between 30-75% (Figure 9B). The response to the different treatments was similar for cell lines with missense mutation in the p53 binding domain and in the HIF1/2α binding domain.
DISCUSSION
By investigating p53 expression in 262 ccRCC, we saw a relationship between p53 expression and the severity of VHL mutations. Our results are consistent with other studies showing that p53 expression was significantly lower in ccRCC than in other RCC subtypes [36-39]. The correlation between severe VHL mutations and negative or low p53 expression suggests a close relationship between loss of function of pVHL and disturbed p53 signaling in ccRCC.
As the location of a VHL missense mutation may specifically affect one of the many binding sites of pVHL, we focused on those missense mutations that alter the p53 binding site. Therefore, we selected four missense mutations identified in the p53 binding domain. The missense mutations were predicted to have different effects on pVHL stability, from highly destabilizing to neutral. [9]. As the p53 binding domain overlaps the ElonginC binding domain [17], the missense mutations in this region should affect pVHL interactions with p53 and/or HIF1/2α.
The VHL mutations Cys162Arg, Arg161Pro and Arg161X were unable to downregulate HIF1/2α at the protein level. This result was expected for the nonsense mutation Arg161X and for Arg161Pro due to its predicted destabilizing effect on pVHL stability but not for Cys162Arg which is predicted to be neutral. As it was demonstrated for the missense mutations Cys162Phe and Cys162Ala [40, 41], Cys162Arg also seems to impair ElonginC binding, thus leading to HIF1/2α accumulation. Mutant Arg161Gln remained only partly functional for HIF1/2α degradation but like VHL30 (wild-type), Leu158Val was able to fully downregulate HIF1/2α. Notably, Arg161Gln had a milder effect on HIF1α degradation than on HIF2α. In contrast to HIF1/2α, p53 expression was hardly affected by the VHL mutations. In a previous study it was shown that pVHL wild-type enhances p53 transcriptional activity for its downstream targets p21 and Bax [17]. We hypothesized that VHL mutations could affect p53 activity rather than stability in our stable cell lines. Therefore, we analyzed the transcription levels of TP53 and its downstream targets p21, Bax and Noxa.
The RNA levels of TP53, p21 and Noxa were lower in all VHL mutant cell lines compared to those in RCC4 VHL30. Bax RNA levels were affected to a lesser extent. We also analyzed the RNA levels in normal kidney and ccRCC tissue. p53 signaling was similar in VHL wild-type ccRCC and in normal tissue. In VHL mutated tumors RNA levels of TP53, p21, Bax and Noxa were significantly lower than in tumors with VHL wild-type, which confirmed the results obtained from the cell lines.
Recent findings suggest complex regulation between p53 and HIF1α. p53 has been described to bind HIF1α [32] which negatively affects its stability and activity [31]. Our results demonstrated that HIF1α knockdown led to an upregulation of TP53 and its downstream targets in both pVHL re-expressing and pVHL deficient RCC4. However, the RCC4 cell lines expressing VHL presented significantly higher RNA levels of TP53, p21, Bax and Noxa compared to the RCC4 pVHL-deficient cell lines, independently of HIF1α knockdown. This led us to the conclusion that pVHL is able to promote p53 signaling even in the presence of HIF1α.
To further prove the association of VHL mutation status with p53 signaling on the protein level in ccRCC tissue, we investigated the expression of the p53 downstream target p21 by immunohistochemistry on the same TMA that was stained for p53. p21 was expressed in only 22% of ccRCC. Similarly to p53, p21 expression was significantly lower in VHL LOF mutants compared to wild-type, thus supporting a potential role of pVHL’s integrity in sustaining functional p53 signaling.
At least 12 p53 isoforms have been reported in the literature [42] and their abnormal expression was observed in a wide range of cancers, including RCC [43, 44]. The p53 antibody (clone DO-7) used in our study detects wild type p53, p53β and p53γ. Therefore, it cannot be excluded that tumors exclusively expressing other p53 isoforms may have been missed. However, mRNA expression data of the different p53 isoforms demonstrated that p53, p53β and p53γ are the main expressed isoforms in RCC [43, 44] suggesting minor impacts of p53 isoform expression on our results.
All our selected VHL mutations led to an attenuated apoptosis compared to VHL wild-type, but no significant change in cell proliferation was seen. Given their apoptosis deficiency, VHL mutant cells seem to have a survival advantage compared to VHL wild-type cells. However, cells with a missense mutation in the p53 binding domain were less apoptotic than cells with missense mutations affecting the HIF1/2α binding domain. Since VHL mutant cells affected apoptosis rather than cell proliferation, we treated the cells both with Camptothecin, a chemotherapeutic drug that stabilizes and activates p53 by inducing DNA damage and decreases cell proliferation and Sunitinib, a TKI which negatively influences cell proliferation and is used to treat ccRCC. Notably, Camptothecin was more effective than Sunitinib because it decreased cell proliferation and increased apoptosis in all VHL mutated cell lines. However, the missense mutation location and the affected binding domains may only partially explain the differing response to treatment. In a previous study, the VHL mutants S111R and S111C, which are located in the HIF1/2α binding domain showed a decrease in apoptosis compared to VHL wild-type via impairment of recruitment of coactivators p300 and Tip60 [21]. The loss of ability to recruit coactivators of p53 may explain the low apoptotic activity of our cell lines with VHL missense mutations located in the HIF1/2α binding domain.
In summary, we found an association of the VHL mutation type and p53 signaling, which was reflected by apoptotic deficiency. We also showed that pVHL integrity had a dominant effect over HIF1α downregulation in enhancing p53 transactivation suggesting disturbed p53 signaling is provoked by mutant pVHL rather than altered pVHL/HIFα axis. Notably, two VHL missense mutations in the p53 binding domain, Leu158Val and Arg161Gln, altered p53 signaling but retained HIF1/2α degradation function, thus confirming our hypothesis of differing effects of missense mutations on pVHL functions (summarized in figure 10) Our results may scrutinize the rationale of the systematic use of anti-angiogenic drugs, particularly for those metastatic ccRCC with VHL missense mutations that specifically affect non HIF1/2α binding sites. A systematic characterization of VHL mutations may help optimizing targeted therapy approaches for patients with metastatic ccRCC.
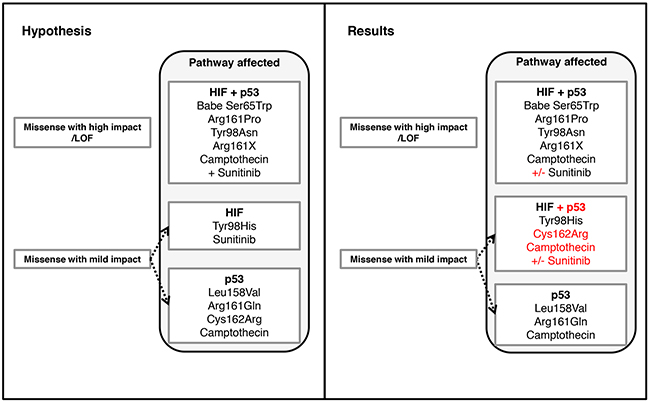
Figure 10: Comparison of predicted effects and own data of VHL mutations affecting the p53 and the HIF1/2α binding sites of pVHL and possible treatment strategies.
MATERIAL AND METHODS
Tissue Microarray and immunohistochemistry
A TMA [45] was stained using the mouse antibody p53 (clone DO-7, dilution 1:150, Dako A/S). As the tumors either showed hardly any or clearly more than ten positive nuclei, cores with up to five nuclei positive for p53 were grouped into “Low content” category and cores showing at least six positive nuclei were grouped into “High content”. The TMA was also stained and analyzed for p21 using the rabbit antibody p21 (C-19) (sc-397, dilution 1:50, Santa-Cruz Biotechnology) and scored following the same criteria used for p53.
In silico selection of VHL mutations
We used the Site Directed Mutator in silico tool [46] to characterize VHL missense mutations in 360 FFPE ccRCC samples [47] which were re-reviewed by two pathologists (H.M., E.D.) according to the new WHO/ISUP grading system [48] (Table 2). The program calculates the thermodynamic change (ddG) occurring after modification of one amino acid according to the main chain conformation, solvent accessibility and hydrogen bonding class. The missense mutations were then classified as follows:
- ddg < -2.0 or >2.0: highly destabilizing or stabilizing referred as “high impact”
- -2.0 ≤ ddg < 2.0: (slightly) destabilizing, neutral, (slightly) stabilizing referred as “mild impact”
Table 2: Grading according to the new WHO/ISUP grading system and tumor stage of 360 ccRCC
ISUP Grade |
N (%) |
pT stage |
N (%) |
|---|---|---|---|
1 |
11 (3.1) |
1 |
147 (40.8) |
2 |
105 (29.2) |
2 |
31 (8.6) |
3 |
115 (31.9) |
3 |
160 (44.4) |
4 |
115 (31.9) |
4 |
8 (2.2) |
unknown |
14 (3.9) |
unknown |
14 (3.9) |
The binding domains of HIF1/2α (amino acid 67-117), p53 (aa 154-163) and Elongin C (aa 157-171) were assessed as described by Leonardi et al. [49]. We selected four mutations that were located in the overlap of p53 and EloC binding domains of pVHL (aa 157-163): three were predicted to have no or little impact on the protein stability (Leu158Val, Arg161Gln, Cys162Arg, mild impact) and one was predicted to highly destabilize pVHL (Arg161Pro, high impact). Controls located in the HIF1/2α binding domain (Tyr98His, Tyr98Asn) and one nonsense mutation (Arg161X) were also included.
Knock down, transfection and transduction experiments
The pVHL deficient cell line RCC4 (expressing VHL Ser65Trp, highly destabilizing mutation) was kindly provided by W. Krek (ETH Zurich, Switzerland) and grown under conditions recommended by ATCC. This cell line has been authenticated as RCC4 plus vector alone (ECACC 03112702) in June 2016. A pcDNA3.1 vector encoding VHL wild-type [9] was used to generate the selected mutants with the Quick Change Lightning site-directed mutagenesis kit (Agilent technologies, United States). Subsequently, pcDNA3.1 was subcloned into pBabe vectors for transduction in mammalian cells.
pBabe empty vector and vectors containing the VHL wild-type or the mutant VHL sequences were transfected into Platinum-A Retroviral Packaging Cell Line (Cell Biolabs, United States) for viral pseudo-particles production according to the X-tremeGENE 9 DNA Transfection Reagent 3:1 protocol (Roche Diagnostics, Switzerland). The viral supernatant was collected and applied to RCC4 for transduction following the manufacturer’s recommendations. Polyclonal batches of the transduced RCC4 were then selected with constant concentration of 4μg/mL puromycin for four weeks and then maintained at 2μg/mL for cell culture.
Knockdown of HIF1α in RCC4 using short hairpin RNA (shHIF1α NM_001530.x_1048s1c1, Sigma) was performed as described previously [50].
Western blot (WB)
WB was performed as described elsewhere [50]. Detailed information of the antibodies used for WB analysis is listed in Table 3.
Table 3: Antibodies used for Western blot analysis
Antigen |
Dilution |
Antibody |
Provider |
|---|---|---|---|
pVHL |
1:1000 |
S2-647 |
BD Biosciences |
p53 |
1:2000 |
Ab1101 |
Abcam |
HIF1α |
1:1000 |
NB100-479 |
Novus Biologicals |
HIF2α |
1:1000 |
PAB12124 |
Abnova |
CA-IX |
1:2000 |
M75 |
J. Zavada, Prague, Czech Republic |
Glut1 |
1:1000 |
07-14-01 |
Millipore |
Actin |
1:2000 |
MAB15-01 |
Millipore |
Goat anti-rabbit |
1:1000 |
7074 |
Cell signaling |
Goat anti-mouse |
1:2000 |
Ab672 |
Abcam |
RNA extraction and quantitative PCR
RNA was extracted from cultured cells or from two core punches of FFPE samples using Maxwell® 16 LEV simplyRNA and RNA FFPE Purification Kit (Promega corporation,USA). RNA was reversely transcribed into cDNA using the High-Capacity cDNA Reverse Transcription Kit. RNA levels were determined by quantitative PCR using TaqMan® Gene Expression Assay on the ViiA7 Real-Time PCR System instrument (Applied Biosystems, United States). The probes used were Hs00184451 (VHL), Hs00153340 (TP53), Hs00355782 (p21), Hs00180269 (Bax), Hs00736699 (Noxa), Hs00936377 (HIF1α) and Hs01026146 (HIF2α). The results were normalized to PPIA (Hs99999904) RNA level in each sample [51]. The results are shown as Relative Quantity (RQ).
Proliferation and apoptosis assays
A colorimetric Cell Proliferation ELISA BrdU assay (Roche diagnostics, Switzerland) and a Caspase-Glo® 3/7 Assay System (Promega corporation, United States) was used to investigate the proliferative and apoptotic behavior of the established cell lines, respectively.
Drug treatment
Camptothecin and Sunitinib (Selleck Chemicals, USA) were diluted in DMSO at 0.05 μM and 10 μM concentration, respectively, and applied to the cells alone or in combination for 48h at a final concentration of 0.3% DMSO following the manufacturer recommendation. The vehicle control with 0.3% DMSO was referred to as “mock”, and “mock VHL30” was used as a reference.
Statistics
Two-tailed Chi square, Fischer’s exact test and Student’s T-tests were performed using the program Graphpad prism 5.04 for Windows (GraphPad Software, San Diego California USA, www.graphpad.com).
ACKNOWLEDGMENTS
We would like to acknowledge Susanne Dettwiler and Fabiola Prutek (Tissue Biobank), the in situ immunohistochemistry laboratory, Adriana von Teichman (VHL sequencing), and Anna Malgorzata Nowicka (drug treatment assays setup) from the Department of Pathology and Molecular Pathology, University Hospital of Zurich.
CONFLICTS OF INTEREST
The authors declare no competing interest.
FUNDING
This study has been funded by the Swiss National Science Foundation, Grant number: 31003A_135792, and by the Zurich Cancer League (to H.M.).
REFERENCES
1. Frew IJ and Moch H. A clearer view of the molecular complexity of clear cell renal cell carcinoma. Annu Rev Pathol. 2015; 10:263-289.
2. Moch H, Cubilla AL, Humphrey PA, Reuter VE and Ulbright TM. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. European urology. 2016; 70:93-105.
3. Banks RE, Tirukonda P, Taylor C, Hornigold N, Astuti D, Cohen D, Maher ER, Stanley AJ, Harnden P, Joyce A, Knowles M and Selby PJ. Genetic and epigenetic analysis of von Hippel-Lindau (VHL) gene alterations and relationship with clinical variables in sporadic renal cancer. Cancer research. 2006; 66:2000-2011.
4. Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, Matveev V, Janout V, Kollarova H, Bencko V, Navratilova M, Szeszenia-Dabrowska N, Mates D, Mukeria A, Holcatova I, Schmidt LS, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008; 14:4726-4734.
5. Kondo K, Yao M, Yoshida M, Kishida T, Shuin T, Miura T, Moriyama M, Kobayashi K, Sakai N, Kaneko S, Kawakami S, Baba M, Nakaigawa N, Nagashima Y, Nakatani Y and Hosaka M. Comprehensive mutational analysis of the VHL gene in sporadic renal cell carcinoma: relationship to clinicopathological parameters. Genes, chromosomes & cancer. 2002; 34:58-68.
6. Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, Lane WS, Kaelin WG, Elledge SJ, Conaway RC, Harper JW and Conaway JW. Rbx1, a Component of the VHL Tumor Suppressor Complex and SCF Ubiquitin Ligase. Science. 1999; 284:657-661.
7. Iwai K, Yamanaka K, Kamura T, Minato N, Conaway RC, Conaway JW, Klausner RD and Pause A. Identification of the von Hippel-lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proceedings of the National Academy of Sciences of the United States of America. 1999; 96:12436-12441.
8. Shen C and Kaelin Jr WG. The VHL/HIF axis in clear cell renal carcinoma. Seminars in Cancer Biology. 2013; 23:18-25.
9. Rechsteiner MP, von Teichman A, Nowicka A, Sulser T, Schraml P and Moch H. VHL gene mutations and their effects on hypoxia inducible factor HIFalpha: identification of potential driver and passenger mutations. Cancer research. 2011; 71:5500-5511.
10. Hoffman MA, Ohh M, Yang H, Klco JM, Ivan M and Kaelin Jr WG. von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Human molecular genetics. 2001; 10:1019-1027.
11. Zhou MI, Wang H, Foy RL, Ross JJ and Cohen HT. Tumor Suppressor von Hippel-Lindau (VHL) Stabilization of Jade-1 Protein Occurs through Plant Homeodomains and Is VHL Mutation Dependent. Cancer research. 2004; 64:1278-1286.
12. Yi Y, Mikhaylova O, Mamedova A, Bastola P, Biesiada J, Alshaikh E, Levin L, Sheridan RM, Meller J and Czyzyk-Krzeska MF. von Hippel-Lindau–Dependent Patterns of RNA Polymerase II Hydroxylation in Human Renal Clear Cell Carcinomas. Clinical Cancer Research. 2010; 16:5142-5152.
13. Li Z, Na X, Wang D, Schoen SR, Messing EM and Wu G. Ubiquitination of a Novel Deubiquitinating Enzyme Requires Direct Binding to von Hippel-Lindau Tumor Suppressor Protein. Journal of Biological Chemistry. 2002; 277:4656-4662.
14. Khacho M, Mekhail K, Pilon-Larose K, Pause A, Côté J and Lee S. eEF1A Is a Novel Component of the Mammalian Nuclear Protein Export Machinery. Molecular Biology of the Cell. 2008; 19:5296-5308.
15. Feldman DE, Spiess C, Howard DE and Frydman J. Tumorigenic Mutations in VHL Disrupt Folding In Vivo by Interfering with Chaperonin Binding. Molecular cell. 2003; 12:1213-1224.
16. The p53 web site, http://p53.free.fr/.
17. Roe JS, Kim H, Lee SM, Kim ST, Cho EJ and Youn HD. p53 stabilization and transactivation by a von Hippel-Lindau protein. Molecular cell. 2006; 22:395-405.
18. Galban S, Martindale JL, Mazan-Mamczarz K, Lopez de Silanes I, Fan J, Wang W, Decker J and Gorospe M. Influence of the RNA-binding protein HuR in pVHL-regulated p53 expression in renal carcinoma cells. Mol Cell Biol. 2003; 23:7083-7095.
19. Stickle NH, Cheng LS, Watson IR, Alon N, Malkin D, Irwin MS and Ohh M. Expression of p53 in renal carcinoma cells is independent of pVHL. Mutation research. 2005; 578:23-32.
20. Roe JS and Youn HD. The positive regulation of p53 by the tumor suppressor VHL. Cell cycle. 2006; 5:2054-2056.
21. Roe JS, Kim HR, Hwang IY, Ha NC, Kim ST, Cho EJ and Youn HD. Phosphorylation of von Hippel-Lindau protein by checkpoint kinase 2 regulates p53 transactivation. Cell cycle. 2011; 10:3920-3928.
22. Wallace-Brodeur RR and Lowe SW. Clinical implications of p53 mutations. Cell Mol Life Sci. 1999; 55:64-75.
23. Semenza GL. VHL and p53: tumor suppressors team up to prevent cancer. Molecular cell. 2006; 22:437-439.
24. Tomasino RM, Morello V, Tralongo V, Nagar C, Nuara R, Daniele E, Curti M and Orestano F. p53 expression in human renal cell carcinoma: an immunohistochemical study and a literature outline of the cytogenetic characterization. Pathologica. 1994; 86:227-233.
25. Vasavada SP, Novick AC and Williams BR. P53, bcl-2, and Bax expression in renal cell carcinoma. Urology. 1998; 51:1057-1061.
26. COSMIC database, http://cancer.sanger.ac.uk/cosmic.
27. Gurova KV, Hill JE, Guo C, Prokvolit A, Burdelya LG, Samoylova E, Khodyakova AV, Ganapathi R, Ganapathi M, Tararova ND, Bosykh D, Lvovskiy D, Webb TR, Stark GR and Gudkov AV. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-kappaB-dependent mechanism of p53 suppression in tumors. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102:17448-17453.
28. Linehan WM, Srinivasan R and Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nature reviews Urology. 2010; 7:277-285.
29. Yu JL, Rak JW, Coomber BL, Hicklin DJ and Kerbel RS. Effect of p53 Status on Tumor Response to Antiangiogenic Therapy. Science. 2002; 295:1526-1528.
30. Pawlowski R, Muhl SM, Sulser T, Krek W, Moch H and Schraml P. Loss of PBRM1 expression is associated with renal cell carcinoma progression. International journal of cancer Journal international du cancer. 2013; 132:E11-17.
31. Sermeus A and Michiels C. Reciprocal influence of the p53 and the hypoxic pathways. Cell death & disease. 2011; 2:e164.
32. Sánchez-Puig N, Veprintsev DB and Fersht AR. Binding of Natively Unfolded HIF-1α ODD Domain to p53. Molecular cell. 2005; 17:11-21.
33. Conley S, Baker T, Burnett J, Theisen R, Lazarus D, Peters C, Clouthier S, Eliasof S and Wicha M. CRLX101, an investigational camptothecin-containing nanoparticle-drug conjugate, targets cancer stem cells and impedes resistance to antiangiogenic therapy in mouse models of breast cancer. Breast Cancer Res Treat. 2015; 150:559-567.
34. Pham E, Birrer MJ, Eliasof S, Garmey EG, Lazarus D, Lee CR, Man S, Matulonis UA, Peters CG, Xu P, Krasner C and Kerbel RS. Translational Impact of Nanoparticle–Drug Conjugate CRLX101 with or without Bevacizumab in Advanced Ovarian Cancer. Clinical Cancer Research. 2015; 21:808-818.
35. Selvarajah J, Nathawat K, Moumen A, Ashcroft M and Carroll VA. Chemotherapy-mediated p53-dependent DNA damage response in clear cell renal cell carcinoma: role of the mTORC1/2 and hypoxia-inducible factor pathways. Cell death & disease. 2013; 4:e865.
36. Hodorova I, Solar P, Mihalik J, Vecanova J, Adamkov M and Rybarova S. Investigation of tumour supressor protein p53 in renal cell carcinoma patients. Biomedical papers of the Medical Faculty of the University Palacky, Olomouc, Czechoslovakia. 2014; 158:44-49.
37. Zigeuner R, Ratschek M, Rehak P, Schips L and Langner C. Value of p53 as a prognostic marker in histologic subtypes of renal cell carcinoma: a systematic analysis of primary and metastatic tumor tissue. Urology. 2004; 63:651-655.
38. Mombini H, Givi M and Rashidi I. Relationship between expression of p53 protein and tumor subtype and grade in renal cell carcinoma. Urol J. 2006; 3:79-81.
39. Noroozinia F, Fahmideh AN, Yekta Z, Rouhrazi H and Rasmi Y. Expression of CD44 and P53 in renal cell carcinoma: association with tumor subtypes. Saudi journal of kidney diseases and transplantation : an official publication of the Saudi Center for Organ Transplantation, Saudi Arabia. 2014; 25:79-84.
40. Knauth K, Bex C, Jemth P and Buchberger A. Renal cell carcinoma risk in type 2 von Hippel-Lindau disease correlates with defects in pVHL stability and HIF-1[alpha] interactions. Oncogene. 2005; 25:370-377.
41. Ohh M, Takagi Y, Aso T, Stebbins CE, Pavletich NP, Zbar B, Conaway RC, Conaway JW and Kaelin WG, Jr. Synthetic peptides define critical contacts between elongin C, elongin B, and the von Hippel-Lindau protein. J Clin Invest. 1999; 104:1583-1591.
42. Khoury MP and Bourdon JC. p53 Isoforms: An Intracellular Microprocessor? Genes Cancer. 2011; 2:453-465. doi: 10.1177/1947601911408893.
43. van den Berg L, Segun AD, Mersch S, Blasberg N, Grinstein E, Wai D, Anlauf M, Gabbert HE, Mahotka C and Heikaus S. Regulation of p53 isoform expression in renal cell carcinoma. Frontiers in bioscience (Elite edition). 2010; 2:1042-1053.
44. Song W, Huo SW, Lu JJ, Liu Z, Fang XL, Jin XB and Yuan MZ. Expression of p53 isoforms in renal cell carcinoma. Chin Med J (Engl). 2009; 122:921-926.
45. Mertz KD, Demichelis F, Kim R, Schraml P, Storz M, Diener P-A, Moch H and Rubin MA. Automated immunofluorescence analysis defines microvessel area as a prognostic parameter in clear cell renal cell cancer. Human pathology. 2007; 38:1454-1462.
46. Site Directed Mutator, http://mordred.bioc.cam.ac.uk/~sdm/links.php.
47. Razafinjatovo C, Bihr S, Mischo A, Vogl U, Schmidinger M, Moch H and Schraml P. Characterization of VHL missense mutations in sporadic clear cell renal cell carcinoma: hotspots, affected binding domains, functional impact on pVHL and therapeutic relevance. BMC cancer. 2016; 16:638.
48. Delahunt B, Cheville JC, Martignoni G, Humphrey PA, Magi-Galluzzi C, McKenney J, Egevad L, Algaba F, Moch H, Grignon DJ, Montironi R and Srigley JR. The International Society of Urological Pathology (ISUP) grading system for renal cell carcinoma and other prognostic parameters. Am J Surg Pathol. 2013; 37:1490-1504.
49. Leonardi E, Murgia A and Tosatto SC. Adding structural information to the von Hippel-Lindau (VHL) tumor suppressor interaction network. FEBS letters. 2009; 583:3704-3710.
50. Boysen G, Bausch-Fluck D, Thoma CR, Nowicka AM, Stiehl DP, Cima I, Luu VD, von Teichman A, Hermanns T, Sulser T, Ingold-Heppner B, Fankhauser N, Wenger RH, Krek W, Schraml P, Wollscheid B, et al. Identification and functional characterization of pVHL-dependent cell surface proteins in renal cell carcinoma. Neoplasia (New York, NY). 2012; 14:535-546.
51. Jung M, Ramankulov A, Roigas J, Johannsen M, Ringsdorf M, Kristiansen G and Jung K. In search of suitable reference genes for gene expression studies of human renal cell carcinoma by real-time PCR. BMC Molecular Biology. 2007; 8:1-13.