
INTRODUCTION
The immunosuppressive tumor microenvironment usurps host anti-tumor immune responses making cancer treatment extremely complex and challenging. Therapeutic approaches that subvert tumor-induced immune suppression and control tumor metastasis are highly sought after in a hope to provide relapse-free survival in cancer patients. A predominance of immune stimulatory cytokines over a suppressive milieu in the tumor microenvironment decides the outcome of therapeutic protocols. Tumors negatively impact effector functions of immune cells through immunosuppressive molecules, such as PD-L1 (B7-H1) and soluble factors, such as transforming growth factor beta (TGFβ), interleukin-10 (IL-10) and vascular endothelial growth factor (VEGF) [1]. Among multiple tumor-associated suppressive mechanisms, tumors also downregulate the evolutionarily conserved Notch signaling, crucial in several biological processes including immune cell differentiation and effector function [2, 3]. Evidence is emerging to support that a functional crosstalk between Notch and nuclear factor-κB (NFκB) signaling pathways are necessary for antitumor CD8+T cell effector and memory differentiation [4–7].
Immunostimulatory cytokines (e.g., IL-2, IL-12, IL-15) support lymphocyte differentiation and survival enabling tumor control [3]. Various approaches to overcome tumor-induced immunosuppression using exogenous cytokines as well as immune adjuvants and modulators, dendritic cell vaccines, and adoptive immune cell transfers have not been successful clinically, albeit promising initial results. Bortezomib (VelcadeTM/PS-341) is a FDA-approved drug for the treatment of multiple myeloma and mantle cell lymphoma [8–14]. It is a dipeptidyl boronate that inhibits the ubiquitin-proteasome proteolytic pathway. The boronic acid moiety of bortezomib with its transient (half-life of 9-15 h) and reversible inhibition of the proteasome set it apart from other proteasome inhibitors as a therapeutic agent. Unlike other non-boronic acid proteasome inhibitors, the boronic acid moiety of bortezomib prevents it from being transported out of the cell by the multidrug resistance system [15]. It has been observed that bortezomib administration enhances the expression of TNF-R1 and IFN-γ-Rα in TBJ neuroblastoma cells [16] suggesting that bortezomib may potentially modulate the tumor microenvironment. In our previous studies, we showed that bortezomib can also sensitize various mouse and human solid tumor cells to apoptosis by tumor necrosis factor family members [17, 18]. Although there are reports of negative side effects of bortezomib on some immune cells, such as a reduction in dendritic cell and leukocyte numbers [19–24], studies have lately suggested that bortezomib, directly or indirectly, may enhance immune functions under optimized conditions [25–32]. We also observed that treatment with bortezomib was associated with an increased crosstalk between Notch and NFκB signaling pathways in effector T cells [7], and an improved response to adoptive T cell therapy by enhanced FasL cytotoxicity [33]. However, it is not well understood how bortezomib affects the cytokine milieu in the tumor microenvironment that could influence antitumor T cell immunity and tumor burden.
In this study, using a therapeutic regimen established by us previously [17], we explored the effects of bortezomib administered at a dose of 1 mg/kg body weight on the cytokine milieu and CD8+T cell function in mice bearing murine mammary and renal adenocarcinomas that present a defined low-avidity MHC-I-restricted epitope (IYSTVASSL) derived from a hemagglutinin (HA) model antigen, or lung fibrosarcoma that expresses human Ras and mutant p53 as xenogenic antigens. We show that bortezomib administration in tumor-bearing mice modulates the levels of immunostimulatory cytokines IL-2, IL-12, and IL-15 and enhances their downstream signaling by increasing the expression of their receptors on tumor-infiltrating CD8+T cells in endogenous or adoptive HA518-526-specific CD8+T cell transfer set ups resulting in reduced pulmonary metastatic nodules. Data also show that these effects of bortezomib treatment are mediated via the activation of PI3K/Akt/STAT5 pathways in CD8+T cells enhancing their effector function. These findings suggest that besides bortezomib’s established role in sensitizing tumors to apoptosis, it also has immunostimulatory potential to therapeutically modulate the tumor microenvironment with a carefully optimized bortezomib regimen to sustain lymphocytic effector function, and overcome tumor-associated immunosuppression.
RESULTS
Bortezomib treatment affects the cytokine milieu in tumor-bearing mice
We investigated the effects of the reversible proteasome inhibitor drug bortezomib on the cytokine milieu in the tumor microenvironment of murine mammary 4T1.2 (representative of stage IV human breast cancer) [34] or Renca adenocarcinomas presenting a low-avidity HA518-526 epitope [35], or lung fibrosarcoma D459. In mice with large established (~125 mm3) orthotopic mammary tumors of 4T1HA cells, MagPix multiplex cytokine bead array showed that bortezomib treatment significantly increased protein levels of immunostimulatory cytokines IL-2, IL-12p40, IL-12p70, and IL-15, and decreased the levels of tumor-promoting cytokines IL-1β and VEGF in the splenic lysates when compared with protein levels in untreated mice with tumor alone (Figure 1 and Table 1). Significantly increased levels of IL-15 were observed in the serum of mice bearing 4T1HA as well as RencaHA or D459 tumors (Table 2). A similar trend of cytokine changes was observed in the lymph node (LN), tumor mass or thymus lysates from mice bearing 4T1HA, RencaHA or D459 tumors (data not shown). An increase in mRNA levels of IL-2, IL-12p40, IL-12p70, and IL-15 correlated with their increased protein levels in splenocytes of bortezomib-treated tumor-bearing mice compared with untreated tumor-bearing mice (Figure 2). Moreover, assessment of cytokine protein levels over the course of 72 h in naïve WT mice showed that expression of the immunostimulatory cytokines IL-2, IL-12p40, IL-12p70, and IL-15 (Figure 3) reached to their peak at 4 h after bortezomib administration.
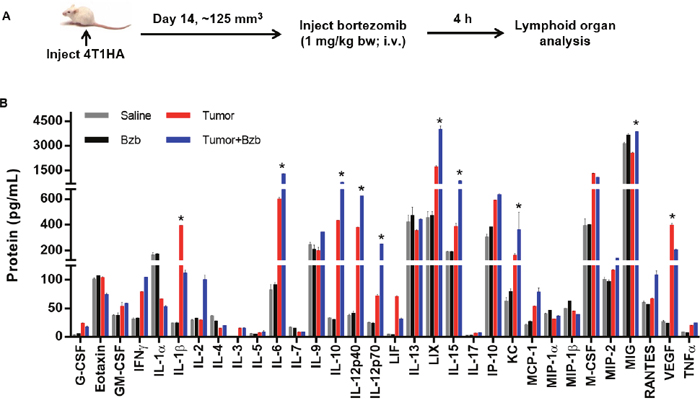
Figure 1: Modulation of cytokine/chemokine expression by bortezomib in 4T1HA tumor-bearing mice. A. Orthotopic tumors were established in mammary pads of Balb/c wild type mice following injection of 2 x 106 4T1HA tumor cells. On day 14 (after the tumor reached at least a size of 125 mm3), mice were injected intravenously with 1 mg/kg body weight of bortezomib (Bzb, ~20 nM by total blood volume in 8-wk mouse). After 4 h, the mice were euthanized and total cell lysates were made from the RBC-depleted splenocytes and 245 μg of protein from these lysates were analyzed for cytokine/chemokine concentration by MagPix array (Millipore). B. MagPix output data constitutes protein concentration (pg/mL) of cytokines/chemokines modulated in the spleen of 4T1HA tumor-bearing mice. Four experimental groups were compared: Saline control (gray bar), naïve mice treated with Bzb alone (black bar), mice with tumor alone (red bar), and tumor-bearing mice treated with Bzb (blue bar). Protein concentration of analytes compared among the 4 groups is shown as means ± SD from 4 independent experiments. *p values are representative as *p<0.05 (ANOVA, one-way) and used to compare tumor alone to tumor+Bzb group.
Table 1: Expression of cytokines/chemokines in splenic lysates of 4T1HA tumor-bearing mice following bortezomib treatment
Protein (pg/mL) |
Saline |
Bzb |
Tumor |
Tumor+Bzb |
|---|---|---|---|---|
Immunostimulatory Cytokines |
||||
IL-2 |
27.9 ± 11.4 |
38.5 ± 13.9 |
26.2 ± 10.1 |
102.1 ± 17.8* |
IL-12p40 |
38.0 ± 12.5 |
48.2 ± 15.7 |
375.8 ± 60.1 |
638.7 ± 58.4* |
IL-12p70 |
27.0 ± 14.1 |
27.0 ± 10.2 |
83.4 ± 16.7 |
254.5 ± 28.5* |
IL-15 |
180.2 ± 31.9 |
173.7 ± 43.0 |
408.1 ± 57.6 |
794.9 ± 90.3* |
Chemoattractants |
||||
MIG |
3133.9 ± 126.9 |
3671.7 ± 134.4 |
2586 ± 30.4 |
3853 ± 44.0* |
RANTES |
59.9 ± 3.7 |
56.0 ± 1.7 |
66.6 ± 2.5 |
106.6 ± 9.0 |
MCP-1 |
21.1 ± 2.2 |
26.9 ± 1.7 |
54.0 ± 1.0 |
77.0 ± 10.4 |
MIP-1α |
39.8 ± 2.7 |
46.3 ± 1.2 |
30.4 ± 0.4 |
35.9 ± 2.9 |
MIP-1β |
49.6 ± 0.2 |
62.6 ± 1.2 |
44 ± 0.9 |
38.6 ± 0.3 |
MIP-2 |
100.1 ± 5.6 |
96.9 ± 3.4 |
117.1 ± 2.0 |
137.3 ± 0.4 |
Effector Cytokines |
||||
IFNγ |
31.5 ± 2.7 |
32.5 ± 0.8 |
78.6 ± 1.7 |
104.4 ± 0.2 |
TNFα |
8.5 ± 0.1 |
6.4 ± 0.2 |
19.6 ± 0.4 |
23.7 ± 0.5 |
Tumor-Promoting Cytokines/Factors |
||||
G-CSF |
3.0 ± 0.5 |
5.6 ± 0.3 |
23.1 ± 1.7 |
17.2 ± 1.9 |
IL-1β |
23.7 ± 1.1 |
24.1 ± 3.2 |
392.2 ± 3.3 |
111.5 ± 8.0* |
M-CSF |
388.4 ± 74.4 |
402.0 ± 4.6 |
1299.0 ± 40.3 |
1061.0 ± 45.5 |
VEGF |
26.8 ± 2.4 |
23.1 ± 0.8 |
397.6 ± 21.4 |
201.5 ± 9.5* |
4T1HA tumor cells (0.5 x 106) were injected orthotopically in mammary pads to establish tumors approximately 125 mm3 in size. Bortezomib (1 mg/kg body weight) was administered intravenously in mice. Bortezomib (1 mg/kg body weight) was administered intravenously in mice, and protein array of various analytes in total splenic lysates was performed 4 h later by MagPix multiplex bead assay. Protein values are presented as mean ± SD. *p<0.05 (ANOVA, one-way), comparing Tumor vs Tumor+Bzb.
Table 2: Expression of cytokines in the serum of tumor-bearing mice following bortezomib treatment
Protein (pg/mL) |
Saline |
Bzb |
Tumor |
Tumor+Bzb |
|---|---|---|---|---|
Immunostimulatory Cytokines: |
||||
4T1HA |
||||
IL-2 |
BDR |
6.8 ± 1.1 |
BDR |
3.4 ± 0.9 |
IL-12p40 |
12.5 ± 0.8 |
BDR |
BDR |
4.6 ± 1.6 |
IL-12p70 |
2.2 ± 0.7 |
BDR |
BDR |
BDR |
IL-15 |
6.3 ± 4.9 |
11.1 ± 0.7 |
80.2 ± 25.9 |
313.4 ± 21.8* |
RencaHA |
||||
IL-2 |
BDR |
19.5 ± 20.9 |
BDR |
11.75 ± 15.2 |
IL-12p40 |
BDR |
6.8 ± 8.2 |
16.2 ± 21.4 |
11.9 ± 1.6 |
IL-12p70 |
BDR |
6.4 ± 7.6 |
BDR |
15.2 ± 20.1 |
IL-15 |
BDR |
33.9 ± 6.7* |
27.1 ± 8.5 |
601.4 ± 19.5* |
D459 |
||||
IL-2 |
5.7 ± 0 |
10.8 ± 2.6 |
9.9 ± 4.4 |
10.3 ± 1.4 |
IL-12p40 |
5.2 ± 1.1 |
9.9 ± 4.8 |
17 ± 5.8 |
17.3 ± 5.4 |
IL-12p70 |
10.3 ± 4.1 |
20 ± 4.5 |
23.7 ± 2.3 |
22.2 ± 2.3 |
IL-15 |
17.6 ± 9.6 |
38.7 ± 5.5* |
38 ± 1.8 |
134.1 ± 3.9* |
4T1HA cells (0.5 x 106) were injected orthotopically and RencaHA or D459 cells subcutaneously to establish tumors approximately 125 mm3 in size. Bortezomib (1 mg/kg body weight) was administered intravenously in mice and protein array of immunostimulatory analytes in serum was performed 4 h later by MagPix multiplex bead assay. Protein values are presented as mean ± SD. *p<0.05 (ANOVA, one-way), comparing Tumor vs Tumor+Bzb. BDR: Below the minimum detection range (3.2 pg/mL).
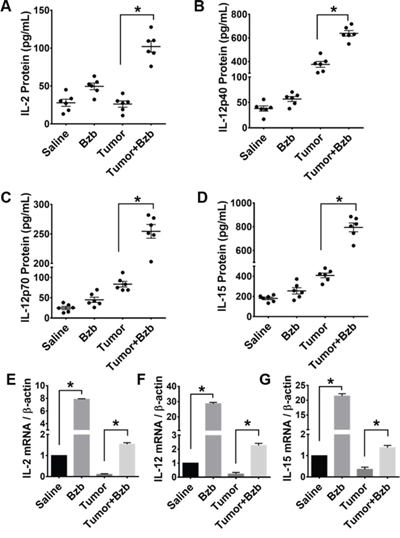
Figure 2: Effect of bortezomib administration on IL-2, IL-12, and IL-15 proteins and mRNA expression in vivo. Protein expression of cytokines IL-2 A. IL-12p40 B. IL-12p70 C. IL-15 D. in total cell lysates of RBC-depleted splenocytes from Balb/c WT mice were quantified 4 h after Bzb (1 mg/kg body weight) administration by MagPix multiplex array. Cytokine mRNA expression levels measured by RT-PCR are shown as the ratio of target gene expression to housekeeping gene, β-actin, for IL-2 E. IL-12 F. IL-15 G. The bar graph is shown as mean ± SD from 2 independent experiments. *p < 0.05; (ANOVA, one-way).
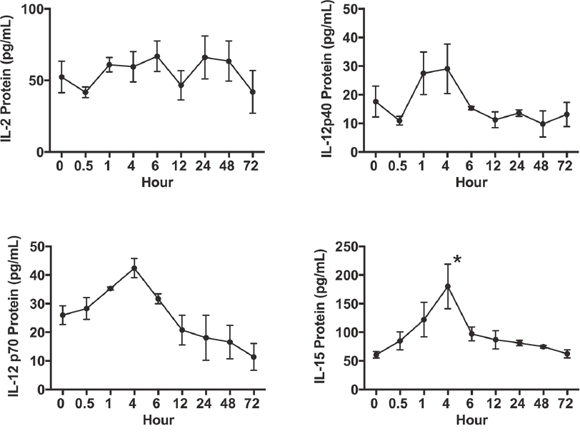
Figure 3: Time kinetics of splenic IL-2, IL-12 and IL-15 expression in vivo following bortezomib treatment. Balb/c WT mice were administered intravenously with Bzb (1 mg/kg body weight) at the indicated time points over a 72 h period. Total cell lysate samples of RBC-depleted splenocytes were prepared from a group of mice at different time points. Protein levels for IL-2, IL-12p40, IL-12p70, and IL-15 were quantified using the MagPix multiplex array. Values are presented as mean ± SEM; ANOVA, one-way; *p<0.05 compared to other time points.
These data suggest that bortezomib administration increases the expression of immunostimulatory cytokines IL-2, IL-12, and IL-15 at both the transcriptional and translational levels in tumor-bearing mice. The effects of bortezomib treatment on these cytokines, which are key players in the cytotoxic and memory response mediated by CD8+ T cells and NK cells [36–38], suggest that bortezomib has the potential to influence the tumor microenvironment and host antitumor immunity.
Bortezomib treatment reduces tumor metastatic nodules in the lung
Administration of a therapeutic regimen of bortezomib [17] given intravenously at 1 mg/kg body weight (~20 nM by blood volume) on days 4, 7, 11, and 15 after the intravenous injection of 4T1HA tumor cells in mice showed significant reduction in metastatic pulmonary nodules (Figure 4). To further understand bortezomib’s effect on enhancing immune mechanisms and reducing tumor burden by modulating immunostimulatory cytokines, we investigated a therapeutic setup where we adoptively transferred Cln4 CD8+T cells specific to the HA518-526 epitope (Vβ8.1 clonotype) into 4T1HA tumor-bearing mice. In these mice, IL-12 and IL-15 cytokines were neutralized with a regimen of blocking antibodies pre- and post-adoptive transfer of HA518-526-specific CD8+T cells. Neutralization of IL-12 and IL-15 cytokines following intravenous injection of 4T1HA cells increased the number of tumor pulmonary nodules. This neutralization effect was counteracted by the tumor metastasis-suppressing action of bortezomib treatments (Figure 5A, 5B).
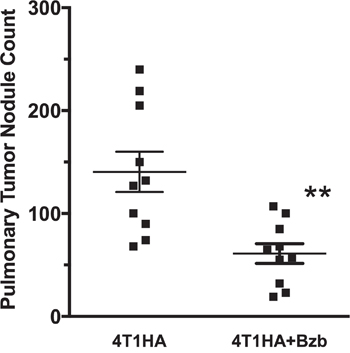
Figure 4: Effect of bortezomib on lung metastases of 4T1HA mammary tumor cells. Balb/c wild type (WT) mice injected with 4T1HA tumor cells (0.5 x 106; i.v.; day 0) were administered intravenously with bortezomib (1 mg/kg body weight, ~20 nM by total blood volume) on days 4, 7, 11, and 15. Tumor metastases pulmonary nodules were counted on day 18. Nodule counts are presented as mean ± SEM; **p <0.01; two-tailed t-test.
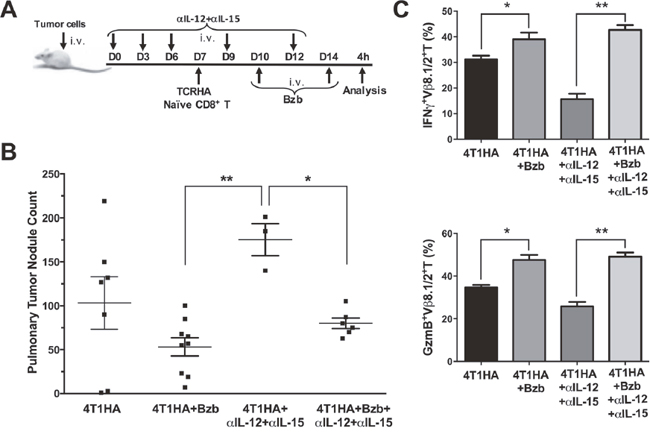
Figure 5: Bortezomib counteracts IL-12 and IL-15 neutralization by enhancing CD8+T cell effector molecules and reducing pulmonary nodules of 4T1HA tumor cells. A. 0.5 x 106 4T1HA tumor cells were injected intravenously in Balb/c WT mice along with intravenous injections of IL-12 and IL-15 neutralizing antibodies on days 0, 3, 6, 9, and 12. On day 7, adoptive transfer of TCRHA CD8+T cells (2 x 106/mouse, i.v.) was performed. Bzb (1 mg/kg body weight) was administered on days 10 and 14. B. Lungs were harvested on day 14 to count tumor pulmonary nodules. Central lines depict values as means ± SEM. C. Bar graphs present data for IFNγ+Vb8.1/2+ and granzyme-B+Vβ8.1/2+ CD8+T cells. Values are depicted as means ± SD. *p<0.05, **p<0.01; (ANOVA, one-way).
We also analyzed the expression of effector molecules on HA518-526-specific CD8+T cells in these mice following bortezomib treatments. We observed that IFNγ and granzyme-B expression on Vβ8.1/2+CD8+T cells in the spleens of tumor-bearing mice injected with IL-12 and IL-15 neutralization antibodies was significantly reduced compared with the expression in unneutralized tumor-bearing mice. Bortezomib administration increased IFNγ and granzyme-B expression on Vβ8.1/2+CD8+T cells in tumor-bearing mice even in the presence of IL-12 and IL-15 neutralization antibody treatments, and restored them to the levels comparable to tumor-bearing mice treated with bortezomib only (Figure 5C). These results suggest bortezomib administration overcomes IL-12 and IL-15 deficiency by increasing the expression of effector molecules and, thus, aids in reducing tumor pulmonary metastasis in 4T1HA-bearing mice.
Bortezomib enhances production of IL-2 from CD4+T cells, and IL-12 and IL-15 from CD11c+ cells
To determine the cellular source of cytokines modulated by bortezomib in tumor-bearing mice, we screened lymphoid and myeloid populations for the intracellular secretion of immunostimulatory cytokines. Although CD8+T cells or myeloid CD11b+ or CD11c+ cells did not show any noticeable increase in the intracellular production of IL-2 in the presence of bortezomib administration in 4T1HA tumor-bearing mice (data not shown), we observed a significant increase in the lymphoid CD4+T cells producing higher levels of IL-2 in bortezomib-treated mice (Figure 6A). Moreover, gated CD11chigh dendritic cells showed a remarkable increase in the production of intracellular IL-12 and IL-15 in bortezomib-treated mice (Figure 6B). The increase in these cytokines was not due to an increase in the frequency of CD4+T or CD11chigh cells, but was due to increase in the cytokine production per cell. We observed that bortezomib given at our tested therapeutic dose (1 mg/kg body weight) had no significant effects either on total leukocyte counts in control or tumor-draining LN, tumor-infiltrating lymphocytes, and spleen, or in subsets of CD4+T, CD8+T, and CD11c+ cells [33]. Further, following bortezomib treatment (10 nM; 4 h) in vitro, macrophage cell line RAW264.7 showed significant increases in both IL-12 and IL-15 mRNA expression and Jurkat (human CD4+T cell line), and T1 (human CD8+T cell line) showed an increase in IL-2 mRNA levels when compared with untreated cells (Figure 7). These results suggest that bortezomib administration can enhance adaptive immune mechanisms against tumor by increasing the secretion of IL-2, IL-12 and IL-15 from CD4+T helper cells and antigen-presenting dendritic cells, respectively.
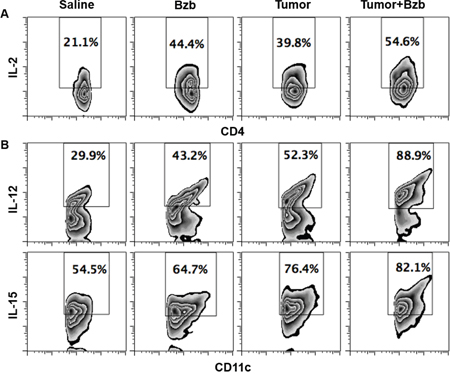
Figure 6: Bortezomib-mediated modulation of IL-2 secretion in CD4+T cells and IL-12 and IL-15 cytokines in CD11c+ cells of tumor-bearing mice. 0.5 x 106 4T1HA tumor cells were injected orthotopically under mammary pads to establish tumors approximately 125 mm3 in size. Bzb (1 mg/kg body weight) was administered intravenously in mice. Spleens were harvested 4 h later for analysis. Intracellular secretion of IL-2 from gated CD4+T cells A. or IL-12 and IL-15 from CD11c+ cells B. is shown by flow cytometry. Numbers inside plots depict % positive cells. Representative data are shown from 2 independent experiments.
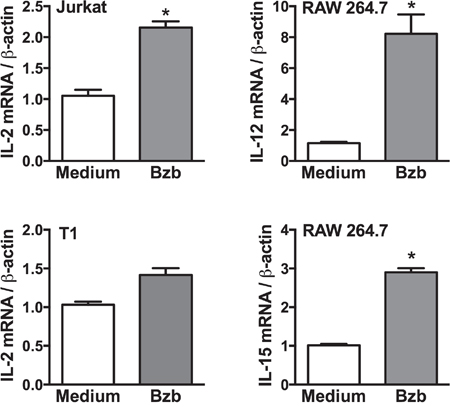
Figure 7: Cytokine expression in lymphoid and myeloid cell lines following bortezomib treatment. Expression of target cytokine mRNA was analyzed by RT-PCR in cell lines: Jurkat (CD4+T cell), T1 (CD8+T cell), and RAW264.7 (macrophage) 4 h after 10 nM Bzb treatment. Data are presented as the ratio of target gene expression to housekeeping gene β-actin from two independent experiments. Values are means ± SD; *p <0.05; two-tailed t-test.
Bortezomib treatment increases cytokine receptor expression on tumor-infiltrating CD8+T lymphocytes
Following our observation that bortezomib increases the secretion of immunostimulatory cytokines from helper T cells and antigen-presenting cells, we next wanted to determine if bortezomib treatment modulates the expression of these cytokine receptors on cytotoxic T cells at the site of tumor. We harvested the tumor mass from 4T1HA tumor-bearing mice and assessed the expression of cytokine receptors on tumor-infiltrating CD8+T cells. We observed that following bortezomib treatment, tumor-infiltrating HA518-526-specific CD8+T cells showed an enhanced expression of IL-2Rα, IL-2Rβ and IL-2Rγ compared to tumor-infiltrating CD8+T cells without bortezomib treatment (Figure 8A-8C). A trend of increased expression of IL-12β, and IL-15Rα was also observed on tumor-infiltrating CD8+T cells following bortezomib treatment compared to tumor-infiltrating CD8+T cells without bortezomib treatment (Figure 8D, 8E).
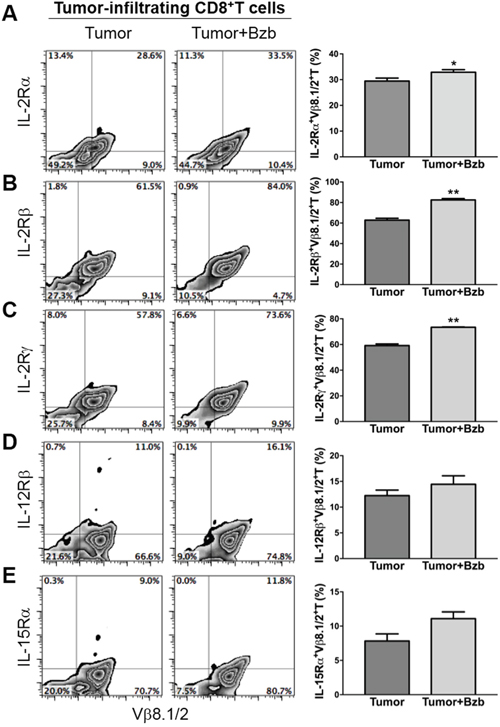
Figure 8: Bortezomib-mediated modulation of IL-2 receptor chains, IL-12Rβ, and IL-15Rα on tumor-infiltrating CD8+T cells. Orthotopic tumors were established under mammary pads in Balb/c WT mice following injection of 2 x 106 4T1HA tumor cells. On day 14 (after the tumor reached an approximate size of 125 mm3), mice were injected intravenously with 1 mg/kg body weight of bortezomib. After 4 h, the mice were euthanized and single cell suspensions were obtained from the tumor mass harvested. The expression of cytokine receptors IL-2Rα A. IL-2Rβ B. IL-2Rγ C. IL-12Rβ D. and IL-15Rα E. is shown on tumor-infiltrating CD8+T cells by flow cytometry, with corresponding bar graphs presenting values as means ± SD. Numbers inside plots depict % positive cells. Representative data are shown from 2 independent experiments with 3 mice per group. *p<0.05, **p<0.01; two-tailed t-test.
Bortezomib augments PI3K/Akt/STAT5 signaling to enhance the expression of CD8+T cell effector molecules
We assessed a panel of signaling molecules in CD8+T cells following bortezomib treatment. A time course of p38 and Akt phosphorylation following bortezomib (20 nM) treatment showed a time-dependent increase in their phosphorylation (Figure 9A). A 2-fold increase in p38 phosphorylation and a 7-fold increase in Akt phosphorylation was noted at 4 h post bortezomib treatment. We also observed that incubation of bortezomib-treated CD8+T cells with PI3K inhibitor, Ly294002, abrogated Akt phosphorylation (Figure 9B), suggesting that bortezomib-induced Akt phosphorylation was mediated via the PI3K pathway. We also screened bortezomib-treated CD8+T cells for a phospho-flow panel of all 6 members of STAT transcription factors. We observed a 3-fold increase in STAT5 phosphorylation in CD8+T cells at 4 h post bortezomib treatment (Figure 9C). Upregulation of STAT5 phosphorylation was also observed in Vβ8.1/2+CD8+T cells infiltrating orthotopic 4T1HA mammary tumors in mice treated with bortezomib (1 mg/kg body weight) (Figure 9D). No noticeable change was observed in the levels of other STAT members. We also analyzed the expression of T-box transcription factors eomesodermin and T-bet in CD8+T cells that were stimulated in vitro with soluble anti-mouse CD3 and CD28 antibodies (1 μg/ml each) for 48 h with or without treatment with bortezomib for another 4 h. We observed significant upregulation of intracellular eomesodermin but not T-bet expression in CD8+T cells (Figure 10A). CD4+T cells also did not show any change in T-bet expression (data not shown). Moreover, no change in the expression of FoxP3 transcription factor was observed in tumor-infiltrating CD4+T cells in 4T1HA and RencaHA tumor-bearing mice (Figure 10B). This suggests that bortezomib treatment upregulated signaling pathways leading to an increased expression of CD8+T cell effector molecules, without upregulating CD4+T regulatory immunosuppressive signaling.
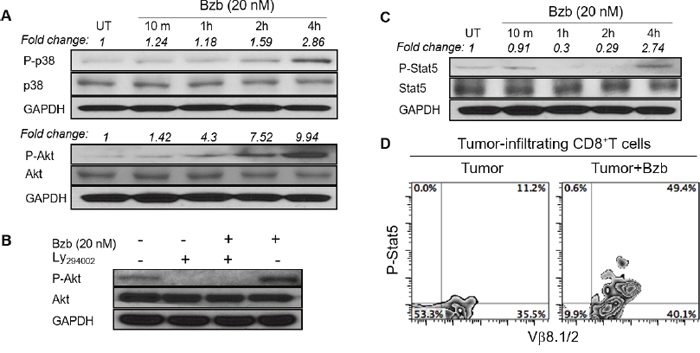
Figure 9: Bortezomib modulates the expression of phosphorylated p38, Akt, and STAT5 in CD8+T cells. Levels of phosphorylated and total proteins were analyzed by Western blot in CD8+T cells following treatment in vitro with Bzb (20 nM) for the indicated duration of time. A. Protein levels of phosphorylated and total p38 and Akt. B. Protein levels of phosphorylated and total Akt following treatment with a combination of Bzb and PI3K inhibitor Ly294002 (50 μM). C. Protein levels of phosphorylated and total STAT5. The values on the Western blots represent fold change in expression analyzed by the densitometry with GAPDH used as a loading control. D. Orthotopic tumors were established in mammary pads of Balb/c WT mice following the injection of 2 x 106 4T1HA tumor cells. On day 14 (after the tumor reached an approximate size of 125 mm3), mice were injected intravenously with 1 mg/kg body weight of bortezomib. After 4 h, the mice were euthanized and single cell suspensions were obtained from the tumor mass harvested. Intracellular expression of phosphorylated STAT5 on tumor-infiltrating CD8+T cells was analyzed by flow cytometry. Numbers inside plots depict % positive cells. Representative data are from 2 individual experiments.
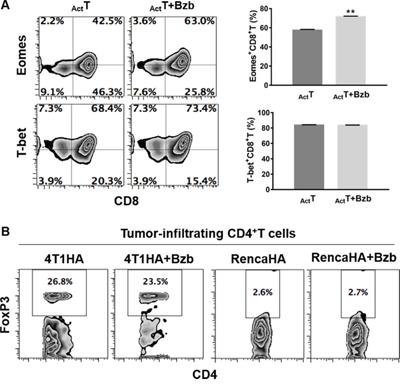
Figure 10: Effect of bortezomib treatment on the expression of transcription factors eomesodermin, T-bet and FoxP3 in T cells. A. Purified CD8+T cells from spleens of naïve Balb/c mice were stimulated in vitro with soluble anti-mouse CD3 and CD28 antibodies (1 μg/ml each) for 48 h with or without treatment with Bzb (10 nM) for another 4 h. Intracellular expression of transcription factors Eomes and T-bet were analyzed by flow cytometry. Numbers inside plots depict % positive cells. Bar graphs present data as means ± SEM for Eomes+CD8+T cells and T-bet+CD8+T cells from 2 independent experiments. **p<0.01; two-tailed t-test. B. Orthotopic (4T1HA) or subcutaneous (RencaHA) tumors were established in Balb/c WT mice following the injection of 2 x 106 tumor cells. On day 14, mice were injected intravenously with 1 mg/kg body weight of bortezomib. After 4 h, the mice were euthanized and single cell suspensions were obtained from the tumor mass harvested. Intracellular expression of FoxP3 on tumor-infiltrating CD8+T cells was analyzed by flow cytometry. Numbers inside plots depict % positive cells. Representative data are from 2 individual experiments.
Altogether, these results suggest the immunostimulatory potential of bortezomib to augment the strength of convergent signals from CD8+T cell signaling molecules such as TCR, IL-2, IL-12 and IL-15 cytokine receptors and downstream PI3K/Akt/STAT5 pathways to sustain the expression of CD8+T cell effector molecules in the tumor microenvironment as illustrated schematically in Figure 11.
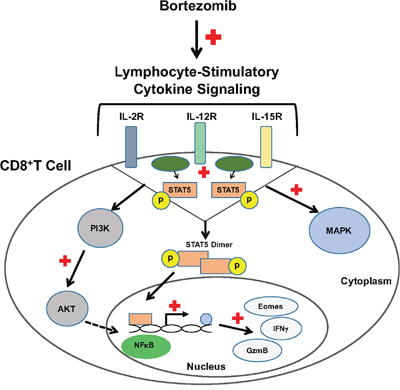
Figure 11: Bortezomib enhances cytokine receptor signaling to enhance the expression of CD8+T cell effector molecules. Bortezomib treatment increases lymphocyte-stimulatory signaling through cytokines, IL-2, IL-12, and IL-15 via p38/PI3K/Akt/STAT5 pathways. This increases the transcription of effector molecules eomesodermin, granzyme-B and IFNγ in CD8+T lymphocytes.
DISCUSSION
We report that in mice bearing kidney or breast adenocarcinomas that present a defined low-avidity epitope derived from hemagglutinin model antigen, bortezomib administration at 1 mg/kg body weight augments the expression of lymphocyte-stimulatory cytokines IL-2, IL-12, and IL-15 and their downstream signaling molecules. We observed that bortezomib treatment increased IL-2 production by CD4+T cells, IL-12 and IL-15 production from CD11c+ dendritic cells, and enhanced the expression of IL-2 receptor-α chain, IFNγ, granzyme-B, as well as the transcription factor eomesodermin on endogenous or adoptively transferred HA-specific CD8+T cells from bortezomib-treated tumor-bearing mice. We also noted increased phosphorylation of mitogen-activated protein kinase p38, Akt and transcription factor STAT5 in CD8+T cells following bortezomib treatment. These observations support the immunostimulatory potential of bortezomib with a capacity to sustain CD8+T cell effector function in the tumor microenvironment of solid tumors.
Tumor-mediated immunosuppression affects immune surveillance mechanisms and impedes the effectiveness of anti-cancer therapy [39]. The expression of immunoinhibitory molecules and cytokines, for example, IL-1β, VEGF, IL-10, and TGFβ and downregulation of immunostimulatory cytokines (e.g., IL-2, IL-12, IL-15) allow tumor to escape from immune control. IL-2 is a cytokine widely known to promote proliferation with a broad range of implications in tumor growth and control [40]. IL-2 promotes the expansion of T-regulatory cells expressing the ICOS molecule due to their increased expression of high-affinity IL-2 receptor CD25, which correlates with worse clinical outcome [41]. On the other hand, IL-15, another member of the IL-2 cytokine family, has more promising antitumor effects since it does not show overt side effects, such as vascular leak syndrome observed for IL-2 [42]. IL-15 promotes differentiation and proliferation of T and NK cells as well as induces long-lasting T cell memory by upregulating anti-apoptotic genes, such as Bcl-xL and Bcl-2 [42]. Thus, IL-2 together with IL-15 sustains productive T cell proliferation, effector differentiation, and survival [43]. IL-12 is another immunostimulatory cytokine produced by primed antigen-presenting cells, and the consensus is that it supports CD8+T cell responses, Th1 lymphocyte differentiation, and NK cell function, all activities necessary for immune surveillance of tumors [44]. IL-12 by itself also has some antitumor activity mediated by its ability to induce IFNγ and elicit anti-angiogenic activity [44].
In our cancer models, we noted that the introduction of HA antigen into the tumor cells elicited some basal immune response. This is seen with increased IL-12 and IL-15 production in tumor-bearing mice as compared to control mice with no tumor. Treatment of tumor-bearing mice with bortezomib further increased the production of these lymphocyte-stimulatory cytokines IL-2, IL-12, and IL-15, which have significant impact on the development of a cytotoxic response mediated by CD8+T cells and NK cells. We also noted that IFNγ synthesis and control of metastatic growth are both dependent on IL-12 and IL-15 signaling since the blocking of these cytokines decreased IFNγ production and increased the number of tumor pulmonary nodules. Of note, bortezomib treatment restored IFNγ synthesis and inhibited metastasis in tumor-bearing mice even in the presence of neutralizing antibodies to IL-12 and IL-15. This indicates that bortezomib can activate alternative pathways leading to IFNγ production that could strengthen tumor immune surveillance. In particular, multiple cytokines such as IL-6, LIX (CXCL5), KC, etc. were upregulated and pro-angiogenic factor VEGF was downregulated following bortezomib treatment. Other studies also indicate that bortezomib inhibits the pro-tumor effects of IL-6, TGFβ, VEGF etc. and decreases tumor angiogenesis [15, 45].
Our data also show that bortezomib treatment increased phosphorylation of mitogen-activated protein kinase p38 and Akt as well as higher levels of phosphorylation of the transcription factor STAT5 in tumor-infiltrating CD8+T cells. Our recent studies showed that bortezomib can also increase the cytoplasmic and nuclear phosphorylation of NFκB p65 in CD8+T cells while maintaining IFNγ secretion to improve FasL-mediated tumor lysis [7, 33]. Recognition of antigen by the TCR activates the Ras-MAPK pathway, and costimulation provided by CD28 activates the phosphatidyl inositol-3-kinase pathway that synergizes to augment TCR-induced transcriptional regulation mediated by AP-1, NF-AT and NFκB [46]. In contrast to signaling by the TCR and CD28, the JAK/STAT pathways play a major role in signaling by cytokine receptors. The phenotype and functions of T cells can be modified by a wide array of cytokines, but IL-12 and/or IFN-α are distinguished by the fact that they can act as the ‘switch’ that determines whether a response to antigen will result in tolerance or full activation of antigen-specific CD8+T cells. Our results show predominant effects of bortezomib on both the p40 and p70 chains of IL-12 produced by dendritic cells. Signals provided by IL-12 and/or IFN-α/β are required for the activation of naïve CD8 T cells, and IL-2 is needed to sustain and further expand the effector cells if antigen persists [46]. The JAKs and STATs involved in the signaling pathways employed by these cytokines are potential targets for immunosuppression. Bortezomib treatment upregulated the expression of STAT5 in antigen-specific tumor-infiltrating CD8+T cells. STAT5 activation contributes to the stabilization of the gene expression program in CD8+T cells. Phosphorylation and nuclear translocation of STAT5 are triggered by IL-2 bound to its high-avidity receptor IL-2Rα (CD25), the expression of which is controlled by TCR stimulation; STAT5 is not activated in naive T cells even when cultured in the presence of IL-2 [47, 48]. This mechanism permits cytokine receptor signaling to rescue abortive TCR signaling, such as that induced in response to weak or partial TCR agonists by complementing it with the activation of STAT5 that binds to TNF receptor family costimulatory molecules and granzyme-B promoters to stabilize the CD8+T cell effector program. Furthermore, bortezomib-increased phosphorylation of Akt in T cells was abrogated by the PI3K inhibitor. The PI3K/Akt signaling pathway controls proliferation, differentiation, and survival in many cell types including T cells [49]. The cumulative strength of convergent signals from signaling molecules, such as TCR, costimulatory molecules, and cytokine receptors governs the magnitude of Akt activation, which in turn dictates the differentiation of cytotoxic T lymphocytes into terminal effectors or memory cells [50]. It is to be noted that in response to IL-15 augmentation upon bortezomib treatment, enhanced PI3K/Akt/STAT5 activation may support the maintenance and survival of effector and memory CD8+T cells as observed during viral infections [51].
Thus, bortezomib may act as a multifaceted immunomodulatory drug in addition to suppressing constitutive activation of NFκB in tumor cells, whereby it abrogates their resistance to cell death triggers [17, 28, 33, 52]. In another study, we have observed that bortezomib treatment in mice bearing various solid tumors resulted in an upregulated expression of Notch signaling components to facilitate interplay between Notch and NFκB signaling pathways in CD8+T lymphocytes [7]. Altogether, these data strongly support the case that bortezomib can augment the strength of activation signals in antigen-specific CD8+T cells in the tumor microenvironment via cytokine receptors and downstream PI3K/Akt/NFκB/STAT5 pathways, which together sustain the differentiation of CD8+T cells into terminal effectors.
It is worth noting that the concentration of bortezomib to a large extent determines the outcome of its effects on the immune system with higher concentrations in excess of 20 nM causing immunosuppressive effects while concentrations lower than 20 nM provide immunostimulatory effects [15, 22, 27, 32, 53-55]. Immunosuppressive effects of bortezomib were observed in in vitro studies where the doses used were high (>100 nM) [56, 57]; doses that cannot be extrapolated to the therapeutic situation in vivo. Only 10–20 nM concentrations of bortezomib are attainable in vivo [58]. For example, Blanco et al observed that bortezomib treatment at high concentrations (>100 nM) generated immunosuppressive CD4+CD25+ regulatory T cells to prevent graft versus host disease [59]. Whereas we observed an immunostimulatory effect in IL-2 secretion by CD4+T cells following low dose (~20 nM) bortezomib treatment that improved anti-tumor immunity. Therefore, therapeutic administration of a carefully optimized, minimal bortezomib dose customized for a disease condition would prove effective in providing immune benefits while limiting the negative immune effects. Considering possible mechanisms of bortezomib action based on available data, we propose that this proteasome inhibitor can affect tumor growth by changing the intrinsic activity of immune cells. It is noteworthy that chronic inflammation has an important impact in the development of tumors by generating a tumor-promoting microenvironment [60]. Bortezomib appears to release breaks on the immune system from the tumor-associated suppressive microenvironment, and thereby allow immune cells to become activated. Our recent studies showed that bortezomib can enhance FasL-mediated tumor lysis [33]. It is also known that bortezomib can make tumor cells sensitive to NK cell-mediated killing [29, 31, 61]. Moreover, bortezomib inhibited TGFβ-mediated suppression of IFNγ and granzyme-B expression in activated CD8+T cells [7]. These studies together with our results show that bortezomib can increase the levels of immunostimulatory cytokines IL-2, IL-12, and IL-15 and enhance their downstream signaling pathways to augment CD8+T cell effector function. Thus, bortezomib, being a reversible proteasome inhibitor with a short half-life, can be combined with other immunotherapies and may have major clinical implications to break tumor-induced tolerance in CD8+T cells and elicit a robust antitumor immune response in cancer patients with solid malignancies.
MATERIALS AND METHODS
Mice
Balb/c mice were purchased from Harlan (Indianapolis, IN) and mice were 6-8-week-old and 25-30 g at the time of use. TCRHA-transgenic Cln4 mice (αβ TCR specific for hemagglutinin HA518–526 peptide restricted to H-2Kd) were provided by Linda A. Sherman (The Scripps Research Institute, La Jolla, CA), and were bred at Meharry Medical College (MMC) animal facility and genotyped according to the methods described earlier [35]. Mice were housed in filter-topped cages under specific pathogen-free conditions in MMC animal facilities. Mice were cared for in accordance with the procedures outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals and Institutional Animal Care and Use Committee (IACUC) at MMC. MMC is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International and follows the Public Health Service Policy for the care and use of laboratory animals under pathogen-free conditions.
Tumor cell lines
The murine mammary adenocarcinoma line 4T1.2HA (courtesy Suzanne Ostrand-Rosenberg, University of Maryland, Baltimore, MD), renal cell adenocarcinoma line RencaHA (courtesy Hyam I. Levitsky, John Hopkins University, Baltimore, MD) and fibrosarcoma line D459 (courtesy David Carbone, Ohio State University, Columbus, OH) were maintained in 10% FCS-supplemented standard RPMI-1640 (Gibco, Invitrogen) culture medium. Low-passage (< 5) tumor cell cultures were used for the experiments, and were regularly authenticated with reference stocks to ensure fidelity; routine sterility and Mycoplasma testing was performed regularly. Solid tumors were induced in syngeneic Balb/c WT by injecting 2 x 106 4T1HA or RencaHA cells orthotopically under mammary pads or subcutaneously, respectively, into the right flank for approximately 14 days. Following the establishment of palpable tumors of approximately 120 mm3 size, mice were injected with therapeutic dose of bortezomib (1 mg/kg body weight) intravenously, as we optimized previously [17], and roughly correlated to a transient 20 nM concentration on the basis of the observation that a mouse of 20-25 g weight has approximately a blood volume of about 1.5 ml. After 4 h lymphoid tissues were harvested for the preparation of single cell suspensions. The right inguinal and brachial LN were used as the tumor-draining LN, while inguinal and brachial LNs of the left side were used as contralateral LN.
Cell harvesting and sample preparation
Tissues were harvested from mice after sedation with isoflourene and cervical dislocation. Single cells suspensions were made from tissues mashed/homogenized on the Falcon 40 μm cell strainers in petri dishes containing complete RPMI media. The media containing cells were transferred to labeled 15 mL conical tubes, then spun down at 1200 rpm for 5 min at 4°C. Cells were washed twice by aspirating media after spin, resuspending cell pellets in media, and centrifuging again. Splenocytes were suspended in 1 ml of ACK buffer (KD Medical, Columbia, MD) for 1 min at room temperature to lyse erythrocytes. The conical tube was filled with complete RPMI media following the 1 min treatment to neutralize the reaction and centrifuged. The pellet was resuspended in complete RPMI media. 10 μL of each sample were mixed with 10μL of trypan blue, placed on a slide, and cells were counted using a Countess (Invitrogen) machine for total cell counts and viability.
Milliplex mouse cytokine/chemokine magnetic bead panel
Cell lysates were prepared from whole tissue or serum samples. Lysates and serum were both quantified by BCA Protein Assay Kit (Pierce), and 250 μg of protein was equalized and used per sample in the cytokine/chemokine magnetic bead panel plate (Millipore) as per the manufacturer’s protocol. The plate was read on the MAGPIX instrument (Millipore) with xPONENT software. The median fluorescent intensity (MFI) data were saved and analyzed using a 5-parameter logistic or spline curve-fitting method for calculating cytokine/chemokines concentrations in samples.
Antibodies and immunofluorescence surface and intracellular staining
RBC-depleted splenocytes and/or lymphocytes (1 x 106) were plated in a 96-well U-bottom plate, then spun down at 1100 rpm for 5 min at 4°C. RPMI medium was flicked off and plate vortexed to break up the pellet. Appropriate dilutions of antibodies were prepared in flow buffer containing 0.5% FBS. 50μL of fluorochrome-labelled anti-mouse antibodies: CD4-PE, CD4-FITC, CD8-APC, CD8-PerCPCy5.5, Vβ8.1/2-PerCPcy5.5, CD11c-PerCPCy5.5, IL-2Rα-APC, IL-2Rβ-FITC, IL-2Rγ-PE, IL-12Rβ-PE and IL-15Rα-APC were added to the designated wells and gently shook. The 96-well plate was placed on ice to incubate for 30 min in the dark. After incubation, 100μL of flow buffer was added to each well and the plate was centrifuged at 1100 rpm for 5 min at 4°C for washing. Cells were washed again with 150μL. Cells were resuspended and fixed in 200μL of paraformaldehyde, covered, and placed in 4°C until acquired. All monoclonal antibodies were purchased from eBioscience and Biolegend.
Following the surface immunofluorescence staining, to measure intracellular protein levels, cells were treated with Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer’s instructions. We used the following intracellular anti-mouse Abs: IL-2-FITC, IL-12-PE, IL-15-APC, IFNγ-PE, granzyme B-FITC, eomesodermin-PE, T-bet-APC, Foxp3-PE and a panel of anti-human/mouse phospho-STAT1-6-PE (eBiosciences and Biolegend). Intracellular staining was performed by the incubation of cells at appropriate dilutions in Permwash buffer for 30 min at 4°C in the dark.
For the cytokine neutralization experiment, anti-human/mouse IL-15 and anti-mouse IL-12 purified Abs as well as their isotype control anti-mouse IgG and anti-mouse IgG1, respectively, were purchased from Abcam.
RNA isolation and quantitative PCR
Total RNA was extracted using an RNeasy mini kit (Qiagen) and quantitated by reading the optical density at 260 nm. Possible genomic DNA contamination was removed by on-column DNase digestion using the RNase-free DNase set. The cDNA was synthesized using iScript cDNA synthesis kit (Bio-Rad). Real-time quantitative RT-PCR (qRT-PCR) was performed using CFX-96 Real Time System (Bio-Rad). The iQ SYBR green supermix (Bio-Rad) and gene-specific PCR primers were used in a 20 μL reaction following protocols recommended by the manufacturer. The conditions used for the PCR were as follows: 95°C for 3 min (1 cycle), 94°C for 20 s, 55°C for 30 s, and 72°C for 40 s (40 cycles). Fold changes in mRNA expression were assessed by the ΔΔCt method. Primer sequences are as follows: Murine IL-2 (forward: 5’-TGAGTCAGCAACTGTGGTGG-3’, reverse: 5’-GCCCTTGGGGCTTACAAAAAG-3’), murine IL-12 (forward: 5’-GGGACCAGGCCCTATTATGC-3’, reverse: 5’-GAC CAAAGCCAGCTCCTCAT-3’), murine IL-15 (forward: 5’-CGCCCAAAAGACTTGCAGTG-3’, reverse: 5’-GGTGGATTCTCTCTGAGCTGT-3’), murine β-actin (forward: 5’-AGTGTGACGTTGACATCCGTA-3’, reverse: 5’-GCCAGAGCAGTAATCTCCTTC-3’), human IL-2 (forward: 5’-AGAATC CCAAACTCACCAGGA-3’, reverse: 5’-TGTTTCAGTTCTGTGGCCTTC-3’), and human β-actin (forward: 5’-CTCGCCTTTGCCGATCC-3’, reverse: 5’-GGGGTACTTCAGGGTGAGGA-3’).
Western blot analysis
Cell pellets from various tissues from tumor-bearing or naïve mice were lysed in complete lysis buffer including protease and phosphatase inhibitors. 50 μg of each protein sample was electrophoresed on NuPage 4-12% Bis-Tris gel (Novex Life Technologies) and transferred to polyvinylidenedifluoride membranes using an iBlot® Dry Blotting system (Life Technologies). The membrane was then blocked in 5% skimmed milk in 1X phosphate-buffered saline-Tween-20 (1X PBST) for 2 h at room temperature with gentle agitation. After blocking, the blots were incubated with specific primary antibodies for the phosphorylated and total levels of STAT5, Akt, and p38 in 1% BSA (in 1X PBST) overnight at 4°C with gentle agitation. After 5 washes of 5 min each in 1X PBST, blots were incubated with goat anti-rabbit horseradish peroxidase (Santa Cruz Biotechnology) at a dilution of 1:4000 in 1X PBST for 1.5 h, with agitation. The blots were rinsed again in 1X PBST, and developed by using chemiluminescence reagent (EMD Millipore, MA) and a Bio-Rad Image Station. The density of each protein band was determined by densitometric analysis using the imageJ software (NIH). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) levels were determined for each condition to verify that equal amounts of protein were loaded. In addition, the density of each protein band was normalized to GAPDH to determine relative protein expression to the internal control.
Acquisition and data analysis
Flow samples were acquired on guava EasyCyte HT (Millipore) instrument with 50-200,000 cells acquired for each sample well. Gates for samples were determined by single color controls. Unstained and isotype controls were used to ascertain if any nonspecific binding of antibodies was present. All data were analyzed on FlowJo 7.6 software (TreeStar). Data were analyzed using either one-way ANOVA or two-tailed t-test using Graphpad Prism 7, with p ≤ 0.05 considered statistically significant.
Abbreviations
HA, Hemagglutinin; 4T1HA, 4T1.2 expressing HA; STAT, Signal transducer and activator of transcription, JAK, Janus kinase; TGFβ, Transforming growth factor beta; VEGF, Vascular endothelial growth factor; NFκB, Nuclear factor kappa-light-chain-enhancer of activated B cells; ICOS, Inducible T-cell costimulator; ACT, Adoptive cell immunotherapy; TIL, Tumor infiltrating lymphocytes; LN, Lymph node; RAG, Recombination activating gene; Eomes, Eomesodermin; MFI, Median fluorescent intensity; qRT-PCR, Real-time quantitative RT-PCR.
ACKNOWLEDGMENTS
We would like to acknowledge and thank Qiujia Shao (Meharry Flow Cytometry Core) and Suzan Vaughn (Vanderbilt Hormone Assay and Analytical Core) for their assistance with instrument operations. We would also like to thank STP’s Committee on Instructions members: Drs. Samuel Adunyah, Fernando Villalta, Bogdan Nowicki, Daniel McVicar, and Jason Grayson for their scientific input and suggestions. The work presented here comprises part of STP’s doctoral dissertation.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
FUNDING
This work was supported by funds to AS by the following National Institutes of Health (NIH) grants U54 CA163069, P50 CA090949, U54 MD007593, SC1 CA182843, and R01 CA175370. STP was supported by NIH training grants T32 5T32HL007737 and R25 GM059994. STP was also supported by Vanderbilt VICTR grant funding VR9111. The Meharry Morphology and Flow Cytometry Cores are supported by NIH grants G12 MD007586, R24 DA036420 and S10 RR0254970. The Vanderbilt Hormone Assay and Analytical Core is supported by NIH grants DK059637 and DK020593.
Author contributions
Conception and design: A Shanker
Development of methodology: A Shanker, ST Pellom, DF Dudimah
Acquisition of data: ST Pellom, DF Dudimah
Analysis and interpretation of data: A Shanker, ST Pellom, DF Dudimah, MC Thounaojam, RV Uzhachenko, A Singhal, A Richmond
Writing and review: A Shanker, ST Pellom, DF Dudimah, MC Thounaojam, RV Uzhachenko, A Singhal
Administrative, technical, or material support: A Shanker
Study supervision: A Shanker
REFERENCES
1. Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013; 39:61-73.
2. Huang Y, Lin L, Shanker A, Malhotra A, Yang L, Dikov MM, Carbone DP. Resuscitating cancer immunosurveillance: selective stimulation of DLL1-Notch signaling in T cells rescues T-cell function and inhibits tumor growth. Cancer Res. 2011; 71:6122-6131.
3. Marcais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, Rabilloud J, Mayol K, Tavares A, Bienvenu J, Gangloff YG, Gilson E, Vivier E, Walzer T. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol. 2014; 15:749-757.
4. Bash J, Zong WX, Banga S, Rivera A, Ballard DW, Ron Y, Gelinas C. Rel/NF-kappaB can trigger the Notch signaling pathway by inducing the expression of Jagged1, a ligand for Notch receptors. The EMBO J. 1999; 18:2803-2811.
5. Guan E, Wang J, Laborda J, Norcross M, Baeuerle PA, Hoffman T. T cell leukemia-associated human Notch/translocation-associated Notch homologue has I kappa B-like activity and physically interacts with nuclear factor-kappa B proteins in T cells. J Exp Med. 1996; 183:2025-2032.
6. Wang J, Shelly L, Miele L, Boykins R, Norcross MA, Guan E. Human Notch-1 inhibits NF-kappa B activity in the nucleus through a direct interaction involving a novel domain. J Immunol. 2001; 167:289-295.
7. Thounaojam MC, Dudimah DF, Pellom ST, Jr., Uzhachenko RV, Carbone DP, Dikov MM, Shanker A. Bortezomib enhances expression of effector molecules in anti-tumor CD8+ T lymphocytes by promoting Notch-nuclear factor-kappaB crosstalk. Oncotarget. 2015; 6:32439-32455. doi: 10.18632/oncotarget.5857.
8. Anguille S, Van Acker HH, Van den Bergh J, Willemen Y, Goossens H, Van Tendeloo VF, Smits EL, Berneman ZN, Lion E. Interleukin-15 Dendritic Cells Harness NK Cell Cytotoxic Effector Function in a Contact- and IL-15-Dependent Manner. PLoS One. 2015; 10:e0123340.
9. Hainsworth JD, Spigel DR, Barton J, Farley C, Schreeder M, Hon J, Greco FA. Weekly treatment with bortezomib for patients with recurrent or refractory multiple myeloma: a phase 2 trial of the Minnie Pearl Cancer Research Network. Cancer. 2008; 113:765-771.
10. Jagannath S, Barlogie B, Berenson J, Siegel D, Irwin D, Richardson PG, Niesvizky R, Alexanian R, Limentani SA, Alsina M, Adams J, Kauffman M, Esseltine DL, Schenkein DP, Anderson KC. A phase 2 study of two doses of bortezomib in relapsed or refractory myeloma. Br J Haematol. 2004; 127:165-172.
11. Yang M, Li D, Chang Z, Yang Z, Tian Z, Dong Z. PDK1 orchestrates early NK cell development through induction of E4BP4 expression and maintenance of IL-15 responsiveness. J Exp Med. 2015; 212:253-265.
12. Shah NN, Baird K, Delbrook CP, Fleisher TA, Kohler ME, Rampertaap S, Lemberg K, Hurley CK, Kleiner DE, Merchant MS, Pittaluga S, Sabatino M, Stroncek DF, Wayne AS, Zhang H, Fry TJ, et al. Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T-cell-depleted stem cell transplantation. Blood. 2015; 125:784-792.
13. Braun M, Bjorkstrom NK, Gupta S, Sundstrom K, Ahlm C, Klingstrom J, Ljunggren HG. NK cell activation in human hantavirus infection explained by virus-induced IL-15/IL15Ralpha expression. PLoS Pathog. 2014; 10:e1004521.
14. Gillgrass A, Gill N, Babian A, Ashkar AA. The absence or overexpression of IL-15 drastically alters breast cancer metastasis via effects on NK cells, CD4 T cells, and macrophages. J Immunol. 2014; 193:6184-6191.
15. Pellom ST, Jr., Dudimah DF, Thounaojam MC, Sayers TJ, Shanker A. Modulatory effects of bortezomib on host immune cell functions. Immunotherapy. 2015; 7:1011-1022.
16. Khan T, Stauffer JK, Williams R, Hixon JA, Salcedo R, Lincoln E, Back TC, Powell D, Lockett S, Arnold AC, Sayers TJ, Wigginton JM. Proteasome inhibition to maximize the apoptotic potential of cytokine therapy for murine neuroblastoma tumors. J Immunol. 2006; 176:6302-6312.
17. Shanker A, Brooks AD, Tristan CA, Wine JW, Elliott PJ, Yagita H, Takeda K, Smyth MJ, Murphy WJ, Sayers TJ. Treating metastatic solid tumors with bortezomib and a tumor necrosis factor-related apoptosis-inducing ligand receptor agonist antibody. J Natl Cancer Inst. 2008; 100:649-662.
18. Brooks AD, Jacobsen KM, Li W, Shanker A, Sayers TJ. Bortezomib sensitizes human renal cell carcinomas to TRAIL apoptosis through increased activation of caspase-8 in the death-inducing signaling complex. Mol Cancer Res. 2010; 8:729-738.
19. Berges C, Haberstock H, Fuchs D, Miltz M, Sadeghi M, Opelz G, Daniel V, Naujokat C. Proteasome inhibition suppresses essential immune functions of human CD4+ T cells. Immunology. 2008; 124:234-246.
20. Feng X, Yan J, Wang Y, Zierath JR, Nordenskjold M, Henter JI, Fadeel B, Zheng C. The proteasome inhibitor bortezomib disrupts tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression and natural killer (NK) cell killing of TRAIL receptor-positive multiple myeloma cells. Molecular Immunol. 2010; 47:2388-2396.
21. Heider U, Rademacher J, Kaiser M, Kleeberg L, von Metzler I, Sezer O. Decrease in CD4+ T-cell counts in patients with multiple myeloma treated with bortezomib. Clin Lymphoma Myeloma Leuk. 2010; 10:134-137.
22. Straube C, Wehner R, Wendisch M, Bornhauser M, Bachmann M, Rieber EP, Schmitz M. Bortezomib significantly impairs the immunostimulatory capacity of human myeloid blood dendritic cells. Leukemia. 2007; 21:1464-1471.
23. Zinser E, Rossner S, Littmann L, Luftenegger D, Schubert U, Steinkasserer A. Inhibition of the proteasome influences murine and human dendritic cell development in vitro and in vivo. Immunobiology. 2009; 214:843-851.
24. Mulder A, Heidt S, Vergunst M, Roelen DL, Claas FH. Proteasome inhibition profoundly affects activated human B cells. Transplantation. 2013; 95:1331-1337.
25. Ames E, Hallett WH, Murphy WJ. Sensitization of human breast cancer cells to natural killer cell-mediated cytotoxicity by proteasome inhibition. Clin Exp Immunol. 2009; 155:504-513.
26. Armeanu S, Krusch M, Baltz KM, Weiss TS, Smirnow I, Steinle A, Lauer UM, Bitzer M, Salih HR. Direct and natural killer cell-mediated antitumor effects of low-dose bortezomib in hepatocellular carcinoma. Clin Cancer Res. 2008; 14:3520-3528.
27. Chang CL, Hsu YT, Wu CC, Yang YC, Wang C, Wu TC, Hung CF. Immune mechanism of the antitumor effects generated by bortezomib. J Immunol. 2012; 189:3209-3220.
28. Hallett WH, Ames E, Motarjemi M, Barao I, Shanker A, Tamang DL, Sayers TJ, Hudig D, Murphy WJ. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol. 2008; 180:163-170.
29. Lundqvist A, Yokoyama H, Smith A, Berg M, Childs R. Bortezomib treatment and regulatory T-cell depletion enhance the antitumor effects of adoptively infused NK cells. Blood. 2009; 113:6120-6127.
30. Seeger JM, Schmidt P, Brinkmann K, Hombach AA, Coutelle O, Zigrino P, Wagner-Stippich D, Mauch C, Abken H, Kronke M, Kashkar H. The proteasome inhibitor bortezomib sensitizes melanoma cells toward adoptive CTL attack. Cancer Res. 2010; 70:1825-1834.
31. Shi J, Tricot GJ, Garg TK, Malaviarachchi PA, Szmania SM, Kellum RE, Storrie B, Mulder A, Shaughnessy JD, Jr., Barlogie B, van Rhee F. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood. 2008; 111:1309-1317.
32. Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood. 2007; 109:4839-4845.
33. Shanker A, Pellom ST, Jr., Dudimah DF, Thounaojam MC, de Kluyver RL, Brooks AD, Yagita H, McVicar DW, Murphy WJ, Longo DL, Sayers TJ. Bortezomib Improves Adoptive T-cell Therapy by Sensitizing Cancer Cells to FasL Cytotoxicity. Cancer Res. 2015; 75:5260-5272.
34. Pulaski BA, Ostrand-Rosenberg S. Mouse 4T1 breast tumor model. Current protocols in immunology / edited by John E Coligan [et al]. 2001; Chapter 20:Unit 20 22.
35. Morgan DJ, Kreuwel HT, Fleck S, Levitsky HI, Pardoll DM, Sherman LA. Activation of low avidity CTL specific for a self epitope results in tumor rejection but not autoimmunity. J Immunol. 1998; 160:643-651.
36. Pilipow K, Roberto A, Roederer M, Waldmann TA, Mavilio D, Lugli E. IL15 and T-cell Stemness in T-cell-Based Cancer Immunotherapy. Cancer Res. 2015; 75:5187-5193.
37. Mao Y, van Hoef V, Zhang X, Wennerberg E, Lorent J, Witt K, Masvidal Sanz L, Liang S, Murray S, Larsson O, Kiessling R, Lundqvist A. IL-15 activates mTOR and primes stress-activated gene-expression leading to prolonged anti-tumor capacity of NK cells. Blood. 2016.
38. Lehmann D, Spanholtz J, Sturtzel C, Tordoir M, Schlechta B, Groenewegen D, Hofer E. IL-12 directs further maturation of ex vivo differentiated NK cells with improved therapeutic potential. PLoS One. 2014; 9:e87131.
39. Wu AA, Drake V, Huang HS, Chiu S, Zheng L. Reprogramming the tumor microenvironment: tumor-induced immunosuppressive factors paralyze T cells. Oncoimmunology. 2015; 4:e1016700.
40. Kamphorst AO, Araki K, Ahmed R. Beyond adjuvants: immunomodulation strategies to enhance T cell immunity. Vaccine. 2015; 33:B21-28.
41. Sim GC, Martin-Orozco N, Jin L, Yang Y, Wu S, Washington E, Sanders D, Lacey C, Wang Y, Vence L, Hwu P, Radvanyi L. IL-2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J Clin Invest. 2014; 124:99-110.
42. Pagliari D, Cianci R, Frosali S, Landolfi R, Cammarota G, Newton EE, Pandolfi F. The role of IL-15 in gastrointestinal diseases: a bridge between innate and adaptive immune response. Cytokine Growth Factor Rev. 2013; 24:455-466.
43. Tamzalit F, Barbieux I, Plet A, Heim J, Nedellec S, Morisseau S, Jacques Y, Mortier E. IL-15.IL-15Ralpha complex shedding following trans-presentation is essential for the survival of IL-15 responding NK and T cells. Proc Natl Acad Sci U S A. 2014; 111:8565-8570.
44. Moreno-Nieves UY, Didier C, Levy Y, Barre-Sinoussi F, Scott-Algara D and Network AHV. NK cells are primed by ANRS MVA(HIV)-infected DCs, via a mechanism involving NKG2D and membrane-bound IL-15, to control HIV-1 infection in CD4+ T cells. Eur J Immunol. 2014; 44:2370-2379.
45. Huntington ND. The unconventional expression of IL-15 and its role in NK cell homeostasis. Immunol Cell Biol. 2014; 92:210-213.
46. Jayaraman A, Jackson DJ, Message SD, Pearson RM, Aniscenko J, Caramori G, Mallia P, Papi A, Shamji B, Edwards M, Westwick J, Hansel T, Stanciu LA, Johnston SL, Bartlett NW. IL-15 complexes induce NK- and T-cell responses independent of type I IFN signaling during rhinovirus infection. Mucosal Immunol. 2014; 7:1151-1164.
47. Lebrec H, Horner MJ, Gorski KS, Tsuji W, Xia D, Pan WJ, Means G, Pietz G, Li N, Retter M, Shaffer K, Patel N, Narayanan PK, Butz EA. Homeostasis of human NK cells is not IL-15 dependent. J Immunol. 2013; 191:5551-5558.
48. Chen Y, Chen B, Yang T, Xiao W, Qian L, Ding Y, Ji M, Ge X, Gong W. Human fused NKG2D-IL-15 protein controls xenografted human gastric cancer through the recruitment and activation of NK cells. Cell Mol Immunol. 2015.
49. Mattiola I, Pesant M, Tentorio PF, Molgora M, Marcenaro E, Lugli E, Locati M, Mavilio D. Priming of Human Resting NK Cells by Autologous M1 Macrophages via the Engagement of IL-1beta, IFN-beta, and IL-15 Pathways. J Immunol. 2015; 195:2818-2828.
50. Guo Y, Luan L, Rabacal W, Bohannon JK, Fensterheim BA, Hernandez A, Sherwood ER. IL-15 Superagonist-Mediated Immunotoxicity: Role of NK Cells and IFN-gamma. J Immunol. 2015; 195:2353-2364.
51. Zanoni I, Spreafico R, Bodio C, Di Gioia M, Cigni C, Broggi A, Gorletta T, Caccia M, Chirico G, Sironi L, Collini M, Colombo MP, Garbi N, Granucci F. IL-15 cis presentation is required for optimal NK cell activation in lipopolysaccharide-mediated inflammatory conditions. Cell Rep. 2013; 4:1235-1249.
52. Richardson PG, Anderson KC. Bortezomib: a novel therapy approved for multiple myeloma. Clin Adv Hematol Oncol. 2003; 1:596-600.
53. Hirai M, Kadowaki N, Kitawaki T, Fujita H, Takaori-Kondo A, Fukui R, Miyake K, Maeda T, Kamihira S, Miyachi Y, Uchiyama T. Bortezomib suppresses function and survival of plasmacytoid dendritic cells by targeting intracellular trafficking of Toll-like receptors and endoplasmic reticulum homeostasis. Blood. 2011; 117:500-509.
54. Mattingly LH, Gault RA, Murphy WJ. Use of systemic proteasome inhibition as an immune-modulating agent in disease. Endocr Metab Immune Disord Drug Targets. 2007; 7:29-34.
55. Moran E, Carbone F, Augusti V, Patrone F, Ballestrero A, Nencioni A. Proteasome inhibitors as immunosuppressants: biological rationale and clinical experience. Semin Hematol. 2012; 49:270-276.
56. Basler M, Lauer C, Beck U, Groettrup M. The proteasome inhibitor bortezomib enhances the susceptibility to viral infection. J Immunol. 2009; 183:6145-6150.
57. Nencioni A, Garuti A, Schwarzenberg K, Cirmena G, Dal Bello G, Rocco I, Barbieri E, Brossart P, Patrone F, Ballestrero A. Proteasome inhibitor-induced apoptosis in human monocyte-derived dendritic cells. Eur J Immunol. 2006; 36:681-689.
58. Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, Anderson KC. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001; 61:3071-3076.
59. Blanco B, Perez-Simon JA, Sanchez-Abarca LI, Caballero-Velazquez T, Gutierrez-Cossio S, Hernandez-Campo P, Diez-Campelo M, Herrero-Sanchez C, Rodriguez-Serrano C, Santamaria C, Sanchez-Guijo FM, Del Canizo C, San Miguel JF. Treatment with bortezomib of human CD4+ T cells preserves natural regulatory T cells and allows the emergence of a distinct suppressor T-cell population. Haematologica. 2009; 94:975-983.
60. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010; 140:883-899.
61. Lundqvist A, Berg M, Smith A, Childs RW. Bortezomib Treatment to Potentiate the Anti-tumor Immunity of Ex-vivo Expanded Adoptively Infused Autologous Natural Killer Cells. J Cancer. 2011; 2:383-385.