
Introduction
The proto-oncogene Ras promotes cell proliferation, survival, migration, and differentiation. It does so through its distinct downstream effectors such as extracellular signal-related kinase (ERK) [1], Jun amino-terminal kinase (JNK), p38-kinase, and phosphatidylinositol 3-kinase (PI3K) and Ral (Ras-like) [2-4]. Ral is coupled to Ras via RalGDS (Ral guanine nucleotide dissociation factors). Ral proteins (RalA and RalB) are major effectors of Ras that have not been explored in human ovarian cancer. The Ras-GTP (active Ras) binds to and recruits RalGDS to conform Ral proteins from Ral-GDP (inactive) to Ral-GTP (active). Activated Ral proteins play an important role in a series of biological functions, such as cell exocytosis and membrane trafficking. They do so by activating various effectors including exocyst complex component 2 (Exoc2), exocyst complex component 8 (Exoc8), Ral binding protein-1 (RalBP1), and zonula occludens-1 (ZO-1)-associated nucleic acid binding protein (Zonab) [5]. Although RalA and RalB are significantly homologous in terms of amino acid identity, differences in their sub-cellular localization and effector binding results in variations in their signaling functionality [6, 7]. Under physiological conditions, only RalA is involved in the delivery of E-cadherin to the basal membrane of epithelial cells [8], and enhances early steps in cytokinesis and cell polarity in neurons through the exocyst subunit Exo84 [9]. RalA regulates synaptic plasticity in partnership with the scaffolding protein postsynaptic density protein-95(PSD-95) [10]. It also promotes insulin exocytosis from islet beta cells through exocyst [11]. In contrast, RalB has discrete functions, such as the regulation of innate immune response through activation of the TANK-binding kinase 1 (TBK1) by the exocyst subunit Exoc2 [12] and promotion of cytokinesis [13].
RalA and RalB are implicated in tumorigenesis and play distinct roles in mediating carcinogenesis. Over-activated Ral proteins confer many properties of malignant phenotypes, such as uncontrolled cell proliferation, survival, invasion, and metastasis, as well as radio-resistance [14]. In particular, RalA is required for anchorage-independent growth. Knockdown of RalA, but not RalB, blocks RalGEF-induced tumorigenesis in primary epithelial cells [15].
Along with others, we revealed that Ral proteins are overactivated and correlate with tumorigenicity of various human cancers, including malignant peripheral nerve sheath tumors [16], cancers of the pancreas [17], bladder[18], melanomas[19], lung[20], and cancer stem cells[21].
In this study, we focused on revealing the contribution of RalA to the biology of ovarian cancer and its potential value as a therapeutic target. Our findings indicated a significant overactivation of RalA and elements of its signaling in human cancer cell lines and tumor tissues. Elements upstream of RalA, such as aurora kinase A (AKA), and effectors downstream of RalA, such as RalBP1, were found to be deregulated in human ovarian cancer cells and ovarian cancer patient tissues. Intervention with the RalA signaling pathway was found to have a significant impact on the growth of ovarian cancer cells in vitro and in vivo. Additionally, we are reporting overactivation of RalA in cells expressing cancer stem cell markers, as well as samples from syngeneic mouse ovarian cancer model for the first time.
Results
RalA and RalB are overactivated in human ovarian cancer cells
Activation of RalA (conversion from GDP- to GTP-bound form) is induced downstream of Ras by mediation of RalGDS proteins and results in signaling to elements such as RalBP1 and cell division cycle 42 (CDC42) (Figure 1A). Phospho-aurora kinase A (P-AKA) also enhances activation of RalA [28], while geranylgeranyltransferase inhibitors (GGTIs) and protein phosphatase 2A (PP2A) reduce its activation [29].
We determined activation of RalA and RalB in selected human ovarian cancer cells and primary cultures of human ovarian surface epithelial cells (hOSEpiCs). The human ovarian cancer cells, OV-177, OV-202, OVCA-429, OVCAR-5, and SKOV-3 contained higher levels of RalA-GTP (active RalA) than hOSEpiCs (Figure 1C). All ovarian cancer cells except OV-202 contained RalB-GTP, while RalB activation was undetectable in hOSEpiC (Figure 1C).
RalA regulators and downstream effectors are deregulated in human ovarian cancer cells
Ras, as the upstream activator of Ral pathway, was found to be overactivated in three out of five ovarian cancer cells lines as compared to hOSEpiCs. The overall expression of Ras remained mainly comparable amongst these cells lines (Figure 1B). RalGDS, as a transmitter of signal from Ras to Ral, was expressed at slightly higher levels in cancer cells than normal cells (Figure 1B). The expression of RalA downstream effectors RalBP1 and CDC42 were also determined. While RalBP1 was undetectable in four of the ovarian cancers cells (Figure 1D), activated GTP-bound CDC42 was detected in OVCAR-5 and SKOV-3 cells. Considering the role of PP2A Aβ in deactivation of RalA [29], we examined the expression level of this tumor suppressor gene in ovarian cells. PP2A Aβ expression was higher in hOSEpiCs as compared to the ovarian cancer cell lines (Figure 1E). Finally, AKA as an activator of RalA was found in active (phosphorylated) form in OV-177, OV-202, and OVCA-429 cells, but not hOSEpiC or OVCAR-5 and SKOV-3 cells (Figure 1E).
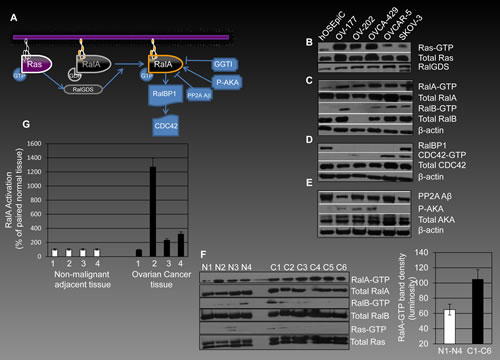
Figure 1: RalA signaling in human ovarian cancer. (A) Activation of RalA, down-stream of Ras, is mediated by RalGDS proteins and leads to conversion of RalA-GDP (inactive form) to RalA-GTP (active form). A series of regulators and down-stream effectors interact with this machinery. AKA phosphorylates RalA and plays a role in its activation, while PP2A Aβ acts as a suppressor of RalA activation. GGTIs down-regulate RalA activation by preventing geranyl-geranylation and therefore its plasma membrane association. One of the most important down-stream effectors of RalA involved in pro-oncogenic events is RalBP1, which signals through CDC42. (B-C) Ras, RalA and RalB in their active and total form were evaluated by affinity precipitation in a panel of human ovarian cancer cells in comparison to hOSEpiCs as their non-malignant counterpart. (D) Expression of RalBP1 and activation levels of CDC42 were also evaluated in ovarian cancer cells. (E) PP2A Aβ is expressed in all cells studied with highest levels observed in hOSEpiC. P-AKA levels were found to be present in three ovarian cancer cells and not in hOSEpiC. (F) The overall theme of overactivation of RalA, RalB and Ras is observed in human ovarian cancer tissues (C1-C6) as compared with non-malignant ovarian tissue samples (N1-N4). The right panel shows the average of band density for RalA-GTP in normal and cancerous tissues analyzed in the left panel (p<0.05). (G) Four independent matched ovarian cancer and adjacent normal tissues were analyzed for RalA-GTP levels using G-Lisa assay. Results are shows as percentage of RalA activation compared to normal tissues for each pair.
RalA activation is increased in human ovarian cancer tumor tissues
In order to further confirm the in vivo relevance of overactivation of RalA in human ovarian cancer, we investigated the levels of RalA-GTP in six human tumor samples and compared them with four non-malignant ovarian tissues. The overall level of RalA-GTP was higher in cancerous tissues. Notably, the cancer tissues (C1, C3, and C2) had increased RalA-GTP as compared to their adjacent normal tissues (N1, N2, and N3) (Figure 1F). RalB-GTP was also increased in cancer tissues although the difference observed was not as drastic as RalA-GTP. The right panel in figure 1F shows average band density for normal and cancerous tissues in the left 1F panel. Additionally, four independent matched ovarian cancer and their adjacent normal tissues were analyzed for the levels of RalA-GTP using G-Lisa assay (Figure 1G). Results are shown in malignant tissues as a percentage of matched normal samples.
Silencing RalA inhibits proliferation and invasion of ovarian cancer cells
To explore the functional role of RalA in proliferation, we infected ovarian cancer cells with lentivirus particles expressing shRNA against RalA (anti-RalA lentivirus, Multiplicity of Infection(MOI)~10 colony forming unit(cfu)/cell) or negative control lentivirus (Neg. Ctrl Virus) expressing scrambled shRNA at the same MOI. Once tested in OVCAR-5 cells, anti-RalA virus repressed RalA protein expression in a significant manner as was shown for days 3 and 4 post-infection (Figure 2A). In response to silencing RalA, a significant decrease in the proliferation rate of ovarian cancer cells was observed as of day 3 post-infection, ending in complete abolishment of OVCAR-5 cells by day 6 (Figure 2B). Panel C in Figure 2 shows the density of cells at day 4 post-infection with anti-RalA or control lentivirus.
Another important biological and translationally relevant characteristic of ovarian cancer cells is their invasiveness. We evaluated the outcome of silencing RalA on in vitro invasiveness of OVCAR-5 cells in a modified Boyden chamber assay. This assay is based on the capability of cells to pass through a matrigel coated base. Upon exposure to anti-RalA lentivirus a ~50% reduction was observed in invasiveness of these cells at day 2 post-infection (Figure 2D, left panel). The right panel in Figure 2D shows the staining of the nuclei of invaded cells using DAPI.
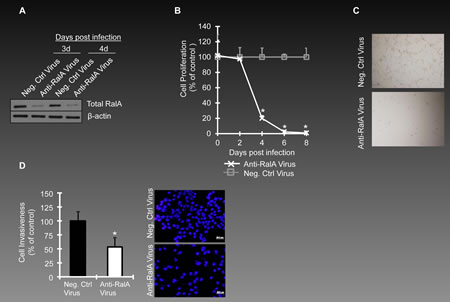
Figure 2: Inhibition of RalA expression in ovarian cancer cells. (A) Ovarian cancer cells OVCAR-5 were infected with lentivirus encoding shRNA against RalA(Anti-RalA Virus) or scrambled shRNA (Neg. Ctrl Virus) at MOI 10(cfu/cells). RalA expression levels are shown for day 3 and 4 post-infection. (B) The effects of anti-RalA or negative control virus (MOI~5) on proliferation of OVCAR-5 cells is studied till day 8th post-infection (p<0.05). (C) Callout panels show the density of cells treated with Anti-RalA or Negative Control virus at day 4 post-infection. (D) For invasion assay, OVCAR-5 cells were transferred to invasion chambers at day 2 post-infection. The nuclei of invaded cells was stained with DAPI and counted under the microscope. The left graph shows the percentage of invaded cells in anti-RalA treated cells as compared to negative control treated virus (p<0.05). The right panel shows one of the microscopy fields used for counting invaded cells.
Pharmacological inhibition of RalA confers decreased proliferation and invasiveness of ovarian cancer cells
Although specific inhibitors of RalA are not available, this pathway can be influenced using pharmacological agents that reduce activation of RalA. Since geranylgeranylation of RalA is necessary for its activation [6], application of geranylgernayltransferase inhibitors (GGTIs) can be considered as one of the strategies to reduce the activation of RalA [30]. We have used GGTIs in our previous studies for such a purpose [16]. GGTI-2147 used in this study is a cell-permeable non-thiol peptidomimetic that acts as a potent and selective inhibitor of geranylgeranyltransferase I (GGTase I) [31, 32]. GGTI-2147 at the concentrations ranging 0.5-50µM inhibited RalA activation; however, Ras activation remained relatively unchanged at these concentrations (Figure 3A). Under such conditions, the proliferation of OVCAR-5 cells was reduced significantly at 48 and 120 h post-exposure to the drug (Figure 3B). Figure 3C shows the morphology of these cells as exposed to different concentrations of GGTI-2147. A progressive loss of viability was observed by increasing concentration of GGTI-2147, while DMSO-treated cells remained healthy. We also studied the invasiveness of these cells once exposed to GGTI-2147 and observed a ~50% loss at 50µM concentration of GGTI-2147 (Figure 3D). Figure 3E shows the DAPI staining of the nuclei of invaded cells. In order to reveal the possible mechanism by which such loss of invasiveness occurs, we investigated the expression levels of two important markers involved in this matter, i.e., α-E-Catenin and N-Cadherin. Interestingly, the expression of α-E-Catenin, an invasion suppressor gene, was significantly elevated by increasing concentrations of GGTI-2147. The expression of N-Cadherin, a pro-invasion marker, was reduced under such conditions (Figure 3F).
In the next step, OVCAR-5 cells were treated with another pharmacological inhibitor influencing RalA activation, Aurora Kinase Inhibitor II (AKI-II) [33]. This compound is a cell-permeable anilinoquinazoline that acts as a potent, selective, and ATP-competitive inhibitor of AK. Once again, cell proliferation was inhibited by AKI-II [25µM] in harmony with a reduction in the activation levels of RalA-GTP (Figure 3G).
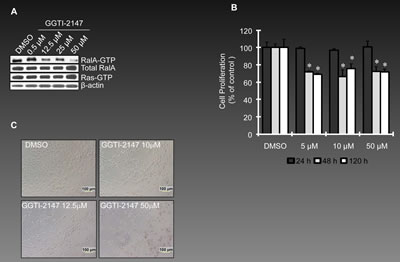
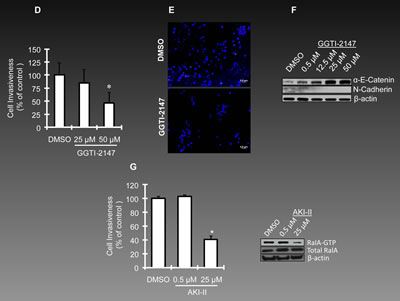
Figure 3: Reduced RalA activation represses ovarian cancer cell growth and invasion. (A) OVCAR-5 cells were treated with GGTI-2147, a potent inhibitor of GTPase geranyl-geranylation resulting in a reduction in RalA-GTP, but not Ras-GTP levels. Cells were treated with GGTI-2147 at the concentrations indicated for 48 h and harvested for pull-down assay to assess RalA or Ras activation. (B) Proliferation of OVCAR-5 cells under treatment with GGTI-2147(5, 10 and 50 µM) was graphed as percentage of control at 24, 48 and 120 hours post-treatment. (C) Increase in concentration of GGTI-2147 resulted in lower density of cultured ovarian cancer as was investigated by light microscopy. (D) Treatment of OVCAR-5 cells with GGTI-2147 at indicated concentrations resulted in loss of invasiveness in a significant manner (p<0.05). (E) Visualization of nuclei of invaded cells (DAPI staining) treated with GGTI-2147 or DMSO at 48 h post-exposure to the chemicals. (F) GGTI-2147 treatment influences the expression levels of the pro-invasion protein, N-Cadherin. On the other hand, the level of anti-invasion protein, α-E-Catenin is increased under the same conditions. (G) Once treated with AKI-II, the invasiveness of OVCAR-5 cells is studied at 0.5 and 25µM concentration of the inhibitor, (left panel, p<0.05). The right panel confirms the effective inhibition of RalA activation at 25µM concentration (but not 0.5 µM) of AKI-II.
RalA activation is increased in CD24+ ovarian cancer cells
Recent theories have presented the field of oncology with the concept of “cancer stem cells (CSCs)” as the main fraction of cells within a heterogenous population capable of repopulating tumor mass following treatment [34, 35]. According to this theory, resistance of CSCs to current modalities is the main reason for the failure of cancer therapy. Therefore, revealing biological and signaling characteristics of CSCs is of significant importance in designing effective cancer therapeutics. A series of markers [36-38] have been suggested to be associated with ovarian CSCs, of which CD24 [39-41] has gained significant attention. CD24+ subset of ovarian cancer cells is shown to possess various cancer stem cell properties, epithelial-mesenchymal transition (EMT) phenotype and high invasive capacity[42]. Therefore, we decided to study the overactivation of RalA in CD24+ cells in an attempt to reveal biological features of these cells in the context of RalA signaling. Using fluorescence activated cell sorting (FACS), both SKOV3 and OV429 cells were shown to express CD24 at high levels (35% and 16% respectively) while hOSEpiCs only expressed CD24 at below 1% range (Figure 4A). In order to evaluate the activation of RalA in CD24+ cells, we further enriched these cells by flow cytometry to ~58%, named as CD24high fraction (Figure 4B, lower panel). A population of cells was also depleted (~0.9%) from expression of this marker, referred to as CD24low fraction (Figure 4B, upper panel). The levels of RalA-GTP in CD24low cells was found to be lower than CD24high fraction, while the basal expression level of RalA was comparable in these cells (Figure 4C). This was taken as an evidence that overactivation of RalA might contribute to the biology of ovarian cancer at the level of cancer stem cells. Band density was also evaluated for RalA-GTP in figure 4C showing ~1.85 fold increase in RalA-GTP in CD24high as compared to CD24low.
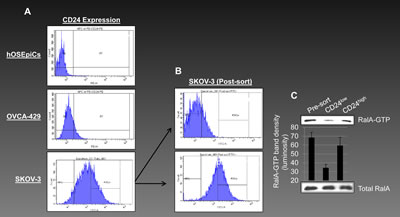
Figure 4: Overactivation of RalA-GTP in CD24+ ovarian cancer cells. (A) CD24 is suggested as a potential marker for ovarian cancer stem cells and is expressed in higher levels in ovarian cancer cells (OVCA-429 and SKOV-3) as compared to hOSEpiCs. (B) SKOV-3 cells were sorted for expression of CD24 resulting in a population with high level of CD24 expression (or CD24high with 57.8% positivity for CD24) and a population negative for CD24 expression (or CD24low). (C) CD24high, CD24low and pre-sort (unsorted) cells were evaluated for RalA activation by affinity precipitation. Density of RalA-GTP bands was also evaluated and graphed.
Reduced RalA activation represses the growth of subcutaneous human ovarian cancer xenografts in nude mice
In order to evaluate the in vivo effects of reducing RalA activation on growth of ovarian cancer cells, we studied the growth of OVCAR-5 cells in subcutaneous (SC) mouse model for ovarian cancer. OVCAR-5 cells were exposed to the GGTI-2147 and AKI-II for 48 h and injected to the right flank of nude hairless mice, while the left flank was injected with the vehicle (DMSO) treated cells. GGTI-2147 treatment resulted in a significantly smaller (p<0.05) volume of tumor than DMSO-treated controls until day 14 post-inoculation (Figure 5A). At day 21 post-inoculation, tumor volumes of GGTI-2147-treated xenografts were similar as vehicle-treated controls, indicating the need for continuous administration of GGTI-2147 in order to gain long-term tumor regression in vivo. We also examined the effects of AKI-II [20µM] in a resembling manner. No tumor growth was observed at day 14 post-inoculation for AKI-II treated cells, while tumor volumes remained significantly (p<0.05) smaller than DMSO-treated tumors even at day 25 post-inoculation (Figure 5B). Figure 5C shows the subcutaneous tumors in GGTI-2147-treated animals at day 14. Figure 5D shows extracted tumors for AKI-II-treated animals at day 25 post-inoculation.
Pre-exposure to anti RalA-virus reduces in-vivo tumorigenesis of ovarian cancer cells
In order to investigate if silencing RalA can inhibit in-vivo tumor growth of ovarian cancer, we mixed OVCAR-5 and OVCA429 cells with anti RalA-lentivirus or negative control lentivirus and injected them via SC to the flank of male athymic nude mice (Left side: Control virus, Right side: Anti-RalA virus, n=5 for each cell line). The SC tumor growth was followed for both groups and the final tumor sizes were recorded at 20 days post-injection for OVCAR-5 cells (Figure 5E) and 14 days post-injection for OVCA429 cells (Figure 5F). In both cases, inhibition of tumor growth was observed. In Figure 5E, we used MOI~10 and MOI~20. Individual tumor volumes are graphed in the upper chart and volumes are mentioned in the middle table. The lower pictures show the animals with SC tumors at the end of the study (Red circles: Control virus injected, Yellow circles: Anti-RalA virus injected) and the extracted tumors.
Figure 5F shows the results from treating OVCA429 cells with MOI~20 of the anti-RalA and control viruses. Once again, a smaller tumor size was observed for the anti-RalA virus treated tumors. The upper graph and chart offer individual tumor sizes. The lower left and right panels show mice bearing tumors at the end of the 20-day course of the study (Red circles: Control virus injected, Yellow circles: Anti-RalA virus injected). Extracted tumors are shown in the lower right panel.
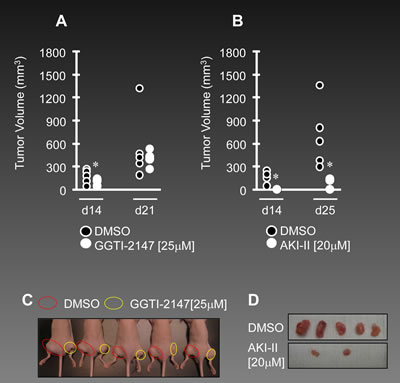
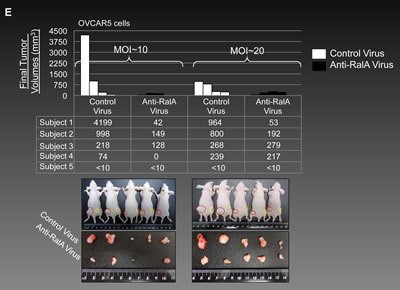
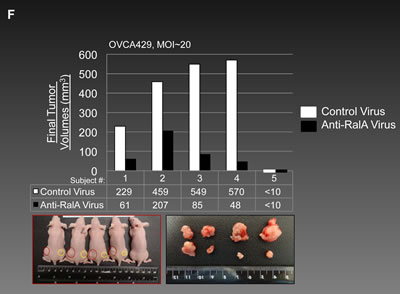
Figure 5: Decreased activation of RalA inhibits tumorigenesis in vivo. (A-B) Male athymic nude mice were subcutaneously injected with the OVCAR-5 cells pre-treated with GGTI-2147 [25µM] or AKI-II [20µM]. Tumor volumes were recorded as tumors became palpable until 21 days post-injection for the GGTI-treated group or 25 days for AKI-II treated group. (C) Subcutaneous tumors in mice are shown at day 14 post-injection. Red circles present tumors developed from DMSO pre-treated cells while yellow circles present tumors developed from GGTI-2147 pre-treated cells (n=5). (D) Subcutaneous tumors from nude mice (n=5) were extracted at day 21 for both DMSO and AKI-II pre-treated cells. (E) 1x106 OVCAR5 cells were mixed with MOI~10 and 20 of Anti-RalA (right side) or negative control lentiviruses (left side) and injected via SC to the nude mice. Tumor volumes were followed and at 20 days post-injection the animals were sacrificed and tumors were extracted and measured. Individual final volumes of each tumor are graphed in the upper chart and table. Lower pictures show SC and extracted tumors at the end of the study. (F) 1x106 OVCA429 cells were mixed with MOI~20 Anti-RalA (right side) or negative control lentiviruses (left side) and injected via SC to the nude mice. Tumor volumes were followed and at 14 days post-injection the animals were sacrificed and tumors were extracted and measured. Individual final volumes of each tumor are graphed in the upper chart and table. Lower pictures show SC and extracted tumors at the end of the study.
Syngeneic mouse model for ovarian cancer shows enhanced activation of RalA
On the basis of the hypothesis that multiple passages of ovarian surface epithelial cells in vitro might induce malignant transformation, a syngeneic mouse model for ovarian cancer has been developed [43, 44]. This model is based on repeated passage of mouse ovarian surface epithelial cells (MOSECs) for more than 20 rounds when cell morphology is drastically changed and contact inhibition of cells is lost. Figure 6A shows the origination of MOSECs from the epithelial crust of ovaries. While low passage MOSECs maintain their normal morphology (Figure 6B), high passage MOSECs exhibit a significant change in morphology as well as loss of contact inhibition (Figure 6C). Further studies showed tumor forming potentials for high passage MOSECs[44]. Intraperitoneal (IP) injection of these cells to C57BL6 mice generated the growth of abdominal cavity tumors as well as hemorrhagic ascites fluid in resemblance to stage III-IV of human ovarian cancer. Figure 6E shows the time course of development of the abdominal cavity tumor and ascites following injection of high passage MOSECs. Figure 6D shows the infiltration of ID8 to mouse ovary (upper panel) and development of mouse ovarian tumor. ID8 is a high passage MOSEC cell which expresses green fluorescent protein, GFP. The middle panel of Figure 6D exhibits the expression of GFP in ID8 cells in the corresponding upper panel. The lower panel of Figure 6D shows a histological section of mouse ovary surrounded by ID8 tumor. Arrowheads show these tumors surrounding the ovary.
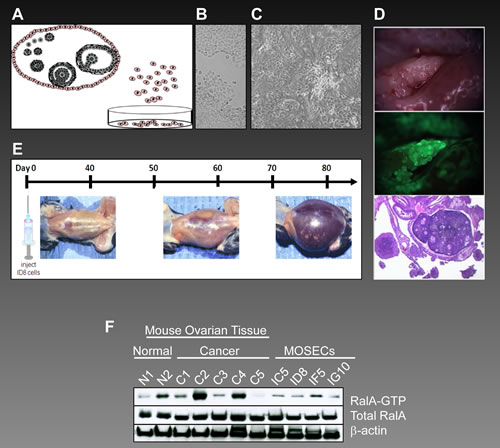
Figure 6: Overactivation of RalA in syngeneic mouse model for ovarian cancer. (A) The origination of MOSECs from epithelial crust of ovaries and their growth in vitro. (B) Low passage MOSECs maintain their normal morphology. (C) High passage MOSECs exhibit a significant change in morphology as well as loss of contact inhibition. (D) Upon intraperitoneal (IP) injection to mouse, ID8 cells (a high passage MOSEC cell which expresses green fluorescent protein or GFP) localize to mouse ovary. Upper panel shows appearance of the tumor while lower panel exhibits GFP expression from ID8 cells. Figure 6D lower panel shows a histological section of mouse ovary surrounded by tumors developed from ID8 cells. Arrowheads show these tumors surrounding the ovary. (E) Once ID8 cells are injected IP to C57BL6 mice, they generate abdominal cavity tumors as well as hemorrhagic ascitis fluid in resemblance to stage III-IV of human ovarian cancer. This figure shows the time frame for development of abdominal tumors. (F) Late passage MOSECs (ID8, IG10, IC5 and IF5) as well as mouse ovarian cancer tissues (C1-C5) along with two normal ovarian tissues (N1-N2) were analyzed for activation of RalA by affinity precipitation assay.
Finally, in order to investigate the levels of RalA activation in this model, a series of late passage MOSECs (ID8, IG10, IC5, and IF5) and mouse ovarian cancer tissues were analyzed. Whereas all MOSECs showed activation of RalA, the level of RalA-GTP was higher in four of five ovarian tumor tissues (C1-C4) as compared to the normal ovarian tissues (N1-N2). This, once again, was taken as evidence for the significant involvement of RalA in the molecular etiology of ovarian cancer.
Discussion
This study reveals the key role of RalA in the biology of human ovarian cancer. We have identified that RalA, RalB, and their upstream activator, Ras, were overactivated in human ovarian cancer cells as compared to non-malignant ovarian surface epithelial cells. The expression of RalA downstream effectors and regulators which modulate RalA activity at post-translational levels was also derailed to enhance Ral signaling in cancer cells and tissues.
In order to further evaluate the role of Ral pathway in ovarian cancer, we focused our efforts on studying RalA in this disease. Targeting RalA for translational studies is justifiable since the studied panel of ovarian cancer cells and tissues, as well as the syngeneic mouse model for ovarian cancer, all exhibited a notable activation level of RalA. It is likely that RalA is the essential small G-protein that confers ovarian cancer cell survival. Notably, OVCAR-5 and SKOV-3 had enhanced RalA activity but reduced Ras activity. This suggests a partial Ras-independent activation of RalA, which has been reported before in other models [7, 12, 45]. At post-translational levels, RalA is regulated by PP2A Aβ [46] and AKA [47, 48]. Tumor suppressor PP2A Aβ de-phosphorylates RalA at Ser183 and Ser194, leads to a reduced activation of RalA [46], whereas AKA-mediated phosphorylation of RalA at Ser194 plays a critical role in RalA activation [48]. Our findings that ovarian cancer cells had reduced expression of PP2A Aβ but enhanced phosphorylated AKA elucidate the mechanism underlying RalA overactivation. Members of the aurora family of serine/threonine kinases are essential for mitotic progression. Particularly, AKA is pivotal for centrosome function and spindle assembly [14]. Over-expression of AKA transforms fibroblasts, whereas selective inhibition of AKA confers abnormal mitotic spindles, chromosome segregation defects [49], and decreases in the growth of human tumor xenografts [49]. Elevated AKA expression is implicated in human ovarian cancer patients [50] and gastroenterological cancers [51, 52]. Our findings about elevated levels of AKA in ovarian cancer and the effects of AKI-II on reduction of RalA activation, and the subsequent decrease in proliferation and tumorigenesis, establishes Ral pathway as a part of the pro-proliferative machinery of AK. Importantly, once cells were exposed to AKI-II, the only effective decline in cell proliferation was observed at concentrations which were inhibitory to RalA activation.
Post-translational modifications, including prenylation, proteolysis, carboxymethylation, and palmitoylation, are essential for correct membrane binding and full biological activities of Ral proteins and other small G-proteins [53]. Prenylation of small G-proteins is catalyzed by farnesyl transferase (FTase) or geranylgeranyltransferases I and II (GGTase I and II). RalA and RalB are extensively geranylgeranylated by GGTase I at C-terminal CAAX motifs that dictates proper localization to specific cellular membranes and enables sufficient small G-proteins signaling transduction and biological activity [32]. We examined whether decreased RalA activation by interfering with geranylgeranylation influences the viability of ovarian cancer cells. As expected, GGTI-2147 at a concentration that inhibits RalA (but not Ras) suppresses ovarian cancer cell proliferation, invasion, and in vivo tumorigenesis. However, the results indicate that repeated administration of GGTI-2147 may be necessary for achieving permanent tumor regression. Also, the compensatory responses by other targets of GGTI, such as members of Rho subfamily (Rac1 and RhoA) and trimeric G-protein γ-subunit [54, 55], might offset the acute suppression of cell survival by GGTI-2147; however, we determined that Ras activation was not affected by GGTI-2147 at the concentration used. In this scenario, AKI-II exerts a more sustained tumor regression. This might also be related to the inhibition of non-RalA related pro-growth effects of AK.
The loss of viability of ovarian cancer cells upon gene specific silencing for RalA not only suggests this pathway as a potential therapeutic target but also presents a novel possibility for gene therapy of ovarian cancer. On the other hand, the suppressive outcome of inhibition of Ral pathway on ovarian cancer cell invasiveness by both gene specific silencing and pharmacological inhibition of RalA is reminiscent of our previous studies about alterations in epithelial-mesenchymal transition (EMT) markers following inhibition of RalA [16]. However, in this work we are also reporting the alteration of two important elements involved in invasiveness (α-E-Catenin and N-Cadherin) as a consequence of GGTI treatment. α-E-Catenin, as a mediator of attachment between Cadherins and cytoskeleton, is usually lost during the progression of human cancers [56]. It is also reported that α-E-Catenin gene (CTNNA1) acts as an invasion suppressor gene [57]. The increase in the expression level of α-E-Catenin in response to GGTI treatment can be considered a novel mechanism for the observed anti-invasion effects. Similarly, down-regulation of N-Cadherin, a classic effector of the pro-invasion “E- to N-Cadherin switch,” plays a role in the reduced invasiveness of ovarian cancer cells.
Finally, we would like to point out the important ramifications of this study for the prevention and/or early diagnosis of ovarian cancer. The first clue for such a claim is obtained from our data regarding increased activation of RalA in CD24+ cells. With an increased level of evidence about this antigen as one of the markers of cancer stem cells, and on the basis of the current theories proposing the preceding role of CSCs in tumor development, the increased level of RalA might be considered an early signaling event in pro-neoplastic transformation of ovarian cells. Therefore, increased RalA activation may serve as an early diagnostic marker. Additionally, intervention with this pathway might provide a method for prevention of ovarian malignant transformation. Increased RalA activation observed in syngeneic mouse model for ovarian cancer offers an interesting possibility for studying the role of RalA signaling in diagnosis, prevention, or therapy of ovarian cancer. Other transgenic mouse models [58] can also be studied in this regards as well .
Materials and Methods
Chemical, Antibodies, and Reagents
Geranylgeranyltransferase inhibitor-2147 (GGTI-2147) (4-[[N-(Imidazol-4-yl) methyleneamino]-2-(1-naphthyl) benzoyl] leucine methyl ester, purity by HPLC 97.6%) and Aurora Kinase Inhibitor II (AKI, 4-(4′-Benzamidoanilino)-6,7-dimethoxyquinazoline) were purchased from Calbiochem (CA, USA). Antibodies specific for RalA, RalB, Ras, CDC42, and tublin were purchased from Millipore (MA, USA). Antibodies against RalBP1, aurora kinase A, phosphorylated aurora kinase A (Thr288)/ aurora B (Thr232)/ aurora C (Thr198), α-E-Catenin, N-Cadherin, and β-actin were purchased from Cell Signaling (MA, USA). Antibody specific for RalGDS was purchased from Santa Cruz Biotechnology (CA, USA). Inhibitors GGTI-2147 and AKI-II (Calbiochem) were dissolved in dimethyl sulfoxide (DMSO). The final concentration of DMSO in medium was 0.1 % for all indications. FITC (Fluorescein isothiocyanate) and PE (R-Phycoerythrin)-labeled antibodies and isotype controls were from BD Biosciences (MD, USA).
Cell culture
OVCAR-5 and SKOV-3 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) (Sigma, MO, USA). OV-177 and OVCA-429 cells (generous gifts from Dr. K.R. Kalli, Mayo Clinic, Rochester, MN, USA) [22, 23] were cultured in Dulbecco’s Modification of Eagle’s Medium (DMEM) (Sigma) supplemented with 10% FBS. OV-202 cells were cultured in Minimum Essential Medium Eagle (Lonza, PA, USA) supplemented with 20% FBS and 2 mM L-glutamine (Sigma). The above mediums were supplemented with penicillin (100 U/ml) and streptomycin (100 mg/ml) (Sigma). Human ovarian surface epithelial cells (hOSEpiC) (ScienCell, CA, USA) were cultured in Ovarian Epithelial Cell Medium supplemented with growth factors provided by the manufacturer. The cells were cultured at 37 ºC in 5% CO2 and 95% relative humidity.
RalA and RalB affinity precipitation assays
Activated RalA and RalB were detected using the Ral Activation Assay Kit (Millipore). Cells were washed with ice-cold phosphate-buffered saline (PBS) and scraped on ice with 1X Ral Activation Buffer (RAB) for RalA or 1X Magnesium-containing Lysis Buffer (MLB) for RalB. Human tissues were homogenized in ice-cold RAB or MLB, then mixed with Protein A/G PLUS agarose beads (Santa Cruz Biotechnology) to reduce non-specific proteins. Protein concentration was determined using the BCA protein assay kit (Pierce, IL, USA). Lysates (250μg) from cell lines and 500μg from tissues were mixed with 10μl or 20μl of RalBP1 agarose beads, respectively. Each reaction mixture was supplemented with RAB up to 1 ml and was gently rocked at 4 ºC for 3 h. Agarose beads were collected by brief centrifugation, washed three times with RAB, and mixed with 6X Laemmli reducing sample buffer and boiled for 5 min. Total beads were loaded onto 4-20% SDS-polyacrylamide gradient gel (Bio-Rad, CA, USA) for electrophoresis. Proteins were transferred onto a nitrocellulose membrane and incubated overnight with anti-RalA or anti-RalB antibodies (Millipore) at 4 ºC. The membrane was washed and incubated with secondary antibody at room temperature for 1 h. After washing, the protein was detected with ECL kit (Millipore). Since only activated RalA or RalB is precipitated during affinity precipitation, western blot results represent activated fractions of RalA or RalB.
Ras pull-down assay
Activated Ras was detected using a Ras Activation Assay Kit (Millipore) according to the manufacturer’s instructions and our previous work [24-26]. All procedures were the same as Ral activation assay except the use of Raf-1 RBD agarose beads and Magnesium lysis buffer optimized for the precipitation of active Ras. An anti-Ras antibody was used for western blotting at a later stage.
Protein Quantification
Malignant and matched normal ovarian tissues were provided by the University of Kansas Cancer Center Tissue Bank. Tissue was homogenized in cell lysis buffer (Cell Signaling) using a gentleMACS Dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany) at manufacturer recommended settings. Homogenized ovarian tissue samples were then used for protein quantification using Coomassie Protein Assay Kit (Thermo Fisher Scientific, MA, USA) and read on a Benchmark Plus Microplate Spectrophotometer (Bio-Rad) using Microplate Manager (Bio-Rad) software to analyze and interpret the absorbance values.
G-LISA Ral Activation Assay
Ral activation activity was measured according to the G-LISA Ral Activation Assay kit (Cytoskeleton Inc., CO, USA). The G-LISA Ral Activation Assay is based on the principle of linked Ral-GTP binding proteins in the provided 96 well plate. Upon adding the tissue lysates to the wells, the active GTP-bound Ral in the tissue lysates will bind to the wells while inactive GDP-bound Ral will be removed through the washing steps. The degree of Ral activation is discerned from the absorbance readings at a wavelength of 490nm with the aid of colorimetric detection substrates that are added to the wells.The ovarian tissues underwent protein equalization in cold lysis buffer and cold binding buffer to yield comparable results when measuring Ral activation activity. The Ral activation assay protocol was performed as outlined in the manufacturer’s protocol. Prior to the addition of our samples to the Ral assay microplate, 100µL of ice cold water was added to dissolve the powder in each well. After the powder had dissolved, we proceeded with the addition of the ovarian tissue lysates, positive Ral control protein, and buffer blank to the wells of the Ral-GTP binding microplate. The microplate was then transferred to a cold orbital shaker at 200 rpm for 20 minutes in 4°C. After 20 minutes, the plate was removed from the cold orbital shaker with the solution discarded before washing twice with 200µL wash buffer. Following the washing steps, 200µL of room temperature Antigen Presenting Buffer was added to each well and left to incubate at room temperature for 2 minutes. The wells were then washed three times with 200 µL of room temperature wash buffer. We then added 50 µL of 1/50 anti-Ral primary antibody diluted in Antibody Dilution Buffer to each well and placed the plate on a room temperature orbital shaker (200 rpm) for 45 minutes. The anti-Ral primary antibody solution in the wells was then discarded and the wells were washed three times with 200 µL of wash buffer. We then prepared 1/100 of secondary HRP labeled antibody in Antibody Dilution Buffer and added 50µL of the diluted secondary antibody to each well and placed the plate on the microplate shaker (200 rpm) for 45 minutes at room temperature. After incubation, we discarded the solution in the wells and proceeded with three wash steps as before. Then we added 50 µL of HRP detection solution to each well and incubated at room temperature for 15 minutes before adding 50 µL HRP Stop solution to all wells. The microplate was then analyzed by an endpoint protocol on Microplate Manager software using a Benchmark Plus Microplate Spectrophotometer. The endpoint protocol was set for a single 490 nm wavelength with 5 seconds of shaking. The resulting Ral activation signal was compared using a template provided by the manufacturer for analyzing the experimental data.
Lentivirus infection
Lentivirus particles carrying shRNA(5’-GATCCGACAGGTTTCTGTAGAAGATTCAAGAGATCTTCTACAGAAACCTGTCTTTTT-3’) against RalA or scrambled shRNA5’-GATCCTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTT-3’)(Santa Cruz Biotechnology) which is not capable of silencing of any known cellular mRNA were used to infect OVCAR-5 cells at MOI~10 cfu/cell . This was done in the presence of 5µg/ml polybrene (Santa Cruz Biotechnology) in accordance to the protocol provided by the manufacturer and our previous work [16, 27]. At the indicated times post-infection, proliferation, invasion and/or western blot assays were performed after harvesting the cells in general lysis buffer (Cell Signaling).
Proliferation assay
OVCAR-5 cells were plated in 96-well plates and cultured in growth medium overnight. On the following day, the cells were infected by lentivirus particles (MOI~10 pfu/cell) or treated with inhibitors. At the indicated time points, a proliferation assay was performed as described previously using the WST-1 assay [24, 27].
Invasion assay
A cell invasion assay was performed using BioCoat Matrigel Invasion Chamber (BD Biosciences) as was previously described [16]. Briefly, OVCAR-5 cells were plated in 24-well plates and cultured overnight followed by lentivirus infection or inhibitor treatment. Forty-eight hours post-infection or post-inhibitor treatment, cells were transferred into Matrigel-coated inserts fitting 24-well plates and cultured for 22 h. After the cells penetrated the layer of Matrigel and accumulated on the external surface of the inserts, the membrane was fixed and the fraction of invaded cells was detected by 4’,6-diamidino-2-phenylindole (DAPI) staining and counted by microscopy.
Western blotting
Cells were washed twice with ice-cold PBS (Sigma), lysed using general cell lysis buffer (Cell Signaling), and normalized for total protein concentration. Proteins (15-150 µg) were separated on 4-20% or 10% Criterion Precast Gel (BD Biosciences) and electro-transferred to nitrocellulose membrane. Membranes were incubated with the appropriate primary antibody overnight at 4 ºC, followed by incubation with a secondary antibody and detected by enhanced chemical luminescence (ECL) kit (Millipore). Membranes were re-probed with β-actin or tubulin antibodies to verify equal loading of protein.
In vivo tumor growth
Athymic nude mice (Hsd: Athymic Nude-Foxn1nu), 5-7 weeks of age, were purchased from Harlan Laboratories (IN, USA). All procedures were conducted in accordance with the National Institute of Health’s Guidelines for the Care and Use of Laboratory Animals, and approved by the University of Kansas Medical Center’s Institutional Animal Care and Use Committee. OVCAR-5 cells were treated with DMSO (0.1%), GGTI-2147 (12.5 µM), or AKI (25µM) for 48 h. The cells were then harvested and cell viability was determined the Countess Automated Cell Counter (Invitrogen, CA, USA). Cells were washed and re-suspended in a 1:1 mixture of growth media/matrigel for a final volume of 100µL and injected subcutaneously (SC) to the flank of the mouse under anesthesia. Tumor diameters were measured using a digital caliper every week and tumor volume was calculated by formula [tumor volume (mm3) = (length [mm]) × (width [mm]) 2 × 0.52]. All animal experiments were performed according to a protocol approved by the University of Kansas Medical Center’s related committees to assure humane treatment of animals.
Flow Cytometry
Cells were harvested using cell dissociation buffer (Sigma), washed and labeled with antibodies under optimized conditions. Antibodies used were FITC- or PE-labeled CD24 and isotype control (BD Biosciences). SKOV-3 cells were incubated with CD24 antibody at 1:10 dilution and a temperature of 4 ºC for 10 min in the darkness. Cells were washed twice and stained with propidium iodide to exclude dead cells from the analysis. The cells were analyzed on a BD Biosciences LSRII flow cytometer and analyses of data were performed using FlowJo (Tree STar, OR, USA) software.
Statistical analysis
All values are expressed as mean ± SD and were analyzed using two-tailed Student’s t-test. A p<0.05 was considered significant. The α-value was set at 0.05.
Acknowledgment:
This work is dedicated to the loving memory of Marsha Rivkin. Research in F. Farassati’s laboratory is supported by funding from the Marsha Rivkin Center for Ovarian Cancer Research (MRF), the Flight Attendant Medical Research Institute (FAMRI), and the University of Kansas Medical School. We are grateful for access to tissues from the tissue bank at the University of Kansas Cancer Center (Dr. A. Godwin, C. Reilly, Z. Naima and J. Ballenger) and Department of Pathology (Dr. O. Tawfik ). We are also grateful for assistance by flow cytometry core staff, R. Hastings, and A. Zeiger as well as microscopy and imaging center, S. Fernald. K. Wang was a recipient of a fellowship award by the American Association for Cancer Research (AACR). We also thank S. Martin for structure and grammatical revisions.
Conflict of interest:
None of the authors above have disclosed a conflict of interest with this submission.
Role of sponsors:
Study sponsors had no role in the study design, collection, analysis, or interpretation of data. They also did not have any role in the writing of the manuscript or in the decision to submit the manuscript for publication.
References
1. Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW, Buchberg AM, Jenkins NA, Parada LF and Copeland NG. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994; 8(9):1019-1029.
2. McCormick F. Cancer therapy based on oncogene addiction. J Surg Oncol. 103(6):464-467.
3. Steelman LS, Chappell WH, Abrams SL, Kempf RC, Long J, Laidler P, Mijatovic S, Maksimovic-Ivanic D, Stivala F, Mazzarino MC, Donia M, Fagone P, Malaponte G, Nicoletti F, Libra M, Milella M, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY). 3(3):192-222.
4. Campbell PM and Der CJ. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin Cancer Biol. 2004; 14(2):105-114.
5. Camonis JH and White MA. Ral GTPases: corrupting the exocyst in cancer cells. Trends Cell Biol. 2005; 15(6):327-332.
6. Bodemann BO and White MA. Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat Rev Cancer. 2008; 8(2):133-140.
7. Chien Y and White MA. RAL GTPases are linchpin modulators of human tumour-cell proliferation and survival. EMBO Rep. 2003; 4(8):800-806.
8. Shipitsin M and Feig LA. RalA but not RalB enhances polarized delivery of membrane proteins to the basolateral surface of epithelial cells. Molecular and cellular biology. 2004; 24(13):5746-5756.
9. Lalli G and Hall A. Ral GTPases regulate neurite branching through GAP-43 and the exocyst complex. J Cell Biol. 2005; 171(5):857-869.
10. Han K, Kim MH, Seeburg D, Seo J, Verpelli C, Han S, Chung HS, Ko J, Lee HW, Kim K, Heo WD, Meyer T, Kim H, Sala C, Choi SY, Sheng M, et al. Regulated RalBP1 binding to RalA and PSD-95 controls AMPA receptor endocytosis and LTD. PLoS Biol. 2009; 7(9):e1000187.
11. Lopez JA, Kwan EP, Xie L, He Y, James DE and Gaisano HY. The RalA GTPase is a central regulator of insulin exocytosis from pancreatic islet beta cells. J Biol Chem. 2008; 283(26):17939-17945.
12. Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL, Balakireva MG, Romeo Y, Kopelovich L, Gale M, Jr., Yeaman C, Camonis JH, Zhao Y and White MA. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006; 127(1):157-170.
13. Cascone I, Selimoglu R, Ozdemir C, Del Nery E, Yeaman C, White M and Camonis J. Distinct roles of RalA and RalB in the progression of cytokinesis are supported by distinct RalGEFs. EMBO J. 2008; 27(18):2375-2387.
14. Lens SM, Voest EE and Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer. 2010; 10(12):825-841.
15. Sowalsky AG, Alt-Holland A, Shamis Y, Garlick JA and Feig LA. RalA function in dermal fibroblasts is required for the progression of squamous cell carcinoma of the skin. Cancer Res. 2011; 71(3):758-767.
16. Bodempudi V, Yamoutpoor F, Pan W, Dudek AZ, Esfandyari T, Piedra M, Babovick-Vuksanovic D, Woo RA, Mautner VF, Kluwe L, Clapp DW, De Vries GH, Thomas SL, Kurtz A, Parada LF and Farassati F. Ral overactivation in malignant peripheral nerve sheath tumors. Mol Cell Biol. 2009; 29(14):3964-3974.
17. Lim KH, O’Hayer K, Adam SJ, Kendall SD, Campbell PM, Der CJ and Counter CM. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr Biol. 2006; 16(24):2385-2394.
18. Smith SC and Theodorescu D. The Ral GTPase pathway in metastatic bladder cancer: key mediator and therapeutic target. Urol Oncol. 2009; 27(1):42-47.
19. Zipfel PA, Brady DC, Kashatus DF, Ancrile BD, Tyler DS and Counter CM. Ral activation promotes melanomagenesis. Oncogene. 2010; 29(34):4859-4864.
20. Male H, Patel V, Jacob MA, Borrego-Diaz E, Wang K, Young DA, Wise AL, Huang C, Van Veldhuizen P, O’Brien-Ladner A, Williamson SK, Taylor SA, Tawfik O, Esfandyari T and Farassati F. Inhibition of RalA signaling pathway in treatment of non-small cell lung cancer. Lung Cancer. 2012; 77(2):252-259.
21. Borrego-Diaz E, Terai K, Lialyte K, Wise AL, Esfandyari T, Behbod F, Mautner VF, Spyra M, Taylor S, Parada LF, Upadhyaya M and Farassati F. Overactivation of Ras signaling pathway in CD133+ MPNST cells. Journal of neuro-oncology. 2012; 108(3):423-434.
22. Kalli KR, Bradley SV, Fuchshuber S and Conover CA. Estrogen receptor-positive human epithelial ovarian carcinoma cells respond to the antitumor drug suramin with increased proliferation: possible insight into ER and epidermal growth factor signaling interactions in ovarian cancer. Gynecol Oncol. 2004; 94(3):705-712.
23. Tsao SW, Mok SC, Fey EG, Fletcher JA, Wan TS, Chew EC, Muto MG, Knapp RC and Berkowitz RS. Characterization of human ovarian surface epithelial cells immortalized by human papilloma viral oncogenes (HPV-E6E7 ORFs). Experimental cell research. 1995; 218(2):499-507.
24. Pan W, Bodempudi V, Esfandyari T and Farassati F. Utilizing ras signaling pathway to direct selective replication of herpes simplex virus-1. PLoS One. 2009; 4(8):e6514.
25. Farassati F, Pan W, Yamoutpour F, Henke S, Piedra M, Frahm S, Al-Tawil S, Mangrum WI, Parada LF, Rabkin SD, Martuza RL and Kurtz A. Ras signaling influences permissiveness of malignant peripheral nerve sheath tumor cells to oncolytic herpes. Am J Pathol. 2008; 173(6):1861-1872.
26. Esfandyari T, Tefferi A, Szmidt A, Alain T, Zwolak P, Lasho T, Lee PW and Farassati F. Transcription factors down-stream of Ras as molecular indicators for targeting malignancies with oncolytic herpes virus. Molecular oncology. 2009; 3(5-6):464-468.
27. Yamoutpour F, Bodempudi V, Park SE, Pan W, Mauzy MJ, Kratzke RA, Dudek A, Potter DA, Woo RA, O’Rourke DM, Tindall DJ and Farassati F. Gene silencing for epidermal growth factor receptor variant III induces cell-specific cytotoxicity. Mol Cancer Ther. 2008; 7(11):3586-3597.
28. Lim KH, Brady DC, Kashatus DF, Ancrile BB, Der CJ, Cox AD and Counter CM. Aurora-A phosphorylates, activates, and relocalizes the small GTPase RalA. Mol Cell Biol. 2009; 30(2):508-523.
29. Sablina AA and Hahn WC. The role of PP2A A subunits in tumor suppression. Cell Adh Migr. 2007; 1(3):140-141.
30. Chiba Y, Sato S and Misawa M. GGTI-2133, an inhibitor of geranylgeranyltransferase, inhibits infiltration of inflammatory cells into airways in mouse experimental asthma. Int J Immunopathol Pharmacol. 2009; 22(4):929-935.
31. Lesh RE, Emala CW, Lee HT, Zhu D, Panettieri RA and Hirshman CA. Inhibition of geranylgeranylation blocks agonist-induced actin reorganization in human airway smooth muscle cells. American journal of physiology Lung cellular and molecular physiology. 2001; 281(4):L824-831.
32. Falsetti SC, Wang DA, Peng H, Carrico D, Cox AD, Der CJ, Hamilton AD and Sebti SM. Geranylgeranyltransferase I inhibitors target RalB to inhibit anchorage-dependent growth and induce apoptosis and RalA to inhibit anchorage-independent growth. Mol Cell Biol. 2007; 27(22):8003-8014.
33. Heron NM, Anderson M, Blowers DP, Breed J, Eden JM, Green S, Hill GB, Johnson T, Jung FH, McMiken HH, Mortlock AA, Pannifer AD, Pauptit RA, Pink J, Roberts NJ and Rowsell S. SAR and inhibitor complex structure determination of a novel class of potent and specific Aurora kinase inhibitors. Bioorganic & medicinal chemistry letters. 2006; 16(5):1320-1323.
34. Schwarz-Cruz-y-Celis A and Melendez-Zajgla J. Cancer stem cells. Rev Invest Clin. 63(2):179-186.
35. Nagler C, Zanker KS and Dittmar T. Cell Fusion, Drug Resistance and Recurrence CSCs. Adv Exp Med Biol. 714:173-182.
36. Guo R, Wu Q, Liu F and Wang Y. Description of the CD133+ subpopulation of the human ovarian cancer cell line OVCAR3. Oncol Rep. 25(1):141-146.
37. Luo L, Zeng J, Liang B, Zhao Z, Sun L, Cao D, Yang J and Shen K. Ovarian cancer cells with the CD117 phenotype are highly tumorigenic and are related to chemotherapy outcome. Exp Mol Pathol. 91(2):596-602.
38. Casagrande F, Cocco E, Bellone S, Richter CE, Bellone M, Todeschini P, Siegel E, Varughese J, Arin-Silasi D, Azodi M, Rutherford TJ, Pecorelli S, Schwartz PE and Santin AD. Eradication of chemotherapy-resistant CD44+ human ovarian cancer stem cells in mice by intraperitoneal administration of clostridium perfringens enterotoxin. Cancer.
39. Gao MQ, Choi YP, Kang S, Youn JH and Cho NH. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene. 29(18):2672-2680.
40. Lim SC. CD24 and human carcinoma: tumor biological aspects. Biomed Pharmacother. 2005; 59 Suppl 2:S351-354.
41. Lim SC and Oh SH. The role of CD24 in various human epithelial neoplasias. Pathol Res Pract. 2005; 201(7):479-486.
42. Kang KS, Choi YP, Gao MQ, Kang S, Kim BG, Lee JH, Kwon MJ, Shin YK and Cho NH. CD24(+) ovary cancer cells exhibit an invasive mesenchymal phenotype. Biochem Biophys Res Commun. 2013; 432(2):333-338.
43. Roby KF, Niu F, Rajewski RA, Decedue C, Subramaniam B and Terranova PF. Syngeneic mouse model of epithelial ovarian cancer: effects of nanoparticulate paclitaxel, Nanotax. Adv Exp Med Biol. 2008; 622:169-181.
44. Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, Persons DL, Smith PG and Terranova PF. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis. 2000; 21(4):585-591.
45. Chien CH, Chang KT and Chow SN. Amplification and expression of c-Ki-ras oncogene in human ovarian cancer. Proc Natl Sci Counc Repub China B. 1990; 14(1):27-32.
46. Sablina AA, Chen W, Arroyo JD, Corral L, Hector M, Bulmer SE, DeCaprio JA and Hahn WC. The tumor suppressor PP2A Abeta regulates the RalA GTPase. Cell. 2007; 129(5):969-982.
47. Lim KH, Brady DC, Kashatus DF, Ancrile BB, Der CJ, Cox AD and Counter CM. Aurora-A phosphorylates, activates, and relocalizes the small GTPase RalA. Molecular and cellular biology. 2010; 30(2):508-523.
48. Wu JC, Chen TY, Yu CT, Tsai SJ, Hsu JM, Tang MJ, Chou CK, Lin WJ, Yuan CJ and Huang CY. Identification of V23RalA-Ser194 as a critical mediator for Aurora-A-induced cellular motility and transformation by small pool expression screening. J Biol Chem. 2005; 280(10):9013-9022.
49. Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, Galvin KM, Hoar KM, Huck JJ, LeRoy PJ, Ray ET, Sells TB, Stringer B, Stroud SG, Vos TJ, Weatherhead GS, et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci U S A. 2007; 104(10):4106-4111.
50. Gritsko TM, Coppola D, Paciga JE, Yang L, Sun M, Shelley SA, Fiorica JV, Nicosia SV and Cheng JQ. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin Cancer Res. 2003; 9(4):1420-1426.
51. Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, Chan CS, Novotny M, Slamon DJ and Plowman GD. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998; 17(11):3052-3065.
52. Sakakura C, Hagiwara A, Shirasu M, Yasuoka R, Fujita Y, Nakanishi M, Aragane H, Masuda K, Shimomura K, Abe T and Yamagishi H. Polymerase chain reaction for detection of carcinoembryonic antigen-expressing tumor cells on milky spots of the greater omentum in gastric cancer patients: a pilot study. Int J Cancer. 2001; 95(5):286-289.
53. Fritz G and Kaina B. Rho GTPases: promising cellular targets for novel anticancer drugs. Curr Cancer Drug Targets. 2006; 6(1):1-14.
54. Zhang FL, Moomaw JF and Casey PJ. Properties and kinetic mechanism of recombinant mammalian protein geranylgeranyltransferase type I. J Biol Chem. 1994; 269(38):23465-23470.
55. Cohen LH, Pieterman E, van Leeuwen RE, Overhand M, Burm BE, van der Marel GA and van Boom JH. Inhibitors of prenylation of Ras and other G-proteins and their application as therapeutics. Biochem Pharmacol. 2000; 60(8):1061-1068.
56. Kobielak A and Fuchs E. Alpha-catenin: at the junction of intercellular adhesion and actin dynamics. Nat Rev Mol Cell Biol. 2004; 5(8):614-625.
57. Vermeulen SJ, Nollet F, Teugels E, Vennekens KM, Malfait F, Philippe J, Speleman F, Bracke ME, van Roy FM and Mareel MM. The alphaE-catenin gene (CTNNA1) acts as an invasion-suppressor gene in human colon cancer cells. Oncogene. 1999; 18(4):905-915.
58. Fong MY and Kakar SS. Ovarian cancer mouse models: a summary of current models and their limitations. J Ovarian Res. 2009; 2(1):12.