
INTRODUCTION
Neuroblastoma is the most common extracranial solid tumor in childhood [1]. It is characterized by a broad biological heterogeneity based on molecular genetic variations in MYCN oncogene copy number, chromosomal ploidy changes or partial losses and gains, alterations in neurotrophin receptor expression that correlate to different degrees with clinical outcome [2] and with recurrent mutations in a few genes [3]. Clinical course ranges from complete spontaneous regression or differentiation of low-stage or stage 4s neuroblastomas, even without therapeutic intervention, to widespread metastatic disease that is refractory to aggressive multimodal therapies. Although treatment of solid tumors in childhood has significantly improved over the past decades, overall survival in high-risk neuroblastoma patients remains less than 40% despite intensive therapy regimens [1]. While 20% of neuroblastomas harbor MYCN amplifications, directly classifying them as high-risk, around 50% of high-risk tumors lack MYCN amplification and display molecular diversity within the high-risk tumor group [1, 4]. Since 40 to 50% of patients initially diagnosed with neuroblastoma must be assigned to the high-risk group [5, 6], treatment of this disease remains challenging for pediatric oncologists and novel therapeutic options are still urgently needed.
Loss of cell cycle regulatory control is a major hallmark of many cancers, including neuroblastoma [7, 8]. The serine/threonine kinase, polo-like kinase 1 (PLK1), is an essential regulator promoting entry into the mitotic phase from the G2/M transition point in the cell cycle of nontransformed cells and after a DNA damage checkpoint arrest [9–11]. There is increasing evidence that elevated PLK1 activity might serve as a tumor-promoting force by stimulating mitotic transcriptional programs to evade the DNA damage checkpoint [12, 13]. PLK1 expression is higher in cancer cells than in nontransformed cells, and promotes G1/S transition and DNA replication in addition to the G2/M phase transition [14–16]. PLK1 dysregulation is initiated early in carcinogenesis, and promotes cellular processes necessary for oncogenesis and enhances pro-oncogenic signaling networks, including TP53 and RB1 [17–20].
A wide variety of cancers including entities predominantly occurring in children overexpress PLK1 [21–29]. PLK1 was identified as one of the most important survival kinases for rhabdomyosarcoma in a genome-wide siRNA library screen [21]. Inhibiting PLK1 in xenografts or cell lines deriving from osteosarcoma and medulloblastoma, another embryonal tumor of childhood, suppressed proliferation and induced apoptosis [22–24, 29]. Abbou and colleagues recently demonstrated preclinical efficacy of PLK1 inhibition in a wide panel of pediatric malignancies independent of tumor histology [30]. PLK1 upregulation in primary neuroblastomas strongly correlates with high-risk disease [6]. We and others have previously demonstrated that PLK1 is a potential therapeutic target in neuroblastoma, and that inhibition with the small molecule, BI2536, effectively decreased growth in cell and mouse models [6, 31]. Normal, but not cancer, cells have previously been shown to survive PLK1 depletion [32]. A small molecule screen to identify kinase inhibitors that suppress neuroblastoma tumor initiating cell (TIC) proliferation identified PLK1 as a promising target whereas only exceedingly high inhibitor concentrations were cytotoxic for neural stem cells in this screen [33]. These results indicate that targeting this aberrant mitotic kinase signaling pathway in precision therapies that combine targeted drugs and standard chemotherapy could benefit patients with high-risk neuroblastoma.
The three PLK1 inhibitors currently furthest in clinical development are the dihydropteridinone derivatives, BI2536 and BI6727 (volasertib), and the imidazotriazine, GSK461364 [34–37]. All three are competitive inhibitors of ATP-kinase binding. GSK461364 treatment produced fewer side effects related to toxicity than BI2536. Side effects in patients treated with GSK461364 included vein thromboses in patients not co-treated with low molecular weight heparin and mild myelotoxicity [36, 38]. GSK461364 treatment at half-maximal inhibitory concentrations (IC50) below 100 nM inhibited proliferation in multiple tumor cell lines [35, 39, 40]. Here we evaluated the ability of GSK461364 to inhibit neuroblastoma cell viability and proliferation and to induce death in cell lines with different MYCN copy number backgrounds, and to suppress xenograft tumor growth in nude mice.
RESULTS
PLK1 pathway inhibition by GSK461364 reduces viability and clonogenicity of neuroblastoma cells
Neuroblastoma cell lines have previously been shown to express high PLK1 protein levels, similar to expression in primary tumors [6]. To survey a broad range of aggressive neuroblastoma subtype backgrounds, we selected neuroblastoma cell lines either lacking (SK-N-AS, SH-SY5Y, SH-EP) or harboring (Kelly, IMR32, SK-N-BE) MYCN amplifications for in vitro analyses. We assessed cellular viability using MTT assays. GSK461364 treatment significantly reduced viability of neuroblastoma cells in culture (Figure 1A and 1B). Fifty percent inhibition of growth (GI50) was observed at inhibitor concentrations below 20 nM in our cell line panel. Viability was comparably suppressed in all cell lines, regardless of whether they harbored MYCN amplifications or TP53 mutations. To insure that GSK461364 was inhibiting PLK1 activity in our experiments, we assessed expression and phosphorylation of target proteins downstream of PLK1 in treated and untreated SK-N-AS and IMR-32 cells comprising MST1, MST2, pMST1/2, WEE1 and pWEE1 (Supplementary Figure 1). We next assessed clonal expansion in the SK-N-AS (MYCN single copy) and IMR32 (MYCN-amplified) cell lines treated with GSK461364 in colony forming assays. Continuous exposure of the SK-N-AS and IMR32 cell lines to GI50 or GI80 GSK461364 concentrations for 10 days fully inhibited colony formation (Figure 1C and 1D). Pulsed GSK461364 treatment for only 24 hours with GI80 still reduced colony formation by more than 50% after 10 days in culture (Figure 1C and 1D). Our experiments show that GSK461364-mediated PLK1 inhibition significantly reduced neuroblastoma cell viability and colony-forming ability in vitro, even when applied in short-term treatment pulses. These effects were independent of the molecular background imparted by MYCN and TP53 status.
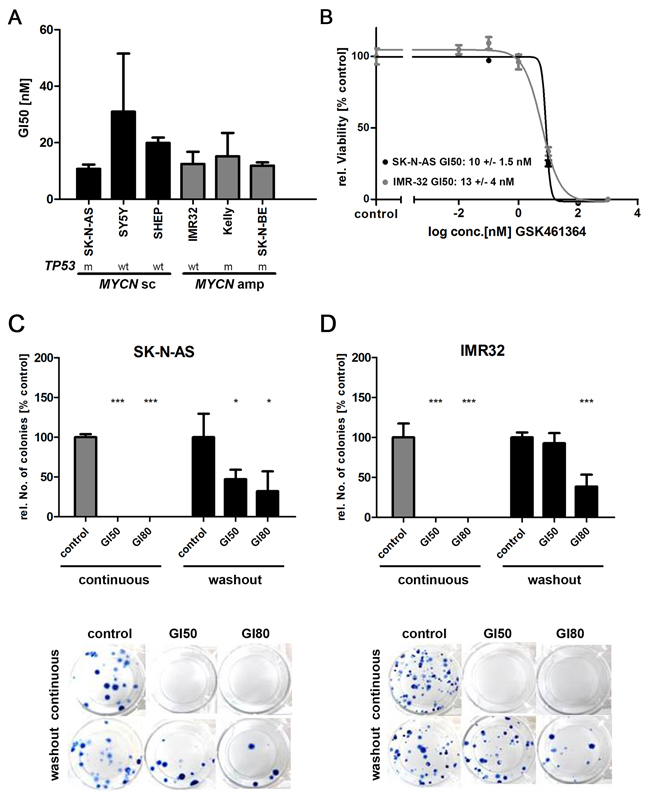
Figure 1: GSK461364-mediated PLK1 inhibition reduces cell viability and clonogenicity in neuroblastoma cell lines. A. Neuroblastoma cell lines were treated with 0.01-1000nM GSK461364 or control medium containing equivalent DMSO carrier concentrations for 72 hours, then cell viability was measured using the MTT assay. Bars represent GI50 values calculated for each cell line. MYCN status (sc = single copy; amp = amplified) and TP53 status (wt = wild-type; m = mutated) of cell lines is indicated. B. Representative dose-response curves of neuroblastoma cell lines SK-N-AS and IMR32, with single copy and amplified MYCN status, respectively. Cells were treated with indicated concentrations of GSK461364 for 72 hours and changes in cell viability relative to solvent-treated cultures was measured by the MTT assay. C. SK-N-AS or D. IMR32 cell lines were either continuously treated over 10 days with GSK461364 at GI50 or GI80 or GSK461364 was relieved from the cells after incubation for 24 hours by repeated replacement of the cell culture medium (wash out). Colony forming ability was assessed for both experimental settings after 10 days using a clonogenic cell survival assay. Solvent-treated cultures (0.1% DMSO) were used as control. ***p<0.001, *p<0.05
PLK1 inhibition reduces proliferation and induces cell cycle arrest and apoptosis in neuroblastoma cells
We next explored how GSK461364 inhibits neuroblastoma cell viability. We assessed proliferation and apoptosis in the SK-N-AS and IMR32 cell lines after short- and long-pulsed GSK461364 treatment, and flow cytometrically monitored cell cycle phase distribution in treated versus control cell cultures. Flow cytometric analyses were conducted after 24 and 72 hours of GSK461364 treatment. A larger proportion of cells treated with GI50 or GI80 concentrations of GSK461364 were in G2/M compared to untreated cultures (Figure 2A). These results are consistent with a previous report that PLK1 activity is necessary for mitotic entry from G2/M arrest induced by DNA damage [41], and indicate that GSK461364-mediated PLK inhibition prevents neuroblastoma cell cycle progression. The effects of GSK461364 on G2/M arrest and apoptosis in neuroblastoma cells did not appear to be influenced by the cellular MYCN background (single copy vs. amplified). BrdU incorporation 24 hours after beginning GSK461364 treatment at GI50 concentrations was significantly reduced in SK-N-AS and IMR32 cells compared with untreated cultures (Figure 2B). Proliferation, as measured by BrdU incorporation, was also further reduced by treating cells with the GI80 or 5-fold GI50 GSK461364 concentrations. GSK461364 treatment also increased the proportion of sub-G1 cells, which includes apoptotic cells (Figure 2A). Treatment at GI80 resulted in slightly more sub-G1 cells than treatment at GI50, and the effect was most pronounced after 72 hours of treatment. We confirmed that GI50 GSK461364 concentrations significantly induced apoptosis in SK-N-AS and IMR32 cells using the cell death detection ELISA™ (Figure 2C), and apoptotic cell fractions rose further when GSK461364 concentrations were increased to GI80 or 5-fold GI50. These data demonstrate that GSK461364 inhibits proliferation and induces both cell cycle arrest at the G2/M restriction point and apoptosis in neuroblastoma cells, regardless of MYCN status.
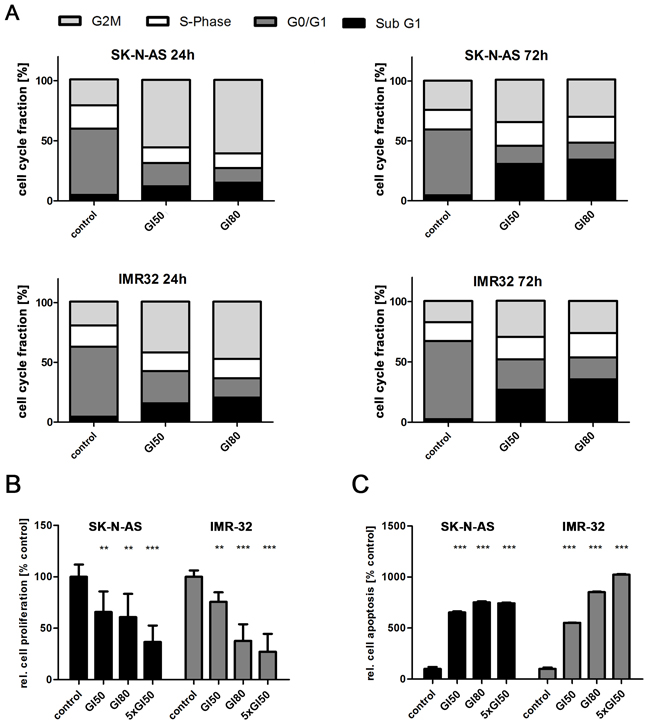
Figure 2: Inhibiting PLK1 with GSK461364 suppresses proliferation and induces both cell cycle arrest and apoptosis in neuroblastoma cells. A. Fractions of SK-N-AS and IMR32 neuroblastoma cells in each point of the cell cycle measured by flow cytometry after 24 and 72 hours of treatment with GSK461364 at GI50 or GI80 or with solvent as control. B. BrdU ELISA performed after GSK461364 treatment of SK-N-AS and IMR32 neuroblastoma cells at GI50, GI80 or 5-fold GI50 for 24 hours. Bar graph shows mean proliferation (±SD) in relation to control (0.1% DMSO). **p<0.01, ***p<0.001 C. Degree of apoptosis in SK-N-AS and IMR32 neuroblastoma cells in relation to control after 24 hours of treatment with GI50, GI80 or 5-fold GI50 of GSK461364 detected by cell death ELISA (mean ±SD). ***p<0.001.
Cell cycle regulators are differentially expressed following PLK1 inhibition by GSK461364
We analyzed gene expression profiles from treated and untreated SK-N-AS cells to investigate molecular mechanisms behind the GSK461364 influence on the neuroblastic cell cycle. Unsupervised hierarchical clustering clearly separated untreated from GSK461364-treated cells (Figure 3A). Gene set enrichment analyses identified increased expression of genes associated with G2/M and a decline in expression of genes essential for S-phase transition (Figure 3B and 3C). Three gene sets associated with the E2F transcription factor family, E2F3 and E2F1 were among the most downregulated in GSK461364-treated cells (Figure 3B, 3C and Supplementary Table 1 and 2). We also validated E2F1 and E2F3 downregulation in GSK461364-treated SK-N-AS and IMR-32 cells using western blotting (Supplementary Figure 1). We also applied GSEA analysis for cancer-related signatures to the expression data from GSK461364-treated and untreated SK-N-AS cells. Genes known to be upregulated by RB1 tumor suppressor loss (gene set RB_DN.V1_UP) were downregulated in GSK461364-treated cells compared to untreated controls (Supplementary Table 2). Since RB1 regulates the G1/S transition via E2F transcription factors [42, 43], these results are in line with our observed drop in gene expression associated with S-phase transition. We conclude that GSK461364 treatment not only induces expression patterns associated with a G2/M arrest, but might also inhibit G1/S transition via the RB1-E2F axis in neuroblastoma cells.
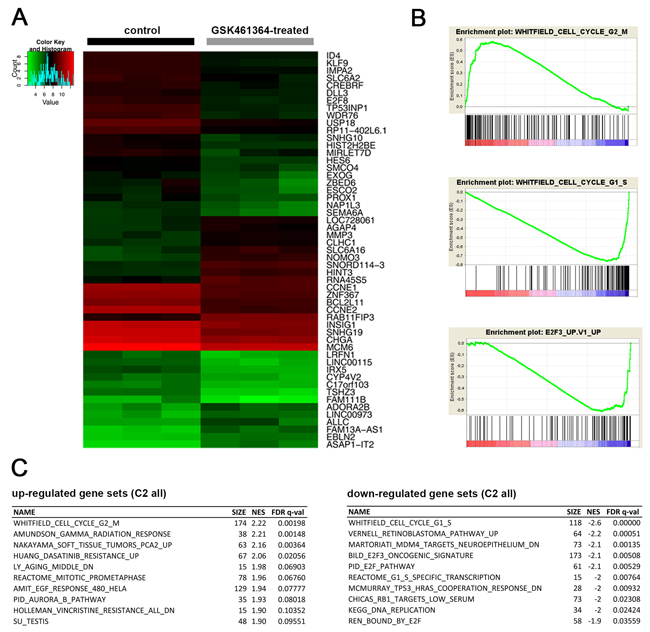
Figure 3: Cell cycle regulators are differentially expressed following GSK461364-mediated PLK1 inhibition. A. Heatmap resulting from unsupervised hierarchical clustering of samples treated with GSK461364 for 24 hours (n = 3) and controls (n = 3). Only the top 50 differentially expressed genes are displayed. B. GSEA plots showing enrichment for genes involved in G2/M-phase (top) and downregulation of genes associated with G1/S-phase (middle) as well as with the E2F3 transcription factor (bottom). C. The top 10 up- and downregulated gene sets.
GSK461364-mediated PLK1 inhibition has antitumoral activity against human neuroblastoma xenografts in mice
To investigate GSK461364 efficacy in vivo, we treated a xenograft model for high-risk neuroblastoma grown subcutaneously in nude mice. The SK-N-AS cell line lacks MYCN amplification and is derived from a bone marrow metastasis from a patient with stage 4 disease [44]. SK-N-AS cells were injected subcutaneously into the flanks of immunocompromised mice, and xenograft tumor development was monitored until they reached 200 μl in volume. GSK461364 (50 mg / kg body weight) or vehicle alone (control group) was intraperitoneally administered once daily to 6 mice per group. GSK461364 treatment significantly reduced tumor volume compared to controls (Supplementary Figure 2A). Kaplan-Meier analysis showed that treatment delayed tumor growth to 2,500 mm3 by 22 days (time when the first tumor in the control group reached 2,500 mm3 compared to when the first tumor in the treatment group reached 2,500 mm3, Figure 4A), thus, significantly increasing survival in the treatment cohort. One of 6 mice in the treatment group had to be euthanized due to cachexia. Mice harboring subcutaneous xenograft tumors formed from MYCN-amplified IMR32 cells were also treated using the same GSK461364 or control injection regimen. IMR32 xenograft tumors developed considerably slower than SK-N-AS xenograft tumors. GSK461364 treatment also significantly reduced IMR32 xenograft tumor growth compared to the control group (Figure 4B, Supplementary Figure 2B). Immunohistological examination of xenograft tumors showed that GSK461364 reduced the number of proliferating cells and increased the number of apoptotic cells in tumors (Figure 4C). Our data clearly demonstrate that GSK461364 treatment in vivo suppresses growth of xenografts derived from neuroblastoma cells with high-risk characteristics regardless of MYCN status.
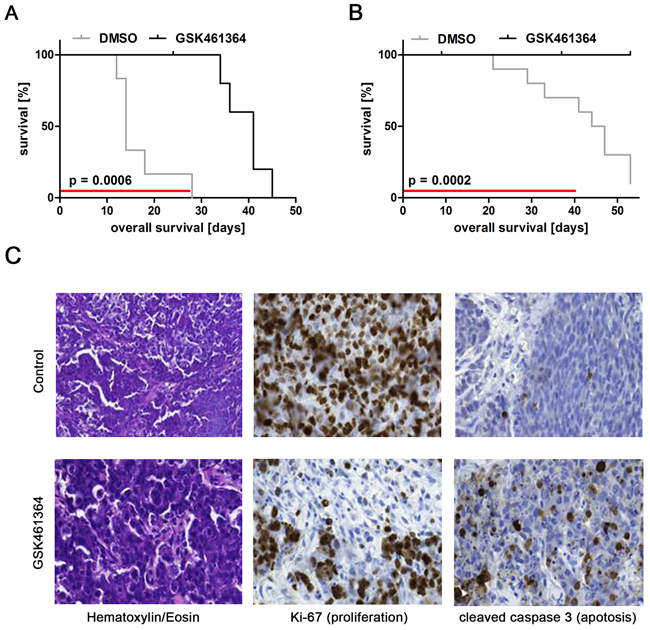
Figure 4: GSK461364-mediated PLK1 inhibition exerts antitumoral activity against human neuroblastoma xenografts in mice. Kaplan-Meier analysis of GSK461364-treated and control mouse cohorts in animals injected with A. SK-N-AS or B. IMR-32 cells. Red lines indicate duration of treatment. C. Micrographs show immunostainings for Ki67 to identify proliferating cells and cleaved caspase 3 to identify apoptotic cells in addition to hematoxylin & eosin staining (HE) for SK-N-AS xenograft tumors 3 days after start of treatment.
DISCUSSION
Treating patients with high-risk neuroblastoma remains challenging, since further escalation of existing therapies is limited by toxicity [45]. Encorporating targeted drugs into existing approaches should lower toxicity while better inhibiting growth characteristics of high-risk neuroblastomas. Here we present preclinical data for the efficacy of the PLK1 inhibitor, GSK461364, against models for high-risk neuroblastoma. GSK461364 inhibited neuroblastoma cell viability in vitro via suppressing proliferation and preventing progression through cell cycle blocks at G2 and possibly G1. GSK461364 induced apoptosis in vitro and suppressed xenograft tumor growth in mice. GSK461364 inhibited neuroblastoma cell growth regardless of MYCN copy number status and presence or absence of TP53 mutations.
High-level PLK1 expression has previously been reported in a variety of pediatric cancers [6, 22, 46]. Aggressive neuroblastoma subtypes overexpressed both PLK1 transcripts and protein [6]. PLK1 overexpression is known to override the G2/M and spindle checkpoints induced by DNA damage, thereby promoting chromosome instability and aneuploidy and fostering cancer progression [47]. We here show that inhibiting PLK1 with GSK461364 effectively reverted these effects, causing G2/M arrest accompanied by reduced proliferation and apoptosis in neuroblastoma cells. The expression profiling we conducted with and without GSK461364 treatment also indicated that PLK1 inhibition represses the E2F1 and E2F3 transcriptional activators, which promote G1/S transition [48]. Interestingly, miR-34a, which acts as a tumor suppressor in neuroblastoma, has also been shown to downregulate E2F factors and is suggested to directly suppress PLK1 translation [49–51]. Effects of miR-34a are comparable to those caused by GSK461364 treatment here. E2F family deregulation especially occurs in recurrent neuroblastomas and tumor-derived cell lines, presumably caused by upregulation of factors, such as CDK4, that inhibit the RB1 tumor suppressor [7, 52, 53]. Our analyses of signaling pathways associated with oncogenesis, also showed that GSK461364 treatment restored gene expression associated with unperturbed RB1 function. We conclude that PLK1 inhibition blocks cell cycle progression in neuroblastic cells by blocking PLK1 canonical regulatory function at the G2/M transition as well as cancer cell-specific blockage of G1/S transition occurring presumably via de-repression of RB1-associated gene expression.
Some genetic backgrounds have been proposed to sensitize cancer cells to PLK1 inhibition. Gorlick and colleagues treated a panel of predominantly MYCN-amplified neuroblastoma cell lines grown as xenograft tumors in CB17SC scid−/− mice with the PLK1 inhibitor, volasertib [54]. They observed a ≥2-fold increase in the time to event in treated mice, and speculated that the enhanced MYCN-driven cell cycle progression might help sensitize neuroblastoma cells to agents that block mitotic progression. In contrast, in our study also GSK461364 treatment of neuroblastoma cells lacking MYCN amplifications and expressing low levels of MYCN significantly inhibited proliferation, induced apoptosis and caused cell cycle arrest in in vitro, and significantly reduced in vivo tumor growth of neuroblastoma cells independent of MYCN copy number status. Degenhardt and colleagues treated a panel of cell lines derived from adult cancers with GSK461364A, and concluded that cancer cell sensitivity to PLK1 inhibition is mediated by loss of TP53 functionality [55]. TP53 is rarely mutated in neuroblastomas. However, a TP53 variant (p53ΔC) lacking the nuclear localization signal and part of the oligomerization domain [56], and a truncated TP53 isoform (p53β) [57] being also present in SK-N-AS cells, were reported for neuroblastoma. The neuroblastoma cell lines tested here harbor both wildtype or mutated TP53 (cf. Figure 1A), but were comparably sensitive to low nanomolar GSK461364 concentrations. We conclude that GSK461364 produces antineoplastic effects in neuroblastoma cells independent of MYCN background or TP53 functional status.
Targeted therapies can be circumvented by resistance mechanisms developed by cancer cells during tumor progression. Gilmartin and colleagues treated mice bearing colorectal (Colo205) xenograft tumors with several GSK461364A dosing schedules [35]. Xenograft tumor volume was reduced by all schedules during the treatment period, but resumed growth afterwards. Antimitotic drugs either enhance antiapoptotic protein expression to keep cell fractions alive during mitotic arrest or act cytotoxically depending on cellular context and inhibitor concentration [58, 59]. Cells in stasis might resume proliferation after treatment. Overexpression of the human multidrug resistance gene, ABCB1, was also recently reported to reduce GSK461364 activity in cancer cells [60]. Future in-depth preclinical studies addressing whether neuroblastic tumors regrow after PLK1-inhibiting treatment ceases and identifying the resistance mechanisms involved should assist schedule refinement for incorporating PLK1 inhibitors into current protocols for high-risk or relapsed disease. Ferrarotto and colleagues recently observed different sensitivities of cancer cell lines comparing treatments with dihydropteridinone derivatives, BI2536 or volasertib, with GSK461364 [34]. They suggested that higher sensitivity to GSK461364 might result from increased PLK1 selectivity of this compound compared to the two other inhibitors. It remains to be further elucidated if one class of PLK1 inhibitors might be substituted by another in case of resistance. To minimize resistance development, PLK1 inhibitors will most likely be combined with other synergizing targeted agents or conventional chemotherapeutics in treatment regimens. To date, synergism has especially been shown for PLK1-inhibitor based combination therapies with microtubule-interfering drugs (e.g. vincristine) [30, 61–63]. In contrast, the PLK1 inhibitor volasertib showed antagonistic effects when combined with etoposide in a cell line panel of pediatric malignancies [30].
Here we explored the feasibility of treating high-risk neuroblastoma patients with GSK461364 in preclinical models. GSK461364 suppressed proliferation, induced apoptosis and caused cell cycle arrest in neuroblastoma cell lines in vitro, and reduced tumor growth and extended survival of mice bearing xenografts of cell lines derived from high-risk neuroblastomas. Our data suggest that GSK461364 has the potential to generate a measurable response in patients with high-risk neuroblastomas and might even be considered for rational combination treatment approaches [31]. GSK461364 should, therefore, be considered for entry into early phase clinical trials in pediatric patients.
MATERIALS AND METHODS
Compound
GSK461364, (R)-5-[6-[(4-methyl-1-piperazinyl)methyl]-1H-benzimidazol-1-yl]-3-[(1r)-1-[2-(trifluoromethyl)phenyl]ethoxy]-2-thioph-enecarboxamide (Axon Medchem BV, Groningen, The Netherlands) was dissolved in DMSO for a 10 mmol/l stock solution for in vitro studies, and stored at -80°C.
Cell culture
The SK-N-AS, SH-SY5Y, SH-EP, IMR32, Kelly and SK-N-BE human neuroblastoma cell lines were cultivated in RPMI-1640 medium (Invitrogen, Karlsruhe, Germany) supplemented with 10% fetal calf serum (Invitrogen), 100 U/ml penicillin/streptomycin (Invitrogen) and 2.5 mg/l amphotericin B (PAA Laboratories GmbH, Egelsbach, Germany) and were incubated in a humidified 5% CO2 atmosphere at 37°C. Cell line identity was verified by the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany).
Cell assays
Neuroblastoma cell lines were seeded onto 96-well plates (2 × 103 cells per well) in triplicate for all assays, and incubated for 24 hours to permit surface adherence. Viability was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Roche, Basel, Switzerland). Apoptosis and proliferation were assessed using the Cell Death and BrdU ELISAs (Roche), respectively. All assays were performed according to the manufacturer’s protocols. For cell cycle analysis, SK-N-AS and IMR32 cells were suspended by trypsinization and washed 3 times with PBS, then incubated with propidium iodide for 15 min to stain DNA. Cellular DNA content was analyzed using a FC500 flow cytometer (Beckman Coulter, Krefeld, Germany). Colony forming ability was assessed in SK-N-AS or IMR32 cells by staining colonies with 1,9-dimethyl-methylene blue (Sigma Aldrich, St. Louis, MO, USA) for 45 minutes, washed twice with PBS and colonies were counted. All experiments were independently performed in triplicates at least 3 times, if not otherwise indicated.
Gene expression analyses
SK-N-AS cells were seeded onto 6-well plates (1 x 105 cells/well), allowed 12 hours to adhere then incubated 24 hours with 500 nM GSK461364 or control medium (containing 0.1% DMSO). Total RNA was extracted using the RNeasyMini kit (Qiagen, Hilden, Germany), and samples were profiled on the Affymetrix Human Gene Expression Array (HG-U133 Plus 2.0, Affymetrix, Santa Clara, CA, USA), both according to manufacturers’ protocols. CEL files were normalized and summarized to gene levels to conduct gcRMA normalization from the Bioconductor repository of statistical tools [64]. Only probes with the highest mean expression for each gene and with a log2 expression >5 were included in the analysis for differential expression using the Rank Product analysis package v2.36.0 in R [65]. Samples were clustered using hierarchical clustering, and dissimilarity calculated using Manhattan distances. Gene set enrichment analysis was performed using GSEA v2.0 software (www. broadinstitute.org/gsea) [66]. Genes were ranked by calculating the difference in population means scaled by the standard deviation to generate a signal to noise ranking metric. We used publicly available curated gene sets (c2.all.v4.0.symbols.gmt), gene motif sets (c3.all.v4.0.symbols.gmt) and oncogenic signatures (c6.all.v4.0.symbols.gmt) in our analyses. Newly generated microarray data were deposited in the GEO database (accession no. GSE67102).
Xenograft model
SK-N-AS and IMR32 neuroblastoma cells were cultured to 80% confluency, harvested and suspended in 200 μL Matrigel™ (BD Bioscience, Heidelberg, Germany) for subcutaneous inoculation of 2 × 107 cells into the left flank of 8-week-old female athymic nu/nu mice (n = 12 mice). Mice were randomly assigned (6 mice per group) to either GSK461364 or vehicle (0.1% DMSO, 5% glucose) control groups after tumors reached 200 mm3. Vehicle control or 50 mg GSK461364 per kg body weight was intraperitoneally injected every second day until all control mice reached the endpoint (tumor volume of 2,500mm3) but not longer than 40 days. The same dose and treatment scheme was chosen as used in previous preclinical GSK461364 studies in various xenograft tumor models, and which was shown to be safe and effective [35]. Tumor growth was monitored using a caliper, and tumor volume was calculated using the formula, (breadth × length × height)/2. Mice were euthanized by cervical dislocation when tumor size exceeded 2,500mm3. In mice, whose xenograft tumors were to be immunohistologically examined for apoptosis and proliferation, 2 doses of 100 mg GSK461364 per kg body weight were administered every 12 hours over a 3-day course (n = 3 mice in both GSK461364 and vehicle control groups). Mice were euthanized 4 hours after the last treatment injection, and xenograft tumors were excised, formalin-fixed and paraffin-embedded. All animal experiments were performed in accordance with the Council of Europe guidelines for accommodation and care of laboratory animals, and protocols were approved by the Ethical Commission for Animal Experimentation at the University Hospital Essen.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections (5 μm) were deparaffinized using routine techniques, and placed in 200 ml of EnVision™ target retrieval solution (pH 6.0; Dako, Hamburg, Germany) for 20 minutes at 100°C. After cooling for 20 minutes, slides were quenched with 3% H2O2 for 5 minutes before incubating with primary antibodies against cleaved caspase 3 (#cat 9661; 1:200; Cell Signaling, Danvers, MA, USA) to detect apoptotic cells or marker of proliferation Ki-67 (#cat M724029-2, 1:25, Dako) using a Dako Autostainer (Dakocytomation). Immunostaining was visualized using the EnVision™+ kit (Dako).
Statistics
Pairs of interval variables were compared with Student’s two-sided t-tests using SPSS Statistics for Windows, v21.0. (IBM, Armonk, NY, USA). Error bars in figures represent standard deviation. Graph Pad Prism 5.0 (GraphPad Software Inc., San Diego, CA, USA) was used to calculate GI50 or 80 concentrations and to perform Kaplan-Meier analyses.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Maris J, Hogarty M, Bagatell R and Cohn S. Neuroblastoma. Lancet. 2007; 369:2106-2120.
2. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003; 3:203-216.
3. Brodeur GM, Iyer R, Croucher JL, Zhuang T, Higashi M and Kolla V. Therapeutic targets for neuroblastomas. Expert opinion on therapeutic targets. 2014; 18:277-292.
4. Kushner BH, Modak S, Kramer K, LaQuaglia MP, Yataghene K, Basu EM, Roberts SS and Cheung NK. Striking dichotomy in outcome of MYCN-amplified neuroblastoma in the contemporary era. Cancer. 2014; 120:2050-2059.
5. Cohn S, Pearson A, London W, Monclair T, Ambros P, Brodeur G, Faldum A, Hero B, Iehara T, Machin D, Mosseri V, Simon T, Garaventa A, Castel V and Matthay K. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol. 2009; 27:289-297.
6. Ackermann S, Goeser F, Schulte JH, Schramm A, Ehemann V, Hero B, Eggert A, Berthold F and Fischer M. Polo-like kinase 1 is a therapeutic target in high-risk neuroblastoma. Clin Cancer Res. 2011; 17:731-741.
7. Gogolin S, Batra R, Harder N, Ehemann V, Paffhausen T, Diessl N, Sagulenko V, Benner A, Gade S, Nolte I, Rohr K, Konig R and Westermann F. MYCN-mediated overexpression of mitotic spindle regulatory genes and loss of p53-p21 function jointly support the survival of tetraploid neuroblastoma cells. Cancer Lett. 2013; 331:35-45.
8. Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW and Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998; 392:300-303.
9. Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB and Medema RH. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008; 455:119-123.
10. Seki A, Coppinger JA, Jang CY, Yates JR and Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science. 2008; 320:1655-1658.
11. van de Weerdt BC, van Vugt MA, Lindon C, Kauw JJ, Rozendaal MJ, Klompmaker R, Wolthuis RM and Medema RH. Uncoupling anaphase-promoting complex/cyclosome activity from spindle assembly checkpoint control by deregulating polo-like kinase 1. Mol Cell Biol. 2005; 25:2031-2044.
12. Fu Z, Malureanu L, Huang J, Wang W, Li H, van Deursen JM, Tindall DJ and Chen J. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat Cell Biol. 2008; 10:1076-1082.
13. Smits VA, Klompmaker R, Arnaud L, Rijksen G, Nigg EA and Medema RH. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat Cell Biol. 2000; 2:672-676.
14. Li H, Wang Y and Liu X. Plk1-dependent phosphorylation regulates functions of DNA topoisomerase IIalpha in cell cycle progression. J Biol Chem. 2008; 283:6209-6221.
15. Song B, Liu XS, Davis K and Liu X. Plk1 phosphorylation of Orc2 promotes DNA replication under conditions of stress. Mol Cell Biol. 2011; 31:4844-4856.
16. Wu ZQ and Liu X. Role for Plk1 phosphorylation of Hbo1 in regulation of replication licensing. Proc Natl Acad Sci U S A. 2008; 105:1919-1924.
17. Ito Y, Miyoshi E, Sasaki N, Kakudo K, Yoshida H, Tomoda C, Uruno T, Takamura Y, Miya A, Kobayashi K, Matsuzuka F, Matsuura N, Kuma K and Miyauchi A. Polo-like kinase 1 overexpression is an early event in the progression of papillary carcinoma. Br J Cancer. 2004; 90:414-418.
18. Petrelli A, Perra A, Schernhuber K, Cargnelutti M, Salvi A, Migliore C, Ghiso E, Benetti A, Barlati S, Ledda-Columbano GM, Portolani N, De Petro G, Columbano A and Giordano S. Sequential analysis of multistage hepatocarcinogenesis reveals that miR-100 and PLK1 dysregulation is an early event maintained along tumor progression. Oncogene. 2012; 31:4517-4526.
19. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr. and Kinzler KW. Cancer genome landscapes. Science. 2013; 339:1546-1558.
20. Weichert W, Schmidt M, Jacob J, Gekeler V, Langrehr J, Neuhaus P, Bahra M, Denkert C, Dietel M and Kristiansen G. Overexpression of Polo-like kinase 1 is a common and early event in pancreatic cancer. Pancreatology. 2005; 5:259-265.
21. Hu K, Lee C, Qiu D, Fotovati A, Davies A, Abu-Ali S, Wai D, Lawlor ER, Triche TJ, Pallen CJ and Dunn SE. Small interfering RNA library screen of human kinases and phosphatases identifies polo-like kinase 1 as a promising new target for the treatment of pediatric rhabdomyosarcomas. Molecular cancer therapeutics. 2009; 8:3024-3035.
22. Harris PS, Venkataraman S, Alimova I, Birks DK, Donson AM, Knipstein J, Dubuc A, Taylor MD, Handler MH, Foreman NK and Vibhakar R. Polo-like kinase 1 (PLK1) inhibition suppresses cell growth and enhances radiation sensitivity in medulloblastoma cells. BMC Cancer. 2012; 12:80.
23. Morales AG, Brassesco MS, Pezuk JA, Oliveira JC, Montaldi AP, Sakamoto-Hojo ET, Scrideli CA and Tone LG. BI 2536-mediated PLK1 inhibition suppresses HOS and MG-63 osteosarcoma cell line growth and clonogenicity. Anticancer Drugs. 2011; 22:995-1001.
24. Triscott J, Lee C, Foster C, Manoranjan B, Pambid MR, Berns R, Fotovati A, Venugopal C, O’Halloran K, Narendran A, Hawkins C, Ramaswamy V, Bouffet E, Taylor MD, Singhal A, Hukin J, et al. Personalizing the treatment of pediatric medulloblastoma: Polo-like kinase 1 as a molecular target in high-risk children. Cancer Res. 2013; 73:6734-6744.
25. Liu L, Zhang M and Zou P. Expression of PLK1 and survivin in diffuse large B-cell lymphoma. Leukemia & lymphoma. 2007; 48:2179-2183.
26. Salvatore G, Nappi TC, Salerno P, Jiang Y, Garbi C, Ugolini C, Miccoli P, Basolo F, Castellone MD, Cirafici AM, Melillo RM, Fusco A, Bittner ML and Santoro M. A cell proliferation and chromosomal instability signature in anaplastic thyroid carcinoma. Cancer Res. 2007; 67:10148-10158.
27. Yamamoto Y, Matsuyama H, Kawauchi S, Matsumoto H, Nagao K, Ohmi C, Sakano S, Furuya T, Oga A, Naito K and Sasaki K. Overexpression of polo-like kinase 1 (PLK1) and chromosomal instability in bladder cancer. Oncology. 2006; 70:231-237.
28. Kanaji S, Saito H, Tsujitani S, Matsumoto S, Tatebe S, Kondo A, Ozaki M, Ito H and Ikeguchi M. Expression of polo-like kinase 1 (PLK1) protein predicts the survival of patients with gastric carcinoma. Oncology. 2006; 70:126-133.
29. Liu X, Choy E, Harmon D, Yang S, Yang C, Mankin H, Hornicek FJ and Duan Z. Inhibition of polo-like kinase 1 leads to the suppression of osteosarcoma cell growth in vitro and in vivo. Anticancer Drugs. 2011; 22:444-453.
30. Abbou S, Lanvers-Kaminsky C, Daudigeos-Dubus E, L LED, Laplace-Builhe C, Molenaar J, Vassal G, Geoerger B, within the IB and Preclinical Evaluation C. Polo-like Kinase Inhibitor Volasertib Exhibits Antitumor Activity and Synergy with Vincristine in Pediatric Malignancies. Anticancer Res. 2016; 36:599-609.
31. Czaplinski S, Hugle M, Stiehl V and Fulda S. Polo-like kinase 1 inhibition sensitizes neuroblastoma cells for vinca alkaloid-induced apoptosis. Oncotarget. 2015; 7: 8700-11. doi: 10.18632/oncotarget.3901.
32. Liu X, Lei M and Erikson RL. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol. 2006; 26:2093-2108.
33. Grinshtein N, Datti A, Fujitani M, Uehling D, Prakesch M, Isaac M, Irwin MS, Wrana JL, Al-Awar R and Kaplan DR. Small molecule kinase inhibitor screen identifies polo-like kinase 1 as a target for neuroblastoma tumor-initiating cells. Cancer Res. 2011; 71:1385-1395.
34. Ferrarotto R, Goonatilake R, Young Yoo S, Tong P, Giri U, Peng S, Minna J, Girard L, Wang Y, Wang L, Li L, Diao L, Peng DH, Gibbons DL, Glisson BS, Heymach JV, et al. Epithelial-Mesenchymal Transition Predicts Polo-Like Kinase 1 Inhibitor-Mediated Apoptosis in Non-Small Cell Lung Cancer. Clin Cancer Res. 2016; 22:1674-1686.
35. Gilmartin AG, Bleam MR, Richter MC, Erskine SG, Kruger RG, Madden L, Hassler DF, Smith GK, Gontarek RR, Courtney MP, Sutton D, Diamond MA, Jackson JR and Laquerre SG. Distinct concentration-dependent effects of the polo-like kinase 1-specific inhibitor GSK461364A, including differential effect on apoptosis. Cancer Res. 2009; 69:6969-6977.
36. Olmos D, Barker D, Sharma R, Brunetto AT, Yap TA, Taegtmeyer AB, Barriuso J, Medani H, Degenhardt YY, Allred AJ, Smith DA, Murray SC, Lampkin TA, Dar MM, Wilson R, de Bono JS, et al. Phase I study of GSK461364, a specific and competitive Polo-like kinase 1 inhibitor, in patients with advanced solid malignancies. Clin Cancer Res. 2011; 17:3420-3430.
37. Schoffski P. Polo-like kinase (PLK) inhibitors in preclinical and early clinical development in oncology. Oncologist. 2009; 14:559-570.
38. Mross K, Frost A, Steinbild S, Hedbom S, Rentschler J, Kaiser R, Rouyrre N, Trommeshauser D, Hoesl CE and Munzert G. Phase I dose escalation and pharmacokinetic study of BI 2536, a novel Polo-like kinase 1 inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2008; 26:5511-5517.
39. Pezuk JA, Brassesco MS, Morales AG, de Oliveira JC, de Paula Queiroz RG, Machado HR, Carlotti CG, Jr., Neder L, Scrideli CA and Tone LG. Polo-like kinase 1 inhibition causes decreased proliferation by cell cycle arrest, leading to cell death in glioblastoma. Cancer gene therapy. 2013; 20:499-506.
40. Brassesco MS, Pezuk JA, Morales AG, de Oliveira JC, Roberto GM, da Silva GN, Francisco de Oliveira H, Scrideli CA and Tone LG. In vitro targeting of Polo-like kinase 1 in bladder carcinoma: comparative effects of four potent inhibitors. Cancer Biol Ther. 2013; 14:648-657.
41. Yata K, Lloyd J, Maslen S, Bleuyard JY, Skehel M, Smerdon SJ and Esashi F. Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Mol Cell. 2012; 45:371-383.
42. Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, Nuckolls F, Giangrande P, Wright FA, Field SJ, Greenberg ME, Orkin S, Nevins JR, Robinson ML and Leone G. The E2F1-3 transcription factors are essential for cellular proliferation. Nature. 2001; 414:457-462.
43. Leone G, DeGregori J, Yan Z, Jakoi L, Ishida S, Williams RS and Nevins JR. E2F3 activity is regulated during the cell cycle and is required for the induction of S phase. Genes Dev. 1998; 12:2120-2130.
44. Thiele CJ. Neuroblastoma. In: Masters J, ed. Human Cell Culture. (Lancaster, UK: Kluwer Academic Publishers), pp. 21-53. (1998).
45. Modak S and Cheung NK. Neuroblastoma: Therapeutic strategies for a clinical enigma. Cancer treatment reviews. 2010; 36:307-317.
46. Amani V, Prince EW, Alimova I, Balakrishnan I, Birks D, Donson AM, Harris P, Levy JM, Handler M, Foreman NK, Venkataraman S and Vibhakar R. Polo-like Kinase 1 as a potential therapeutic target in Diffuse Intrinsic Pontine Glioma. BMC Cancer. 2016; 16:647.
47. Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov. 2010; 9:643-660.
48. Chen HZ, Tsai SY and Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009; 9:785-797.
49. Tivnan A, Tracey L, Buckley PG, Alcock LC, Davidoff AM and Stallings RL. MicroRNA-34a is a potent tumor suppressor molecule in vivo in neuroblastoma. BMC Cancer. 2011; 11:33.
50. Lal A, Thomas MP, Altschuler G, Navarro F, O’Day E, Li XL, Concepcion C, Han YC, Thiery J, Rajani DK, Deutsch A, Hofmann O, Ventura A, Hide W and Lieberman J. Capture of microRNA-bound mRNAs identifies the tumor suppressor miR-34a as a regulator of growth factor signaling. PLoS genetics. 2011; 7:e1002363.
51. Welch C, Chen Y and Stallings RL. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene. 2007; 26:5017-5022.
52. Molenaar JJ, van Sluis P, Boon K, Versteeg R and Caron HN. Rearrangements and increased expression of cyclin D1 (CCND1) in neuroblastoma. Genes Chromosomes Cancer. 2003; 36:242-249.
53. Molenaar JJ, Ebus ME, Koster J, van Sluis P, van Noesel CJ, Versteeg R and Caron HN. Cyclin D1 and CDK4 activity contribute to the undifferentiated phenotype in neuroblastoma. Cancer Res. 2008; 68:2599-2609.
54. Gorlick R, Kolb EA, Keir ST, Maris JM, Reynolds CP, Kang MH, Carol H, Lock R, Billups CA, Kurmasheva RT, Houghton PJ and Smith MA. Initial testing (stage 1) of the Polo-like kinase inhibitor volasertib (BI 6727), by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2014; 61:158-164.
55. Degenhardt Y, Greshock J, Laquerre S, Gilmartin AG, Jing J, Richter M, Zhang X, Bleam M, Halsey W, Hughes A, Moy C, Liu-Sullivan N, Powers S, Bachman K, Jackson J, Weber B, et al. Sensitivity of cancer cells to Plk1 inhibitor GSK461364A is associated with loss of p53 function and chromosome instability. Molecular cancer therapeutics. 2010; 9:2079-2089.
56. Nakamura Y, Ozaki T, Niizuma H, Ohira M, Kamijo T and Nakagawara A. Functional characterization of a new p53 mutant generated by homozygous deletion in a neuroblastoma cell line. Biochem Biophys Res Commun. 2007; 354:892-898.
57. Goldschneider D, Horvilleur E, Plassa LF, Guillaud-Bataille M, Million K, Wittmer-Dupret E, Danglot G, de The H, Benard J, May E and Douc-Rasy S. Expression of C-terminal deleted p53 isoforms in neuroblastoma. Nucleic Acids Res. 2006; 34:5603-5612.
58. Gascoigne KE and Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008; 14:111-122.
59. Tao W, South VJ, Zhang Y, Davide JP, Farrell L, Kohl NE, Sepp-Lorenzino L and Lobell RB. Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage. Cancer Cell. 2005; 8:49-59.
60. Wu CP, Hsiao SH, Luo SY, Tuo WC, Su CY, Li YQ, Huang YH and Hsieh CH. Overexpression of human ABCB1 in cancer cells leads to reduced activity of GSK461364, a specific inhibitor of polo-like kinase 1. Molecular pharmaceutics. 2014; 11:3727-3736.
61. Hugle M, Belz K and Fulda S. Identification of synthetic lethality of PLK1 inhibition and microtubule-destabilizing drugs. Cell Death Differ. 2015; 22:1946-1956.
62. Stehle A, Hugle M and Fulda S. Eribulin synergizes with Polo-like kinase 1 inhibitors to induce apoptosis in rhabdomyosarcoma. Cancer Lett. 2015; 365:37-46.
63. Weiss LM, Hugle M, Romero S and Fulda S. Synergistic induction of apoptosis by a polo-like kinase 1 inhibitor and microtubule-interfering drugs in Ewing sarcoma cells. Int J Cancer. 2016; 138:497-506.
64. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004; 5:R80.
65. Hong F, Breitling R, McEntee CW, Wittner BS, Nemhauser JL and Chory J. RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics. 2006; 22:2825-2827.
66. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES and Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005; 102:15545-15550.