
INTRODUCTION
Melanoma, the deadliest type of skin cancer, is responsible for 75% of skin cancer-related deaths with incidence continually rising [1]. In 2015, the estimated prevalence and mortality of cutaneous melanoma in the USA were 73,870 and 9,940, respectively [2]. Melanoma is driven by the combination of genetic and environmental factors. Ultraviolet (UV)-induced DNA damage [3–5], loss-of-function mutations in CDKN2A, and inactivating variants of melanocortin 1 receptor gene (MC1R) that are associated with poor tanning ability [6], as well as red hair all play roles in increased risk of melanoma occurrence [7]. In addition, evidence is accumulating that defects in vitamin D signaling can affect melanomagenesis, tumor progression, and final disease outcome [8–15].
Current treatment options for advanced cutaneous melanoma include surgical metastasectomy [16], radiation therapy [17], immunotherapy, and targeted therapy against the mitogen-activated protein kinase (MAPK) and c-KIT pathways [18–24]. Although many of these targeted therapeutic modalities are beneficial, they tend to be costly and are associated with many adverse effects. In addition, molecularly targeted therapies require mutations in targeted genes such as BRAF or c-KIT which may not be present in subsets of melanoma patients, rendering these treatments ineffective. Finally, disease recurrence often occurs due to development of resistance (discussed in [24–26]).
Vitamin D3 undergoes a two-step activation via hydroxylation in the liver at C25 and in the kidney at C1α, resulting in the production of its active form, 1,25(OH)2D3 (calcitriol) [27, 28]. The same sequence of D3 activation occurs in the skin [29, 30]. 1,25(OH)2D3 is not only implicated in regulating calcium levels, but also is involved in promoting the expression of genes that are anti-inflammatory, anti-proliferative, anti-angiogenic and anti-carcinogenic [27-29, 31-34]. The anticancer properties of D3 [14, 33, 35, 36] served as the rationale for an ongoing phase II clinical trial in Australia evaluating the use of vitamin D as an adjuvant therapy for treating melanoma with a high risk of recurrence [37].
Having vitamin D levels at the higher end of the physiological range may be beneficial. However, anticancer activities of vitamin D, particularly those of 1,25(OH)2D3, appear to require pharmacological doses that cause hypercalcemia. At a dose as low as 0.1 μg/kg for 1,25(OH)2D3, calcium depositions cause damage to the vital organs [38]. Thus, the hypercalcemic effects of 1,25(OH)2D3 at therapeutic doses severely limit its utility as an anti-cancer agent.
Mammalian CYP11A1, (also known as cytochrome P450scc), encoded by the CYP11A1 gene, can hydroxylate vitamin D3 at positions C17, C20, C22 and C23 to produce several D3 hydroxyderivatives with 20(OH)D3 being a major product of the pathway [39–43]. Many of these novel hydroxylated vitamin D3 derivatives have been found in various organs, including the skin [44–46]. These hydroxyderivatives display anti-tumorigenic and anti-melanoma activities comparable to those of 1,25(OH)2D3 [47–52], and are noncalcemic in rodents at therapeutic doses as high as 60 μg/kg [50, 53, 54].
The main goal of this project was to test the hypothesis that 20(OH)D3 has anticancer activity equivalent to 1,25(OH)2D3on the growth, proliferation and progression of melanoma cells in vitro, and displays such properties at therapeutic doses that are noncalcemic using an in vivo model with immunocompromised mice.
RESULTS
Antimelanoma activity of 20(OH)D3 with in vitro models
Following our previous findings that CYP11A1-derived hydroxysecosteroids can inhibit proliferation of melanoma cell lines [49, 55], we selected 20(OH)D3 for further experimental testing on a SKMel-188 human melanoma line using established in vitro and in vivo assays of tumorigenesis and tumor progression. 20(OH)D3 inhibited colony formation in monolayer culture in a manner similar to 1,25(OH)2D3 (Figure 1A). The cytotoxicity of 20(OH)D3 in comparison to 1,25(OH)2D3 was tested using a novel spheroid toxicity assay, in which the contraction of spheroids formed through magnetic 3D bioprinting, was captured in real-time and utilized as a cytotoxic endpoint. This system was more representative of the 3D tissue environment than viability in 2D seen in an MTT assay. Once spheroids were formed, they began contracting immediately as a function of cell-cell interaction and cell-ECM (extracellular matrix) interaction, but the rate of contraction was slower in the presence of cytotoxic compounds [56]. 20(OH)D3 at a concentration of 10-7 M showed significant inhibition of spheroid contraction (Figure 1B) with 1,25(OH)2D3having no effect (not shown).
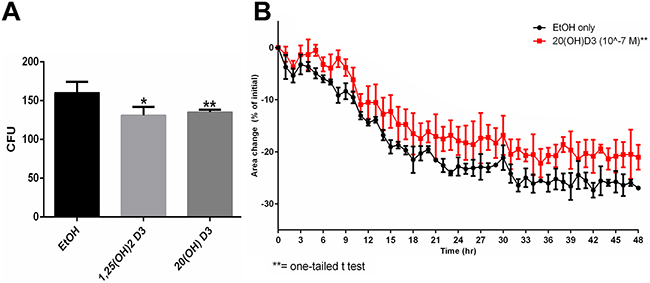
Figure 1: Effects of active forms on vitamin D on colony formation and spheroid toxicity assay in melanoma cells. A. SKMel-188 cells were seeded at 14 cells/cm2 and incubated with 10-7 M 20(OH)D3 or 1,25(OH)2D3(or ethanol vehicle) for 14 days. Colonies > 0.2 mm were counted. B. Assessment 20(OH)D3’s cytotoxicity using the spheroid toxicity assay where area changes are indicative of migratory capability, cell-cell interactions and cell-ECM interactions. A smaller area change is indicative of greater cytotoxicity. SKMel-188 cells (50,000 cells/well) were printed into spheroids. Subsequently, they were incubated in the presence of 1,25(OH)2D3 or 20(OH)D3 (or ethanol vehicle as a control) at concentrations of 10-7 M (only 20(OH)D3 is shown here, since 1,25(OH)2D3 had no significant effect), where *P<0.05, **P<0.01.
Anchorage-independent growth, assessed by the formation of colonies with diameters >0.1mm, >0.15mm and >0.2 mm, was also inhibited by both 20(OH)D3 and 1,25(OH)2D3 in a dose-dependent manner (Figure 2). The IC50 values were lower in the groups treated with 1,25(OH)2D3 than 20(OH)D3 (0.48 x 10-9 M vs. 1.58 x 10-8 M, respectively for colonies > 0.1 mm and 0.89 x 10-9 M vs. 6.91 x 10-9 M, respectively for colonies > 0.2 mm. However, 20(OH)D3 showed higher efficacy in absolute percentage inhibition when comparing the inhibition at 10-7 M to that of 1,25(OH)2D3 (56.0% vs. 17.0%, respectively for colonies > 0.1 mm and 53.1% vs. 46.6%, respectively, for colonies > 0.2 mm). Furthermore, 20(OH)D3 significantly inhibited cell migration with an efficacy comparable to that of 1,25(OH)2D3 using the transwell migration assay at 100,000 and 10,000 cells/well (Figure 3).
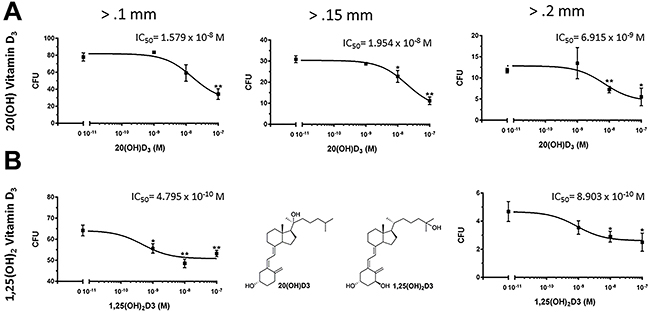
Figure 2: Effects of active forms on vitamin D on anchorage-independent growth of melanoma cells. Both 20(OH)D3 and 1,25(OH)2D3 inhibit anchorage-independent growth in soft agar. Structures of 20(OH)D3 and 1,25(OH)2D3 are shown in panel B. SKMel-188 cells were seeded (500 cells/well) on a 0.8% agar layer and treated with 20(OH)D3 A. or 1,25(OH)2D3 B. (or ethanol vehicle as control) for 12 days. Colonies > 0.1 mm, 0.15 mm, and 0.2 mm were counted, where *P<0.05, **P<0.01.
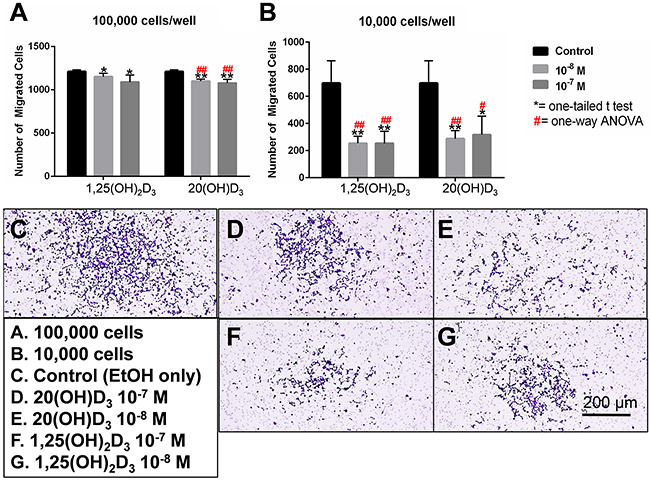
Figure 3: Effects of active forms on vitamin D on chemotactic capacity using a transwell migration assay. SKMel-188 cells were placed on top of a transwell insert (polycarbonate membrane, pore size=8.0 μm) at a density of either 100,000 A. or 10,000 cells/well B. and the number of cells that migrated to the other side of the member in presence of a chemoattractant was quantified. The cells were incubated in the presence of graded concentrations of 20(OH)D3 D, E. or 1,25(OH)2D3 F, G. or ethanol as vehicle C. for 24 h. Top: 20(OH)D3 and 1,25(OH)2D3at 10-7 and 10-8 M inhibited chemotaxis in a dose-dependent manner at both cell densities (10,000 cells/well or 100,000 cells/well). The statistical significance was measured by a student t-test (*) or one way ANOVA (#); where (*, #) P<.05, (**, ##) P<.01. Bottom: A representative image from each treatment group (A-E) is shown.
Antimelanoma activity of 20(OH)D3 in an in vivo model
Our in vivo study showed that the novel hydroxylated vitamin D3 derivative, 20(OH)D3, inhibits the growth of human melanoma in NSG mice. Of the twenty immunocompromised animals injected subcutaneously with SKMel-188 human melanoma cells, only mice that developed tumors were included for further study and analysis. Once the first palpable and measurable tumors (≥ 1 mm3) appeared on the 9th day following implantation in 4 out of 20 mice (equal prevalence in control and treated group), the treatment with either 20(OH)D3 or vehicle began for all animals according to the randomization arm (see M&M). By the 12th day following implantation (day 3 of treatment), 40% of animals had developed palpable tumors. All from both 20(OH)D3- and vehicle-treated mice (n=20) developed tumors by day 14 (5th day of treatment). Sustained growth of tumors was subsequently observed in both groups, but tumor growth was impaired in 20(OH)D3-treated mice as compared to vehicle-treated mice. On day 6 of treatment, the accrued tumor volume was 262.35 mm3 in the 20(OH)D3-treated group versus 674.11 mm3 in the vehicle-treated group, representing a 61% decrease in tumor volume in the treated and control group, respectively. The inhibitory action of 20(OH)D3 on melanoma growth was statistically significant as measured by tumor volume or the geometric mean of the tumor dimensions on days 13, 18 and 21 post-implantation (Figure 4A, 4B).
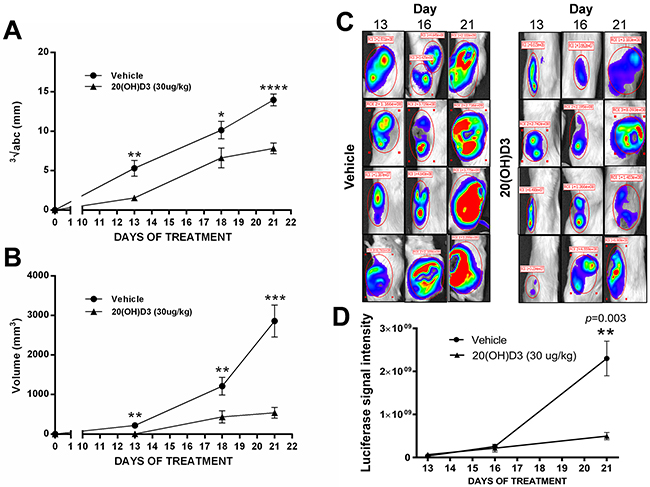
Figure 4: Treatment with 20(OH)D3 leads to an inhibition in growth of human melanoma in a mouse model. The time dependent growth of implanted SKMel-188 melanomas is presented as geometric means of tumor dimensions A. and tumor volumes B. measured with caliper, where *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001; evaluated with a student’s t-test to indicate the differences between control (vehicle) and 20(OH)D3 treatment. Representative examples (n=4 per each group) of luciferase intensity captured on day 13, 16, and 21 after tumor implantation are shown in C. with attendant calculation of the average signal intensity (n=10 from each group) in D.
The results of caliper evaluation of tumor growth in mice were further confirmed using bioluminescent imaging of tumor formation by melanoma cells expressing luciferase. The intensity of the luciferase signal indicates the number of viable melanoma cells and corresponds to tumor burden. Treatment with 20(OH)D3 led to a visible decrease in tumorigenesis when compared to control animals. Figure 4C, 4D shows representative images of 3 measurements (taken on 13, 16, and 21 day after tumor implantation) from 4 representative mice from each group, along with calculations of the signal intensity accumulated as a sum from all animals (n=10) in each group.
Although the survival rate was 90% in both groups on the last day of experiment, the total body score representing the health status of the animals was higher in the 20(OH)D3-treated group compared to controls (2.8 vs 2.55, respectively). Body condition scoring is a practical, rapid, noninvasive method for assessing health status in animals. Changes in body weight, behavior and physical appearances, commonly suggested as standard indicators of health or illness, were recorded during observation and palpation performed by a veterinarian. A mouse of BC3 status was in optimal condition and any increase/decrease in BC value indicated deterioration of health. No significant inhibitory effect on melanoma growth was observed in animals treated with a lower (3 μg/kg) dose of 20(OH)D3 (data not shown).
The morphology of implanted melanomas is shown in Figure 5. H&E stained sections showed that tumors treated either with vehicle or 20(OH)D3 were composed of pleomorphic amelanotic melanoma cells with epithelioid morphology with pleomorphic hyperchromatic nuclei, frequent mitoses and areas of necrosis (Figure 5).
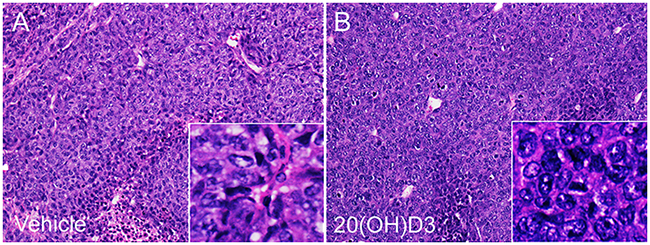
Figure 5: Histology of the implanted tumors. Panels A and B show representative H&E stained sections of tumors from animals treated with either vehicle A. or 20(OH)D3 B. Microscopic magnification of main panels: x 12.5; insert: x 45.
DISCUSSION
20(OH)D3 is the major metabolite of a newly discovered CYP11A1-mediated hydroxylation pathway of vitamin D3 activation [43, 45, 57, 58], which operates in vivo in humans [44]. 20(OH)D3 is noncalcemic and non-toxic at high pharmacological doses (30-60 μg/kg in rodents) [53, 54], which are approximately 300-600 times higher than doses of 1,25(OH)2D3 that produce hypercalcemia [38, 50]. Previously, we reported anticancer and antimelanoma activities of 20(OH)D3 in vitro [48, 49, 51-53, 55]. In accordance with our prior findings, this current study showed that 20(OH)D3 inhibits colony formation by SKMel-188 cells in soft agar assays, indicating that it reduces anchorage-independent growth of melanoma cells. Additionally, we showed that 20(OH)D3 inhibits cell migration, and cell-cell and cell-extracellular matrix interactions in both a transwell migration assay that assesses the chemotactic capability of cells, and a spheroid toxicity assay that assesses the migratory capability, cell-cell interactions and cell-ECM interactions [56, 59]. Lastly, the effects of 20(OH)D3 were observed in vivo since it decreased melanoma tumor in immunocompromised mice, without visible signs of toxicity.
The chemotactic capability and migratory capability are both important metastatic parameters that become unregulated and facilitate cancer cell migration [60]. Furthermore, the loss of intracellular adhesion and abnormalities in cell-matrix interaction are also critical in the progression stage of carcinogenesis [61]. Developing a therapy targeting tumor progression of melanoma is essential since metastatic malignant melanoma has an especially poor prognosis, even with the advent of new molecularly targeted therapeutic modalities. 20(OH)D3 demonstrated antimelanoma activity comparable to 1,25(OH)2D3 in the transwell migration assay, and demonstrated superior antimelanoma activity in the spheroid toxicity assay.
The partial success of the recently introduced treatment, aimed at PD-1/PD-L1 blockade limited by the lack of a sufficient number of activated lymphocytes, encouraged scientists to search for more reliable and efficient models and treatments for melanoma. The gold standard for melanoma study is the patient-derived orthotopic xenograft model which enables the tracking of tumor complexity that mimics the behavior in “real” in vivo environment [62]. The immunocompromised (nude) mouse is a well-established animal model to study human melanoma [63–66]. Tumor growth is a complex process, ultimately dependent on the environment of the proliferating tumor cells in the host tissues. The current view of tumor growth kinetics is based on the general assumption that tumor cells grow exponentially. However, a discrepancy is evident between the exponential tumor growth theory and in vivo experimental data, since tumor doubling times have been found to greatly exceed cell cycle times [67]. This may explain why tumors occurred in our study at a different time (9 to 14 days) after inoculation. Usually it takes 5 to 10 days to reach a volume of 1 mm3 with an injection of approximately 105 – 106 cells [64, 68]. However, the murine melanoma model is unpredictable with regard to the specific timeline of tumor development. This unpredictability is particularly relevant for defining a strict criterion for tumor diagnosis (size or volume) and for setting a starting day for drug treatment. Thus, we employed two independent methods of tumor measurement (caliper and bioluminescence) and two methods of presenting values (tumor volume and a geometric mean of the tumor dimensions) in our experiments to provide a reliable track of tumor growth, as recommended for assuring the validity of an in vivo study [69, 70]. All of these methods showed attenuation of tumor growth by 20(OH)D3 at the dose of 30 μg/kg without any sign of toxicity. Another robust “tool” to track progressiveness of metastatic melanomas is the GFP-transduced melanoma model [71]. Naturally fluorescent proteins have revolutionized biology in the way that single metastatic cell can be observed and tracked even at the subcellular level [72, 73]. Moreover, the combination of GFP-expressing melanomas and transgenic mice displaying traced blood vessels along with the application of the noninvasive imaging [74] may be extremely advantageous method for future studies.
The total body score that assessed animal health, was higher in the treatment compared to the control group (2.8 vs. 2.55). The better overall health condition observed after 20(OH)D3 treatment demonstrated as closer to optimal 3 BC score than for untreated animals and may be a useful paradigm describing the tolerability of novel compounds under investigation. Although the direct extrapolation of rodent models to human well-being is questioned by many investigators, it is worthy of consideration in designing novel preclinical studies.
1,25(OH)2D3, acting through the VDR, causes hypercalcemia leading to failure of multiple organs at doses as low as 0.1 μg/kg [38]. Also, 25(OH)D3 induces hypercalcemia in animals lacking the 1α-hydroxylase (CYP27B1 −/−knock-out) at relatively low doses [38]. 20(OH)D3 at a dose of 30 μg/kg did not cause side effects in our experiment whereas doses of 1,25(OH)2D3 and 25(OH)D3 300 and 30 times lower than this, respectively, caused hypercalcemic (0.1-3 μg/kg), as we previously reported [50]. The lack of side effects of 20(OH)D3 is consistent with our previous testing on mice which showed that 20(OH)D3 is non-calcemic at doses as high as 30-60 μg/kg [53, 54]. These studies identify 20(OH)D3 as an excellent therapeutic or adjuvant agent that is non-toxic and non-calcemic at least at 30 μg/kg.
It is accepted that the phenotypic activity of 1,25(OH)2D3 is mediated by its interaction with the VDR [31, 32, 75, 76] which can be defined as the canonical signal transduction pathway. Similarly, non-calcemic 20(OH)D3 can also activate the VDR on melanoma cells [48, 58, 77] causing downstream anti-proliferative and antitumorigenic effects. Previously, we have demonstrated that 20(OH)D3 and its metabolites can inhibit NF-κB signaling through an interaction with VDR in keratinocytes [78, 79] and melanoma cells [47]. Thus, 20(OH)D3 can inhibit melanoma growth through inhibition of NF-κB in tumor cells and/or by reduction of proinflammatory activity in the stroma. Recently we discoverd that 20(OH)D3 also act on retinoic acid orphan receptors (ROR) α and γ as a inverse agonist [80], and that these receptors are expressed in human melanomas [80, 81], suggesting their involvement in the inhibition of melanoma growth by 20(OH)D3 through a non-canonical pathway. The relative involvement of the VDR or RORα and γ in the inhibition of melanoma growth by 20(OH)D3 represents a challenging goal for future investigation. In this context, it must be noted that clinic-pathological studies have shown a correlation between changes in the expression of VDR or RORs with melanoma progression and also with overall and disease free survival time of the patients [9, 81, 82].
In conclusion, we provided in vitro and in vivo evidence that the CYP11A1-derived 20(OH)D3 is an excellent candidate for further testing as a primary or adjuvant therapeutic agent against human melanoma.
MATERIALS AND METHODS
Vitamin D3 hydroxyderivatives
1,25(OH)2D3 was obtained from Fluka Chemicals (Sigma-Aldrich, St. Louis, MO). 20(OH)D3 was produced by the enzymatic hydroxylation of vitamin D3 by CYP11A1 as described previously [40, 57]. The extracted product was purified by preparative thin-layer chromatography followed by reverse phase HPLC. The structure was confirmed by NMR as detailed in [40, 57]. The D3 hydroxyderivatives were dried and stored at –80oC until use.
Culture of the SKMel-188 melanoma cell line
A human melanoma line (SKMel-188) was obtained from Dr. Chakraborty from Yale University. Since then, it had been characterized and maintained in our laboratory [49, 83, 84]. This cell line was used for assays of plating efficiency, colony formation in soft agar, spheroid toxicity, and transwell migration. The line was grown at 37°C in a humidified atmosphere with 5% CO2. Ham’s F10 medium supplemented with glucose, L-glutamine, pyridoxine hydrochloride (Cellgrow, Manassas, VA), 5% charcoal-treated fetal bovine serum (CS-FBS) (Sigma, St. Louis, MO) and 1% penicillin/ streptomycin/amphotericin antibiotic solution (Sigma, St. Louis, MO) was used to culture the cells. In addition, human SKMEL-188 cells were transduced with a lentiviral luciferase (LUC) construct for live animal bioimaging analysis for tumor formation as described previously [85].
Plating efficiency of SKMel-188 cells
SKMel-188 cells were plated on a 6-well plate at a density of 14 cells/cm2 in Ham’s F10 medium supplemented with 5% CS-FBS. 1,25(OH)2D3 or 20(OH)D3 (or ethanol vehicle as a control) was added to each well every 72 h to a final concentration of 10-7 M. Each condition was tested at least in triplicate. After 14 days, colonies were fixed in 4% paraformaldehyde and stained with 2.3% crystal violet (Sigma-Aldrich, St. Louis, MO). Colonies were imaged using the Cytation 5 Cell Imaging Multi-Mode Reader and quantified using the Gen5 software (Biotek, Winooski, VT). The number of colony-forming units (CFU) was calculated for each condition using the following formula: CFU = 100[(number of colonies)/(number of cells plated)].
Colony formation in soft agar
The melanoma cells were trypsinized, re-suspended in medium containing 0.4% agarose (American Bioanalytical, Natick, MA) and 5% CS-FBS and seeded at 500 cells/well in a 0.8% agar layer on 24-well plates. 20(OH)D3 or 1,25(OH)2D3 (or ethanol vehicle as a control)was added to each well to final concentrations of 10-7 M, 10-8 M, or 10-9 M using 100 μL of media supplemented with 5% CS-FBS. Each condition was tested at least in duplicate. Every 72 h over 12 days, fresh 20(OH)D3or 1,25(OH)2D3 (or ethanol control) in 100 μL medium, was added. Subsequently, resulting colonies were stained with 0.5 mg/ml of 5-(dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent (Promega, Madison, WI), imaged using the Cytation 5 Cell Imaging Multi-Mode Reader in 3 different z-planes and scored using Gen5 software. The number of CFU was calculated for each condition.
Spheroid toxicity
The procedure followed was based on a more detailed protocol (1). Briefly, melanoma cells at 70-80% confluence were incubated with magnetic nanoparticles (NanoShuttle) containing gold, iron oxide, and poly-L-lysine (Nano3D Biosciences, Houston, TX) at a concentration of 50 pg/cell, overnight. This process allowed the cells to become magnetized. Subsequently, the cells were trypsinized, resuspended in medium at a concentration of 1.6 × 106 cells/mL (3.2 × 106 cells in 2 mL) and dispensed into an ultra-low attachment 6-well plate (Corning, Tewksbury, MA). A magnetic drive of 6 neodymium magnets (Nano3D Biosciences) was placed on top of the 6-well plate to allow the cells to levitate and to induce the formation of extracellular matrix (ECM). After 24 h, the levitated cultures were broken up using rigorous pipette action. Melanoma cells (50,000 cells/well) were then dispensed into an ultra-low attachment 96-well plate (Corning). A magnetic drive of 96 neodymium magnets was placed below the 96-well plate to print the cells into spheroids over 1 h. 1,25(OH)2D3 or 20(OH)D3 (or ethanol vehicle as a control) was added to each well at final concentrations of 10-7 or 10-8 M. Each condition was tested at least in triplicate. The magnet was subsequently removed to allow the spheroids to contract for 48 h. Images were automatically taken every hour on a mobile device, iPod touch 5th generation, 16 GB (Apple Computer, Cupertino, CA) using an application (Experimental Assistant, Nano3D Biosciences). The images were then analyzed to calculate the area of each spheroid using a custom analysis code in Python provided by Nano3D Biosciences.
Transwell migration assay
The HTS Transwell-24 System (Corning) was used to carry out the assay as per the manufacturer’s instructions. Serum-free Ham’s F10 medium was added to each well and to each transwell insert; the plate was incubated for 1 h to provide an initial equilibration period. The melanoma cells were harvested, resuspended in Ham’s F10 medium supplemented with 1% CS-FBS and 100 μL of the cell suspension was dispensed in an insert of the transwell plate (polycarbonate membrane, pore size=8.0 μm). Two separate conditions with varying cell densities, 10,000 cells/well and 100,000 cells/well, were tested. 20(OH)D3 or 1,25(OH)2D3 (or ethanol vehicle or medium as controls) was added to each insert to final concentrations of 10-7 M or 10-8 M. Six hundred μL of 10% CS-FBS were then added to the lower chamber to serve as a chemoattractant. After 24 h, a cotton-tipped applicator was used on top of the membrane to remove excess cells and media. The inserts were then washed in chilled PBS for 2 min, fixed with ice-cold methanol for 10 min and stained using 2.3% crystal violet. The inserts were then washed in distilled water twice for 5 min, air-dried and mounted using Permount (Fisher Scientific, Pittsburgh, PA). Each experiment condition was tested in duplicate. The slides were then imaged using the Cytation 5 Cell Imaging Multi-Mode Reader and the migrated cells were quantified using Gen5 software in 3 separate 10X fields in color bright field (6 separate fields/condition).
Animal experiments
All animal experiments were performed in accordance with a study protocol approved by the Institutional Animal Care and Use Committee of the University of Tennessee Health Science Center (UTHSC). Seven-week-old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratory) were placed on a vitamin D-deficient diet (TD.89123, Harlan Laboratories, Madison, WI). After two weeks, animals were randomly divided into 2 groups (n=10) and injected subcutaneously into the flanks with 1x106 luciferase-expressing SKMel-188 cells. Tumor initiation and progression were monitored twice a week after D-luciferin injection using the Xenogen IVIS (Perkin Elmer, Waltham, MA) bioimaging system as well as measured with a caliper. Tumor size, expressed as a total volume, was calculated using the formula: a x b x c, and was provided in mm3 [67] or as (geometric mean) ; where a, b and c represented length, width and depth, respectively [70, 86]. Once implanted tumors became palpable and reached 1 mm3 in size, the treatment was started with intraperitoneal injection of a matched volume of either 30 μg/day of 20(OH)D3 diluted in vehicle (25 % propylene glycol in distilled water) or vehicle, per animal. Ten doses in total, 5 days per week, were applied for 2 weeks. The body score (BS) scale, representing the overall health condition of animal, was performed bi-weekly [87]. Three days following the last treatment (day 21th), animals were sacrificed with CO2 followed by cervical dislocation and their organs were collected for macroscopic and microscopic evaluation.
Statistical analysis
All statistical analyses were performed using GraphPad Prism Version 6.0 (GraphPad Software, San Diego, CA). For the assays of colony formation in soft agar and plating efficiency, the digital data obtained from the Gen5 software were analyzed using a t-test, and an IC50 for each condition was calculated. For the transwell migration assay, the digital data obtained from the Gen5 software was analyzed using a t-test and a one-way ANOVA test with a post hoc Tukey’s test. For the spheroid toxicity assay, the area of each spheroid, directly measured using the images acquired from a mobile device, was plotted and analyzed using a one-tailed t-test. Differences were considered statistically significant when P < 0.05. The data are presented as mean ± standard error. The t-test was employed for comparing the measurement of in vivo tumor growth between the vehicle (control) and 20(OH)D3 treatment, and considered statistically significant at P < 0.05 (*), P < 0.01 (**), P < 0.001 (***), and P < 0.0001 (****).
CONFLICTS OF INTEREST
All authors declare no conflicts of interest.
GRANT SUPPORT
The work was supported in part by grants from the National Institutes of Health (2R01AR052190-A6, R21AR066505-01A1 and 1R01AR056666-01A2) to ATS, NIH R01CA133322, Department of Defense (W81XWH-11-1-0533) and the Muirhead Chair Endowment at the University of Tennessee Health Science Center to LMP), funding from Comprehensive Cancer Center, Cancer Chemoprevention Program and from VA grant 1I01BX003395-01 to ATS and from the University of Western Australia to RCT.
REFERENCES
1. Schadendorf D and Hauschild A. Melanoma in 2013: Melanoma[mdash]the run of success continues. Nat Rev Clin Oncol. 2014; 11:75-76.
2. Siegel RL, Miller KD and Jemal A. Cancer statistics, 2015. CA: A Cancer Journal for Clinicians. 2015; 65:5-29.
3. Jarrett SG, Novak M, Harris N, Merlino G, Slominski A and Kaetzel DM. NM23 deficiency promotes metastasis in a UV radiation-induced mouse model of human melanoma. Clin Exp Metastasis. 2013; 30:25-36.
4. Jarrett SG, Novak M, Dabernat S, Daniel JY, Mellon I, Zhang Q, Harris N, Ciesielski MJ, Fenstermaker RA, Kovacic D, Slominski A and Kaetzel DM. Metastasis suppressor NM23-H1 promotes repair of UV-induced DNA damage and suppresses UV-induced melanomagenesis. Cancer Res. 2012; 72:133-143.
5. Shepelin D, Korzinkin M, Vanyushina A, Aliper A, Borisov N, Vasilov R, Zhukov N, Sokov D, Prassolov V, Gaifullin N, Zhavoronkov A, Bhullar B and Buzdin A. Molecular pathway activation features linked with transition from normal skin to primary and metastatic melanomas in human. Oncotarget. 2016; 7:656-670. doi: 10.18632/oncotarget.6394.
6. Goldstein AM, Landi MT, Tsang S, Fraser MC, Munroe DJ and Tucker MA. Association of MC1R Variants and Risk of Melanoma in Melanoma-Prone Families with CDKN2A Mutations. Cancer Epidemiol Biomarkers Prev. 2005; 14:2208-2212.
7. Schadendorf D, Fisher DE, Garbe C, Gershenwald JE, Grob J-J, Halpern A, Herlyn M, Marchetti MA, McArthur G, Ribas A, Roesch A and Hauschild A. Melanoma. Nat Rev Dis Primers. 2015; 1:15003.
8. Brozyna AA, Jozwicki W, Janjetovic Z and Slominski AT. Expression of the vitamin D-activating enzyme 1alpha-hydroxylase (CYP27B1) decreases during melanoma progression. Hum Pathol. 2013; 44:374-387.
9. Brozyna AA, Jozwicki W and Slominski AT. Decreased VDR expression in cutaneous melanomas as marker of tumor progression: new data and analyses. Anticancer Res. 2014; 34:2735-2743.
10. Slominski AT, Brozyna A, Jozwicki W and Tuckey RC. Vitamin D as an adjuvant in melanoma therapy. Melanoma Manag. 2015; 2:1-4.
11. Zeljic K, Kandolf-Sekulovic L, Supic G, Pejovic J, Novakovic M, Mijuskovic Z and Magic Z. Melanoma risk is associated with vitamin D receptor gene polymorphisms. Melanoma Res. 2014; 24:273-279.
12. Newton-Bishop JA, Davies JR, Latheef F, Randerson-Moor J, Chan M, Gascoyne J, Waseem S, Haynes S, O’Donovan C and Bishop DT. 25-Hydroxyvitamin D2 /D3 levels and factors associated with systemic inflammation and melanoma survival in the Leeds Melanoma Cohort. Int J Cancer. 2015; 136:2890-2899.
13. Wyatt C, Lucas RM, Hurst C and Kimlin MG. Vitamin D deficiency at melanoma diagnosis is associated with higher Breslow thickness. PLoS One. 2015; 10:e0126394.
14. Reichrath J and Reichrath S. Sunlight, vitamin D and malignant melanoma: an update. Adv Exp Med Biol. 2014; 810:390-405.
15. Field S and Newton-Bishop JA. Melanoma and vitamin D. Mol Oncol. 2011; 5:197-214.
16. Wasif N, Bagaria SP, Ray P and Morton DL. Does metastasectomy improve survival in patients with stage IV melanoma? a cancer registry analysis of outcomes. J Surg Oncol. 2011; 104:111-115.
17. Seegenschmiedt MH, Keilholz L, Altendorf-Hofmann A, Urban A, Schell H, Hohenberger W and Sauer R. Palliative radiotherapy for recurrent and metastatic malignant melanoma: prognostic factors for tumor response and long-term outcome: a 20-year experience. Int J Radiat Oncol Biol Phys. 1999; 44:607-618.
18. Ribero S, Longo C, Glass D, Nathan P and Bataille V. What is new in melanoma genetics and treatment?. Dermatology. 2016.
19. Schadendorf D, Fisher DE, Garbe C, Gershenwald JE, Grob J-J, Halpern A, Herlyn M, Marchetti MA, McArthur G, Ribas A, Roesch A and Hauschild A. Melanoma. Nat Rev Dis Primers. 2015.
20. Lo JA and Fisher DE. The melanoma revolution: from UV carcinogenesis to a new era in therapeutics. Science. 2014; 346:945-949.
21. Schadendorf D and Hauschild A. Melanoma in 2013: Melanoma-the run of success continues. Nat Rev Clin Oncol. 2014.
22. Shah DJ and Dronca RS. Latest advances in chemotherapeutic, targeted, and immune approaches in the treatment of metastatic melanoma. Mayo Clin Proc. 2014; 89:504-519.
23. Kwong LN and Davies MA. Targeted therapy for melanoma: rational combinatorial approaches. Oncogene. 2014; 33:1-9.
24. Rajkumar S and Watson IR. Molecular characterisation of cutaneous melanoma: creating a framework for targeted and immune therapies. Br J Cancer. 2016; 115:145-155.
25. Nikolaou V and Stratigos AJ. Emerging trends in the epidemiology of melanoma. Br J Dermatol. 2014; 170:11-19.
26. Slominski AT and Carlson JA. Melanoma resistance: a bright future for academicians and a challenge for patient advocates. Mayo Clinic proceedings. 2014; 89:429-433.
27. Holick MF. Vitamin D: A millenium perspective. J Cell Biochem. 2003; 88:296-307.
28. Holick MF. Vitamin D deficiency. N Engl J Med. 2007; 357:266-281.
29. Bikle DD. Vitamin D metabolism and function in the skin. Mol Cell Endocrinol. 2011; 347:80-89.
30. Bikle DD. Vitamin D: an ancient hormone. Exp Dermatol. 2011; 20:7-13.
31. Christakos S, Dhawan P, Verstuyf A, Verlinden L and Carmeliet G. Vitamin D: metabolism, molecular mechanism of action, and pleiotropic effects. Physiol Rev. 2016; 96:365-408.
32. Bouillon R, Carmeliet G, Verlinden L, van Etten E, Verstuyf A, Luderer HF, Lieben L, Mathieu C and Demay M. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev. 2008; 29:726-776.
33. Bikle DD, Elalieh H, Welsh J, Oh D, Cleaver J and Teichert A. Protective role of vitamin D signaling in skin cancer formation. J Steroid Biochem Mol Biol. 2013; 136:271-279.
34. Mason RS and Reichrath J. Sunlight vitamin D and skin cancer. Anticancer Agents Med Chem. 2013; 13:83-97.
35. Feldman D, Krishnan AV, Swami S, Giovannucci E and Feldman BJ. The role of vitamin D in reducing cancer risk and progression. Nat Rev Cancer. 2014; 14:342-357.
36. Grant WB. Roles of Solar UVB and Vitamin D in Reducing Cancer Risk and Increasing Survival. Anticancer Res. 2016; 36:1357-1370.
37. Saw RPM, Armstrong BK, Mason RS, Morton RL, Shannon KF, Spillane AJ, Stretch JR and Thompson JF. Adjuvant therapy with high dose vitamin D following primary treatment of melanoma at high risk of recurrence: a placebo controlled randomised phase II trial (ANZMTG 02.09 Mel-D). BMC Cancer. 2014; 14:780.
38. Deluca HF, Prahl JM and Plum LA. 1,25-Dihydroxyvitamin D is not responsible for toxicity caused by vitamin D or 25-hydroxyvitamin D. Arch Biochem Biophys. 2011; 505:226-230.
39. Slominski A, Zjawiony J, Wortsman J, Semak I, Stewart J, Pisarchik A, Sweatman T, Marcos J, Dunbar C and Tuckey RC. A novel pathway for sequential transformation of 7-dehydrocholesterol and expression of the P450scc system in mammalian skin. Eur J Biochem. 2004; 271:4178-4188.
40. Tuckey RC, Li W, Zjawiony JK, Zmijewski MA, Nguyen MN, Sweatman T, Miller D and Slominski A. Pathways and products for the metabolism of vitamin D3 by cytochrome P450scc. FEBS J. 2008; 275:2585-2596.
41. Tuckey RC, Li W, Shehabi HZ, Janjetovic Z, Nguyen MN, Kim TK, Chen J, Howell DE, Benson HA, Sweatman T, Baldisseri DM and Slominski A. Production of 22-hydroxy metabolites of vitamin D3 by cytochrome p450scc (CYP11A1) and analysis of their biological activities on skin cells. Drug Metab Dispos. 2011; 39:1577-1588.
42. Slominski AT, Li W, Kim TK, Semak I, Wang J, Zjawiony JK and Tuckey RC. Novel activities of CYP11A1 and their potential physiological significance. J Steroid Biochem Mol Biol. 2015; 151:25-37.
43. Guryev O, Carvalho RA, Usanov S, Gilep A and Estabrook RW. A pathway for the metabolism of vitamin D3: unique hydroxylated metabolites formed during catalysis with cytochrome P450scc (CYP11A1). Proc Natl Acad Sci U S A. 2003; 100:14754-14759.
44. Slominski AT, Kim TK, Li W, Postlethwaite A, Tieu EW, Tang EK and Tuckey RC. Detection of novel CYP11A1-derived secosteroids in the human epidermis and serum and pig adrenal gland. Sci Rep. 2015; 5:14875.
45. Slominski AT, Kim TK, Shehabi HZ, Semak I, Tang EK, Nguyen MN, Benson HA, Korik E, Janjetovic Z, Chen J, Yates CR, Postlethwaite A, Li W and Tuckey RC. In vivo evidence for a novel pathway of vitamin D(3) metabolism initiated by P450scc and modified by CYP27B1. FASEB J. 2012; 26:3901-3915.
46. Slominski AT, Kim TK, Li W and Tuckey RC. Classical and non-classical metabolic transformation of vitamin D in dermal fibroblasts. Exp Dermatol. 2016; 25:231-232.
47. Janjetovic Z, Brozyna AA, Tuckey RC, Kim TK, Nguyen MN, Jozwicki W, Pfeffer SR, Pfeffer LM and Slominski AT. High basal NF-kappaB activity in nonpigmented melanoma cells is associated with an enhanced sensitivity to vitamin D3 derivatives. Br J Cancer. 2011; 105:1874-1884.
48. Slominski AT, Kim TK, Janjetovic Z, Tuckey RC, Bieniek R, Yue J, Li W, Chen J, Nguyen MN, Tang EK, Miller D, Chen TC and Holick M. 20-Hydroxyvitamin D2 is a noncalcemic analog of vitamin D with potent antiproliferative and prodifferentiation activities in normal and malignant cells. Am J Physiol Cell Physiol. 2011; 300:C526-541.
49. Slominski AT, Janjetovic Z, Kim TK, Wright AC, Grese LN, Riney SJ, Nguyen MN and Tuckey RC. Novel vitamin D hydroxyderivatives inhibit melanoma growth and show differential effects on normal melanocytes. Anticancer Res. 2012; 32:3733-3742.
50. Slominski AT, Janjetovic Z, Fuller BE, Zmijewski MA, Tuckey RC, Nguyen MN, Sweatman T, Li W, Zjawiony J, Miller D, Chen TC, Lozanski G and Holick MF. Products of vitamin D3 or 7-dehydrocholesterol metabolism by cytochrome P450scc show anti-leukemia effects, having low or absent calcemic activity. PLoS One. 2010; 5:e9907.
51. Wasiewicz T, Szyszka P, Cichorek M, Janjetovic Z, Tuckey RC, Slominski AT and Zmijewski MA. Antitumor effects of vitamin D analogs on hamster and mouse melanoma cell lines in relation to melanin pigmentation. Int J Mol Sci. 2015; 16:6645-6667.
52. Wierzbicka JM, Binek A, Ahrends T, Nowacka JD, Szydlowska A, Turczyk L, Wasiewicz T, Wierzbicki PM, Sadej R, Tuckey RC, Slominski AT, Chybicki J, Adrych K, Kmiec Z and Zmijewski MA. Differential antitumor effects of vitamin D analogues on colorectal carcinoma in culture. Int J Oncol. 2015; 47:1084-1096.
53. Wang J, Slominski A, Tuckey RC, Janjetovic Z, Kulkarni A, Chen J, Postlethwaite AE, Miller D and Li W. 20-hydroxyvitamin D(3) inhibits proliferation of cancer cells with high efficacy while being non-toxic. Anticancer Res. 2012; 32:739-746.
54. Chen J, Wang J, Kim TK, Tieu EW, Tang EK, Lin Z, Kovacic D, Miller DD, Postlethwaite A, Tuckey RC, Slominski AT and Li W. Novel vitamin D analogs as potential therapeutics: metabolism, toxicity profiling, and antiproliferative activity. Anticancer Res. 2014; 34:2153-2163.
55. Tieu EW, Tang EK, Chen J, Li W, Nguyen MN, Janjetovic Z, Slominski A and Tuckey RC. Rat CYP24A1 acts on 20-hydroxyvitamin D3 producing hydroxylated products with increased biological activity. Biochem Pharmacol. 2012; 84:1696-1704.
56. Tseng H, Gage JA, Shen T, Haisler WL, Neeley SK, Shiao S, Chen J, Desai PK, Liao A, Hebel C, Raphael RM, Becker JL and Souza GR. A spheroid toxicity assay using magnetic 3D bioprinting and real-time mobile device-based imaging. Sci Rep. 2015; 5:13987.
57. Slominski A, Semak I, Zjawiony J, Wortsman J, Li W, Szczesniewski A and Tuckey RC. The cytochrome P450scc system opens an alternate pathway of vitamin D3 metabolism. FEBS J. 2005; 272:4080-4090.
58. Slominski AT, Kim TK, Li W, Yi AK, Postlethwaite A and Tuckey RC. The role of CYP11A1 in the production of vitamin D metabolites and their role in the regulation of epidermal functions. J Steroid Biochem Mol Biol. 2014; 144 Pt A:28-39.
59. Justus CR, Leffler N, Ruiz-Echevarria M and Yang LV. In vitro cell migration and invasion assays. J Vis Exp. 2014;
60. Ruddon RW. (2007). Cancer biology: Oxford University Press.
61. Okegawa T, Pong RC, Li Y and Hsieh JT. The role of cell adhesion molecule in cancer progression and its application in cancer therapy. Acta Biochim Pol. 2004; 51:445-457.
62. Yamamoto M, Zhao M, Hiroshima Y, Zhang Y, Shurell E, Eilber FC, Bouvet M, Noda M and Hoffman RM. Efficacy of Tumor-Targeting Salmonella A1-R on a Melanoma Patient-Derived Orthotopic Xenograft (PDOX) Nude-Mouse Model. PLoS One. 2016; 11:e0160882.
63. Pollack VA, Alvarez E, Tse KF, Torgov MY, Xie S, Shenoy SG, MacDougall JR, Arrol S, Zhong H, Gerwien RW, Hahne WF, Senter PD, Jeffers ME, Lichenstein HS and LaRochelle WJ. Treatment parameters modulating regression of human melanoma xenografts by an antibody-drug conjugate (CR011-vcMMAE) targeting GPNMB. Cancer Chemother Pharmacol. 2007; 60:423-435.
64. Overwijk WW and Restifo NP. B16 as a mouse model for human melanoma. Curr Protoc Immunol. 2001; Chapter 20:Unit 20 21.
65. Hoffman RM. Orthotopic metastatic mouse models for anticancer drug discovery and evaluation: a bridge to the clinic. Invest New Drugs. 1999; 17:343-359.
66. Hoffman RM. Patient-derived orthotopic xenografts: better mimic of metastasis than subcutaneous xenografts. Nat Rev Cancer. 2015; 15:451-452.
67. Gerlee P. The model muddle: in search of tumor growth laws. Cancer Res. 2013; 73:2407-2411.
68. Dietrich A, Kraus K, Brinckmann U, Friedrich T, Muller A, Liebert UG and Schonfelder M. Complex cancer gene therapy in mice melanoma. Langenbecks Arch Surg. 2002; 387:177-182.
69. Perez-Torres A, Vera-Aguilera J, Hernaiz-Leonardo JC, Moreno-Aguilera E, Monteverde-Suarez D, Vera-Aguilera C and Estrada-Barcenas D. The synthetic parasite-derived peptide GK1 increases survival in a preclinical mouse melanoma model. Cancer Biother Radiopharm. 2013; 28:682-690.
70. Schrek R. Further Quantitative Methods for the Study of Transplantable Tumors. The Growth of R39 Sarcoma and Brown-Pearce Carcinoma. The American Journal of Cancer. 1936; 28:345-363.
71. Yang M, Jiang P, An Z, Baranov E, Li L, Hasegawa S, Al-Tuwaijri M, Chishima T, Shimada H, Moossa AR and Hoffman RM. Genetically fluorescent melanoma bone and organ metastasis models. Clin Cancer Res. 1999; 5:3549-3559.
72. Hoffman RM. The multiple uses of fluorescent proteins to visualize cancer in vivo. Nat Rev Cancer. 2005; 5:796-806.
73. Hoffman RM and Yang M. Subcellular imaging in the live mouse. Nat Protoc. 2006; 1:775-782.
74. Tsai MH, Aki R, Amoh Y, Hoffman RM, Katsuoka K, Kimura H, Lee C and Chang CH. GFP-fluorescence-guided UVC irradiation inhibits melanoma growth and angiogenesis in nude mice. Anticancer Res. 2010; 30:3291-3294.
75. Bikle DD. Vitamin D receptor, a tumor suppressor in skin. Can J Physiol Pharmacol. 2015; 93:349-354.
76. Carlberg C. What do we learn from the genome-wide perspective on vitamin D3? Anticancer Res. 2015; 35:1143-1151.
77. Kim TK, Wang J, Janjetovic Z, Chen J, Tuckey RC, Nguyen MN, Tang EK, Miller D, Li W and Slominski AT. Correlation between secosteroid-induced vitamin D receptor activity in melanoma cells and computer-modeled receptor binding strength. Mol Cell Endocrinol. 2012; 361:143-152.
78. Janjetovic Z, Zmijewski MA, Tuckey RC, DeLeon DA, Nguyen MN, Pfeffer LM and Slominski AT. 20-Hydroxycholecalciferol, product of vitamin D3 hydroxylation by P450scc, decreases NF-kappaB activity by increasing IkappaB alpha levels in human keratinocytes. PLoS One. 2009; 4:e5988.
79. Janjetovic Z, Tuckey RC, Nguyen MN, Thorpe EM, Jr. and Slominski AT. 20,23-dihydroxyvitamin D3, novel P450scc product, stimulates differentiation and inhibits proliferation and NF-kappaB activity in human keratinocytes. J Cell Physiol. 2010; 223:36-48.
80. Slominski AT, Kim TK, Takeda Y, Janjetovic Z, Brozyna AA, Skobowiat C, Wang J, Postlethwaite A, Li W, Tuckey RC and Jetten AM. RORalpha and ROR gamma are expressed in human skin and serve as receptors for endogenously produced noncalcemic 20-hydroxy- and 20,23-dihydroxyvitamin D. FASEB J. 2014; 28:2775-2789.
81. Brozyna AA, Jozwicki W, Skobowiat C, Jetten A and Slominski AT. RORalpha and RORgamma expression inversely correlates with human melanoma progression. Oncotarget. 2016; 7:63261-63282. doi: 10.18632/oncotarget.11211.
82. Brozyna AA, Jozwicki W, Janjetovic Z and Slominski AT. Expression of vitamin D receptor decreases during progression of pigmented skin lesions. Hum Pathol. 2011; 42:618-631.
83. Chakraborty AK, Funasaka Y, Slominski A, Ermak G, Hwang J, Pawelek JM and Ichihashi M. Production and release of proopiomelanocortin (POMC) derived peptides by human melanocytes and keratinocytes in culture: regulation by ultraviolet B. Biochim Biophys Acta. 1996; 1313:130-138.
84. Slominski A, Ermak G and Wortsman J. Modification of melanogenesis in cultured human melanoma cells. In Vitro Cell Dev Biol Anim. 1999; 35:564-565.
85. Yang CH, Yue J, Sims M and Pfeffer LM. The curcumin analog EF24 targets NF-kappaB and miRNA-21, and has potent anticancer activity in vitro and in vivo. PLoS One. 2013; 8:e71130.
86. Scislowski PW, Slominski A and Bomirski A. Biochemical characterization of three hamster melanoma variants--II. Glycolysis and oxygen consumption. Int J Biochem. 1984; 16:327-331.
87. Ullman-Cullere MH and Foltz CJ. Body condition scoring: a rapid and accurate method for assessing health status in mice. Lab Anim Sci. 1999; 49:319-323.